

Abstract
The main purpose of the present study was to investigate the effects of the neuroprotective agent riluzole on the electrically evoked release of [3H]-glutamate ([3H]-Glu) in mouse neocortical slices. The reported selectivity of riluzole for excitatory amino acids was tested in release experiments with further neurotransmitters. Also distinct species, mouse, rat and man were compared.
[3H]-Glu was formed endogenously during incubation of slices with [3H]-glutamine ([3H]-Gln). Released [3H]-Glu and tissue [3H]-Glu was separated by anion exchange chromatography. Electrically evoked [3H]-Glu release was strongly diminished by tetrodotoxin (TTX) and Ca2+-withdrawal.
Riluzole (100 μM) depressed the release of [3H]-Glu up to 77% (IC50=19.5 μM). Riluzole was also able to inhibit strongly the electrically evoked release of [3H]-acetylcholine ([3H]-ACh) (at 100 μM by 92%, IC50=3.3 μM, and [3H]-dopamine ([3H]-DA) (at 32 μM by 72%, IC50=6.8 μM). However, the release of [3H]-serotonin ([3H]-5-HT) was less diminished (at 100 μM by 53%, IC50=39.8 μM). Riluzole up to 100 μM did not affect [3H]-noradrenaline ([3H]-NA) release.
Between species, i.e. in mouse, rat and human neocortex, no significant differences between the effects of riluzole could be observed.
The NMDA-receptor blocker MK-801 (1 μM) and the AMPA/Kainate-receptor blocker NBQX (1 μM) did neither affect the electrically evoked [3H]-ACh release nor its inhibition by riluzole, indicating that effects of riluzole on transmitter release were neither due to modulation of ionotropic Glu receptors, nor due to indirect inhibition of Glu release through these receptors.
Taken together, riluzole inhibits the release of distinct neurotransmitters differently, but is not selective for the excitatory amino acid Glu.
Keywords: Superfusion, neurotransmitter release, riluzole, human, rat, mouse neocortex
Introduction
Riluzole (2 - amino - 6 - [trifluoromethoxy] - benzothiazole) is known as a neuroprotective agent with anticonvulsive, anxiolytic and anaesthetic properties (Doble, 1996). Clinical trials have shown that riluzole prolongs the life of patients with amyotrophic lateral sclerosis (ALS), a rapidly progressive and fatal disease in which both upper and lower motoneurons degenerate (Lacomblez et al., 1996; Bensimon et al., 1994). As a result of these studies, riluzole has been approved and marketed for the treatment of ALS in many countries. In addition, it proved effective in animal models of Parkinson's disease (Barneoud et al., 1996), Chorea Huntington (Mary et al., 1995) and cerebral and retinal ischaemia (Malgouris et al., 1989; Pratt et al., 1992; Lagreze et al., 1999).
According to the literature, these effects of riluzole are due to it's ability to diminish excitatory neurotransmission. Riluzole has often been outlined as an antiGlu substance, a Glu release inhibitor and an antagonist of Glu neurotransmission (Benavides et al., 1985; Malgouris et al., 1989; Martin et al., 1993; for review see Doble, 1996). In vitro, riluzole inhibited K+-evoked release of endogenous Glu and aspartate from hippocampal slices (Martin et al., 1993). In vivo, riluzole antagonized the spontaneous release of Glu (Chramy et al., 1992), as well as the Glu-induced DA release from the rat caudate nucleus (Chramy et al., 1986). Although riluzole did not bind to any of the known recognition sites of Glu receptors, it was shown to attenuate cation influx initiated by NMDA and kainate (Debono et al., 1993; Hubert et al., 1994).
Obrenovitch & Urenjak (1998) raised the question, which kind of Glu release may be modulated by riluzole, and whether it inhibits Glu release as a Na+-channel blocker selectively or not. Our aims were (1) to determine whether riluzole does also inhibit the electrically evoked release of [3H]-Glu; (2) to compare its effects on [3H]-Glu release with those on other neurotransmitters: i.e. [3H]-ACh, [3H]-NA, [3H]-DA and [3H]-5-HT; and (3) to gain information about the efficacy of riluzole in human neocortex in comparison to mouse and rat brain tissue.
Methods
Tissue preparation
Six black mice (25–30 g) and Wistar rats (250–350 g) were decapitated, brains were quickly removed and hemispheres were placed in cold oxygenated physiological buffer. Composition of the buffer was (mM): NaCl 121, KCl 1.8, CaCl2 1.3, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, Glucose 11, pH 7.4. When experiments were performed in Ca2+-free medium, ethylenediaminetetraacetic acid (EDTA, 100 μM) was added to the superfusion buffer. Tissue removal, dissection and slicing were accomplished rapidly and the tissue was placed in cold buffer between steps. All efforts were made to minimize animal suffering, to reduce the number of animals used, according to the obtained licence of the local ethical committee on animal care.
Specimens of human neocortical tissue were obtained from patients undergoing surgical treatment of epilepsy. The procedure was approved by the local Ethical Committee of the University of Freiburg. The tissue was instantly immersed in ice-cold oxygen-saturated buffer and processed immediately. The patients obtained antiepileptic drugs (lamotrigine and carbamazepine) or the psychopharmacon clomipramine which were stopped 1 day before surgery. Mannit and dexamethasone was applied before neurosurgery if signs of elevated intracranial pressure were present. After premedication with flunitrazepam, anaesthesia was performed with thiopental, fentanyl or flunitrazepam. To obtain muscular relaxation pancuronium was used. These drugs given were assumed to be washed out of the slices and not to affect the superfusion experiments.
Slices of mice neocortex (300 μm-thick) were washed three times in 5 ml cold oxygen-saturated buffer and then incubated with either [3H]-glutamine, [3H]-choline, [3H]-NA or [3H]-5-HT. Slices of mice caudatoputamen (300 μm-thick) washed as described above, were incubated with [3H]-DA. Rat and human neocortex slices, 350 μm-thick and washed as described, were incubated in [3H]-choline.
[3H]-Glu release
Slices were incubated in 1 ml buffer containing 2 μM [3H]-Gln and the Glu uptake blocker L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC, 3 μM) for 60 min at 37°C. Then they were rinsed and transferred (two slices per chamber) to superfusion chambers (dead volume 100 μl) and superfused at 22°C at a flow rate of 0.4 ml per minute. After preperfusion for 50 min, 2 ml per 5 min fractions of superfusion fluid were collected and mixed with 13 ml H2O. [3H]-Glu release was stimulated twice with a series of rectangular, unipolar pulses at 65 (S1) and 105 (S2) min (parameters: 28 mA, 2 ms, 3 Hz, 240 pulses). After superfusion the slices were homogenized by sonification in 1 ml H2O. This homogenate was centrifuged at 10,000 r.p.m. for 5 min and the supernatant was mixed with 14 ml H2O. [3H]-Glu was separated from [3H]-Gln and other [3H]-labelled metabolites, mainly [3H]-GABA, by anion exchange chromatography using Amberlite IRA-67 resin. Chromatography columns (6×50 mm) were charged with HCO3−-loaded Amberlite IRA-67. Columns were washed with 30 ml H2O and [3H]-Glu was eluted in three steps each covering 3 ml of 1 M NaCl. Eluats of both slices and superfusion samples were mixed with 10 ml of liquid scintillation cocktail (Ultima Gold XR®) and quantified by liquid scintillation counting.
Validation of anion exchange chromatography
Separation of Glu by anion exchange chromatography was validated by high performance liquid chromatography (HPLC) and also by use of [3H]-labelled Glu, Gln and GABA and liquid scintillation counting. HPLC was performed with precolumn derivatization and gradient elution using a modification of the methods of Graser and Godel (Graser et al., 1985; Godel et al., 1984). Glu, Gln and GABA were mixed in a concentration of 20 μM each and separated by anion exchange chromatography as described above. Each fraction (sample application, rinsing and eluation step) was collected and analysed by HPLC. [3H]-labelled compounds were applied separately and collected fractions were quantified by liquid scintillation counting.
[3H]-ACh release
Slices were incubated for 30 min in buffer containing 0.1 μM [3H]-choline at 37°C. Subsequently they were rinsed, transferred to superfusion chambers (one slice in each) and superfused with buffer at a rate of 0.4 ml min−1. Hemicholinium-3 (10 μM) was added in order to inhibit uptake of [3H]-choline. After 60 min of preperfusion, the superfusate was collected in 2 ml samples. At 75 min (S1), 105 min (S2), 135 min (S3) and 165 min (S4) slices were depolarized by electrical pulses (depending on the experimental question, parameters were either 28 mA, 2 ms, 3 Hz, 240 pulses, 22°C or 60 mA, 2 ms, 3 Hz, 90 pulses, 37°C).
It has been shown earlier that the depolarization-evoked [3H]-overflow from brain slices incubated with [3H]-choline represents the transmitter [3H]-ACh (Richardson & Szerb, 1974; Feuerstein et al., 1998).
[3H]-NA release
Slices were incubated for 30 min in buffer containing 0.1 μM [3H]-NA at 37°C and L(+)-ascorbic acid (0.1 g l−1). In the superfusion fluid, oxaprotiline (1 μM) was additionally present in order to inhibit uptake of [3H]-NA. At 75 min (S1), 105 min (S2), 135 min (S3) and 165 min (S4) slices were depolarized by electrical pulses (depending on the experimental question, parameters were either 28 mA, 2 ms, 3 Hz, 240 pulses, 22°C; 60 mA, 2 ms, 3 Hz, 90 pulses, 37°C or 60 mA, 2 ms, 100 Hz, 4 pulses, 37°C).
[3H]-DA release
Slices were incubated for 30 min in buffer containing 0.1 μM [3H]-DA at 37°C and L(+)-ascorbic acid (0.1 g l−1). In the superfusion fluid, nomifensine (3.2 μM) was additionally present in order to inhibit uptake of [3H]-DA. At 75 min (S1), 105 min (S2) and 135 min (S3) slices were depolarized by electrical pulses (parameters: 28 mA, 2 ms, 3 Hz, 240 pulses, 22°C).
[3H]-5-HT release
Slices were incubated for 60 min in buffer containing 0.1 μM [3H]-5-HT at 37°C and L(+)-ascorbic acid (0.1 g l−1). In the superfusion fluid, fluvoxamine (1 μM) was additionally present in order to inhibit uptake of [3H]-5-HT. After 60 min of preperfusion, the superfusate was collected in 2 ml samples. At 75 min (S1), 105 min (S2) and 135 min (S3) slices were depolarized by electrical pulses (parameters: 28 mA, 2 ms, 3 Hz, 240 pulses, 22°C).
In all experiments, except those with [3H]-Glu, the slices were removed from the chambers after superfusion and solubilized in 0.5 ml of Soluene®. The solubilized slices were mixed with 10 ml, the superfusate fractions with 3 ml, of liquid scintillation cocktail (Ultima Gold®), and tritium content was determined by liquid scintillation counting.
Calculation
The outflow of tritiated compounds was expressed as fractional rate, i.e. the radioactivity of the superfusates was divided by the radioactivity in the tissue at the beginning of the corresponding collection period. Effects of drugs or Ca2+-withdrawal on basal [3H]-outflow were evaluated by the ratio of the fractional rate before the second, third and fourth stimulation (b2, b3, b4) and that before the first stimulation (b1). The bX/b1 ratios were then compared between treatments and controls using the 95% confidence intervals (CI95) of the bX/b1 ratios (see legend to Table 3). The stimulation-evoked overflow of tritium of the first (S1) and the following (SX) stimulations are calculated by subtracting the basal radioactivity from the total radioactivity in the stimulation fractions (basal outflow was assumed to decline linearly). The difference was expressed as percentage of the radioactivity in the slice at the beginning of the respective stimulation period. Drugs to be tested were given 15 min before SX; their effects were evaluated by comparing the ratios SX/S1. Control experiments were always run in parallel to drug experiments. The SX/S1 ratios were normalized by dividing each individual ratio by the mean ratio of the corresponding control stimulation. All results were expressed as means with CI95s.
Table 3.
Effects of riluzole, TTX and Ca2+-withdrawal on the basal, unstimulated [3H]-transmitter outflow
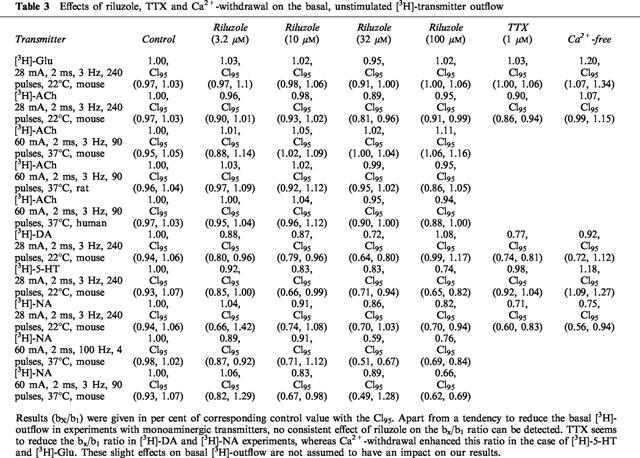
Concentration-inhibition curves of riluzole for each transmitter (except [3H]-NA) were evaluated using the logistic function, SX/S1=Imax*10Lril/(10−pIC50+10Lril) with Lril=logarithm of the applied concentration of riluzole (independent variable) and the individual SX/S1 ratios (dependent variable), yielding the parameter estimates Imax=maximum inhibition of [3H]-transmitter release due to riluzole and pIC50=negative logarithm of the riluzole concentration at half-maximum inhibition (Feuerstein & Limberger, 1999).
Materials
Soluene® and Ultima Gold® (XR) were obtained from Packard, Frankfurt, Germany. [3H]-glutamine L-[3.4-3H(N)] (49.9 Ci mmol−1) was purchased from NEN, Du Pont, Bad Homburg, Germany; [methyl-3H]-choline chloride (81 Ci mmol−1); [7.8-3H]-dopamine (47.0 Ci mmol−1); [3H]-noradrenaline (55 Ci mmol−1) from Amersham Buchler, Braunschweig, Germany; hydroxytryptamine binoxalate 5-[1.2-3H] and TTX from Bio Trend, Köln, Germany (27.5 Ci mmol−1); (+)-oxaprotilin HCl from Novartis Pharma, Nürnberg, Germany; fluvoxamine maleate from Solvay, Hannover, Germany; EDTA, nomifensine and hemicholinium from Sigma, Deisenhofen, Germany and PDC, MK 801 maleate and NBQX from Tocris, Cookson LTD, Bristol, U.K. Riluzole was a gift from Rhône-Poulenc-Rorer, Köln, Germany.
Results
The major focus of the present study was the effect of riluzole on electrically evoked [3H]-Glu release in mouse neocortex. As a prerequisite of this, [3H]-Glu was successfully separated from other [3H]-compounds by anion exchange chromatography, which was validated HPLC and also by use of [3H]-labelled Glu, Gln and GABA (Table 1). For comparison with glutamate experiments, other neurotransmitters and other species, rat and human neocortex, were used.
Table 1.
Validation of the anion exchange chromatography

Effect of TTX, Ca2+-withdrawal and riluzole on [3H]-Glu release
In mouse neocortical slices, electrical field stimulation (2 ms, 28 mA, 240 pulses, 3 Hz, 22°C) evoked [3H]-Glu efflux beyond basal levels (Figure 1). The mean of S1 was 0.65% of tissue [3H]-Glu, CI95 (0.58%, 0.72%). Resulting S2/S1 ratios of control experiments amounted to 0.74, CI95 (0.64, 0.85), to be compared with ratios obtained under drug treatment. In order to prove the physiological relevance of the electrical stimulation paradigm used, the modulation of [3H]-Glu release by the Na+-channel blocker TTX at 1 μM, and by withdrawal of Ca2+ was investigated. TTX and Ca2+ withdrawal strongly reduced the electrically evoked release (Figure 2a). The basal [3H]-Glu efflux was neither affected by TTX nor by riluzole at the concentrations used. Withdrawal of Ca2+, however, led to a slight increase of basal [3H]-Glu efflux (Table 3).
Figure 1.
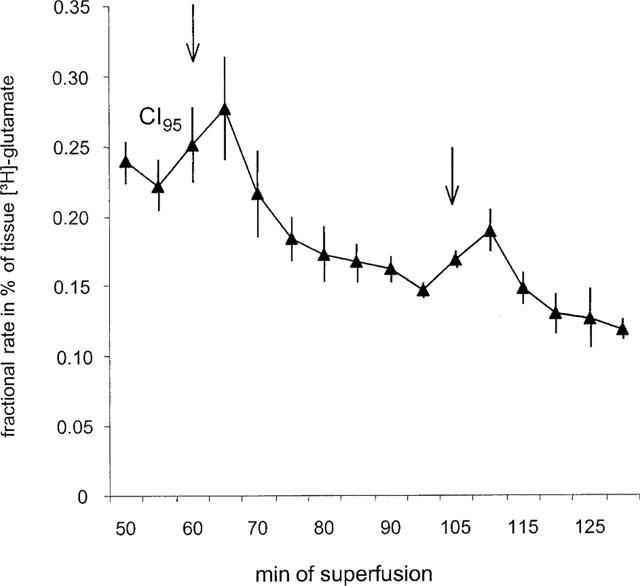
The electrically evoked [3H]-Glu release as fractional rates (see Methods) after [3H]-Gln incubation of mouse neocortex slices. Each value is the mean value of nine controls with corresponding CI95s. Series of electrical pulses (parameters: 28 mA, 2 ms, 3 Hz, 240 pulses, at 22°C) were applied at 60 and 105 min. The Glu uptake inhibitor PDC (3 μM) was present throughout the experiment.
Figure 2.

(a) Effects of TTX (1 μM) and Ca2+-free medium on electrically evoked neurotransmitter release of mouse neocortex. Stimulation was induced by the following parameters: 28 mA, 2 ms, 3 Hz, 240 pulses, at 22°C. Columns represent mean SX/S1 values in per cent of corresponding controls with CI95s. TTX or Ca2+ withdrawal was applied from 15 min before the second stimulation onwards (n=7–10 per column). The S1 as well as the control SX/S1 values are given in the text. (b) Effects of riluzole on electrically evoked neurotransmitter release in mouse neocortex. Stimulation was induced by the following parameters: 28 mA, 2 ms, 3 Hz, 240 pulses, at 22°C. Columns represent mean SX/S1 values in per cent of corresponding controls (0 μM riluzole) with CI95s. Riluzole was given (at increasing concentrations during S2, S3 and S4 in the case of [3H]-ACh, [3H]-DA, [3H]-NA and [3H]-5-HT) from 15 min before the second stimulation onwards (n=6–20 per column). Since at least three controls were running in each superfusion experiment the number of controls accumulated was up to 20 per column whereas the number of drug experiments was only 6–10 per column. The S1 as well as the control SX/S1 values are given in the text.
[3H]-Glu release was diminished by riluzole in a concentration-dependent fashion as shown in Figure 2b. Nonlinear regression analysis using the logistic function given in Methods yielded a pIC50 value of 4.71, CI95 (4.25, 5.18), and an Imax estimate of 93.4%, CI95 (68.5%, 136.1%).
Effect of TTX, Ca2+-withdrawal and riluzole on [3H]-ACh release, [3H]-NA, [3H]-5-HT, [3H]-DA in mouse neocortex and caudatoputamen
To compare effects of riluzole in different transmitter systems, we needed to use the same experimental parameters for each investigated transmitter. Therefore, the experimental conditions of release were based on the parameters used for [3H]-Glu experiments. Electrical stimulation using these parameters (2 ms, 28 mA, 240 pulses, 3 Hz, 22°C) increased [3H]-efflux from mouse neocortical slices prelabeled with [3H]-choline. S1 was 1.54% of tissue tritium, CI95 (1.41%, 1.66%); in controls the S2/S1 ratio was 1.05, CI95 (0.87, 1.24), S3/S1 was 1.33, CI95 (1.18, 1.47), and S4/S1 was 1.43, CI95 (1.23, 1.62).
Under the same parameters the mean S1 value of electrically evoked [3H]-NA was 7.94% of tissue tritium, CI95 (7.27%, 8.60%); in controls the S2/S1 ratio was 0.95, CI95 (0.92, 0.99), S3/S1 was 0.93, CI95 (0.88, 0.98), and S4/S1 was 0.90, CI95 (0.85, 0.95).
Under the same parameters the mean S1 value of electrically evoked [3H]-5-HT was 2.09% of tissue tritium, CI95 (1.75%, 2.44%); in controls S2/S1 was 0.70, CI95 (0.60, 0.79) and S3/S1 was 0.68, CI95 (0.57, 0.80).
Under the same parameters the mean S1 value of electrically evoked [3H]-DA was 3.14% of tissue tritium, CI95 (2.72%, 3.58%); in controls the S2/S1 ratio 0.76, CI95 (0.72, 0.81) and S3/S1 was 0.64, CI95 (0.60, 0.69).
TTX and Ca2+ withdrawal abolished the electrically evoked release of [3H]-ACh, [3H]-NA, [3H]-5-HT, and [3H]-DA (Figure 2a). Figure 2b shows that riluzole concentration-dependently diminished the release of [3H]-ACh and [3H]-DA, but was less potent in reducing [3H]-5-HT release. Accordingly, the pIC50 estimates by nonlinear regression analysis of the inhibition of [3H]-ACh, [3H]-DA, and [3H]-5-HT release were, 5.48, CI95 (5.26, 5.72), 5.17, CI95 (5.00, 5.34), and 4.40, CI95 (4.03, 4.74), respectively. For the concentration-response relationship of [3H]-ACh and [3H]-DA the Imax value was estimated to 93.1%, CI95 (84.4%, 102.3%), and 89.2%, CI95 (79.6%, 101.2%), respectively, which was very similar to the maximum inhibition seen with [3H]-Glu release. For the inhibition of [3H]-5-HT release, however, Imax could not be estimated from the data points, but was assumed to be 50% from Figure 2b in order to roughly assess the corresponding pIC50 value mentioned above. [3H]-NA release was not changed by riluzole.
[3H]-NA release using other experimental conditions
Since riluzole, even at the highest concentration, did not affect [3H]-NA release under the conditions used for [3H]-Glu release, other parameters were tested. Electrical field stimulation with 60 mA, 2 ms, 90 pulses, 3 Hz, at a superfusion temperature of 37°C (parameters usually used for NA experiments) led to S1=7.98% of tissue tritium, CI95 (7.48%, 8.47%). In controls, the S2/S1 ratio of controls was 0.92, CI95 (0.84, 1.01), and the S3/S1 ratio was 0.90, CI95 (0.79, 1.01). We also examined pseudo-one pulse stimulation parameters for autoinhibition-free release (Singer, 1988; 60 mA, 2 ms, 4 pulses, 100 Hz, 37°C), resulting in S1-values of 2.18% of tissue tritium, CI95 (1.96%, 2.40%). In the controls the S2/S1 ratio was 0.96, CI95 (0.88, 1.04), and the S3/S1 ratio was 1.00, CI95 (0.92, 1.07). The effects of riluzole under these conditions are shown in Table 2. It remained ineffective under autoinhibition-free release conditions, but showed significant inhibitory effects at 32 and 100 μM when autoinhibition was operative at 3 Hz stimulation frequency. However, the magnitude of these effects of riluzole were less than those observed with all other transmitters.
Table 2.
Effects of riluzole on electrically evoked [3H]-NA release in the absence and presence of autoinhibition in mouse neocortex

Comparison of riluzole effects on [3H]-ACh release in mouse, rat and human neocortex tissue
Investigating the question, whether the efficacy of riluzole was similar within different species we found the S1-values to be in mouse 2.22% of tissue tritium, CI95 (2.11%, 2.32%), in rat 1.42%, CI95 (1.28%, 1.56%), and in human neocortex 1.03%, CI95 (0.87%, 1.19%). In controls, the S2/S1 ratio was 1.03, CI95 (0.98, 1.07), and the S3/S1 ratio was 1.08, CI95 (1.02, 1.14) in mouse tissue, in rat tissue the S2/S1 ratio was 0.93, CI95 (0.89, 0.97), and the S3/S1 was 0.84, CI95 (0.79, 0.89) and in human tissue the S2/S1 ratio was 1.04, CI95 (0.90, 1.18) and S3/S1 was 1.02, CI95 (0.92, 1.13). The concentration-dependent inhibition of [3H]-ACh release by riluzole in human neocortical slices was not significantly different from that seen in mouse and rat (Figure 3). Note that the maximum inhibition by riluzole in the present experimental setup was significantly less than that observed with [3H]-ACh release under the conditions of [3H]-Glu release (Figure 2b).
Figure 3.
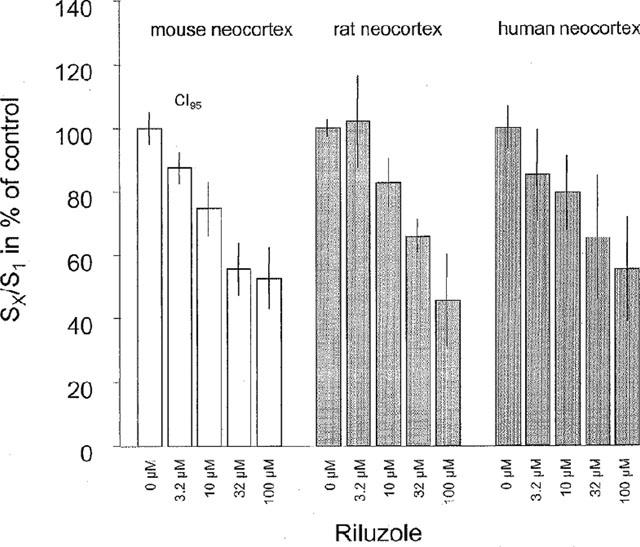
Effects of riluzole on electrically evoked [3H]-ACh release. Stimulation was induced by the following parameters: 60 mA, 2 ms, 3 Hz, 90 pulses, at 37°C. Columns represent mean SX/S1 values in per cent of corresponding controls (0 μM riluzole) with CI95s. Riluzole was given (at increasing concentrations during S2 and S3) from 15 min before the second stimulation onwards (n=6–9 per column). The S1 as well as the control SX/S1 values are given in the text.
Effects of PDC, NBQX+MK 801, and riluzol+NBQX+MK 801 on [3H]-ACh release
To investigate whether Glu itself might influence [3H]-ACh release, we increased extracellular Glu with PDC (100 μM). [3H]-ACh release was unchanged in the presence of PDC (Figure 4). To exclude the effects of riluzole on [3H]-ACh release through ionotropic Glu receptors these receptors were blocked by MK 801 and NBQX (1 μM each). The inhibition due to riluzole was not changed by MK 801 and NBQX (Figure 4).
Figure 4.

Effects of MK 801+NBQX, riluzole, MK 801+NBQX+riluzole and PDC on electrically evoked [3H]-ACh release of mouse neocortex. Stimulation was induced by the following parameters: 60 mA, 2 ms, 3 Hz, 90 pulses, at 37°C. Columns represent mean SX/S1 values in per cent of corresponding controls with CI95s. Drugs were given from 15 min before the second stimulation onwards (n=6–20 per column, see comment in the legend of Figure 2b). The corresponding S1 as well as the control SX/S1-values are given in the text.
Discussion
The present study examined the effects of riluzole on electrically evoked neurotransmitter release in mouse, rat and human brain tissue. We chose an electrical stimulation paradigm because electrical pulses, according to their TTX sensitivity, simulate action potentials to induce Ca2+-dependent, i.e. exocytotic, transmitter release.
According to Waldmeier et al. (1993), [3H]-Glu release experiments were performed at 22°C, to reduce both the spontaneous activity of neurons and the reuptake of released Glu. Reuptake was additionally suppressed by PDC (3 μM). Thus, the signal to noise ratio (evoked to basal transmitter efflux) was improved. Our model of [3H]-Glu release from slices preloaded with [3H]-Gln mainly reflects release from the neuronal Glu pool, since Gln, the major precursor of the neurotransmitter Glu, is metabolized predominantly in neurons (Bradford et al., 1978; Reubi, 1980). Because of the [3H]-Gln incubation, [3H]-Glu had to be separated by anion-exchange chromatography from other [3H]-compounds including [3H]-Gln that were excreted in the superfusion medium. At variance to the present approach, information about the source of Glu is vague if endogenous Glu is determined, e.g. in microdialysis studies (Timmerman & Westerink, 1997).
Because the Na+ channel blocker TTX as well as Ca2+-free medium inhibited the electrically evoked [3H]-Glu release, we assume that mainly action potential-induced, exocytotic Glu release occurred in our experiments. With this model, we were able to investigate the effects of substances on release of neuronal Glu.
The first question to answer was whether or not riluzole is able to suppress electrically evoked [3H]-Glu release. As expected from other studies, using potassium depolarization, which demonstrated riluzole to inhibit Glue release (e.g. Martin et al., 1993) this drug did also suppress electrically evoked [3H]-Glu release in mouse neocortical slices in a concentration-dependent manner and even more efficaciously. Obrenovitch & Urenjak (1998) presumed for experiments using potasium depolarization that release of Glu might be due to a reversal of the Glu transporter and/or due to osmotic pressure (see also Szatkowski et al., 1990). We assume that in our hands electrically evoked [3H]-Glu release is of neuronal origin and, to a large extent, exocytotically released. Thus, we demonstrated that riluzole can efficiently suppress this electrically stimulated release of Glu.
In earlier articles (Doble, 1996; Martin et al., 1993) riluzole was suggested to be a selective Glu release inhibitor. Therefore, we investigated riluzole effects in five different transmitter systems to test this proposed selectivity. We chose the same parameter constellation in all experiments, namely that for [3H]-Glu release, to be sure that distinct riluzole effects were not due to differences in parameters, but to the transmitter systems themselves. The physiological relevance of these rather unusual release conditions, including temperature, was assured by experiments with TTX and Ca2+-free medium for all transmitter systems. Riluzole inhibited [3H]-Glu, [3H]-ACh and [3H]-DA release to a similar extent and with similar potency, in contrast to its minor inhibition of the release of [3H]-5-HT. The inhibition of [3H]-ACh by riluzole was also minor when stimulation conditions with less electrical pulses were applied, as in the case of the comparison of mouse, rat and human neocortical tissue. This may be explained by the smaller number of pulses and, correspondingly, the smaller use-dependent action of riluzole, as described by MacIver et al. (1996).
Concerning the lack of effects of riluzole on [3H]-NA release, we further tested other parameter constellations (4 pulses, 60 mA, 100 Hz and 90 pulses, 60 mA, 3 Hz). We found only a weak inhibitory effect of riluzole on the release of [3H]-NA evoked by 90 pulses at 3 Hz. The fact that high concentrations of riluzole significantly depressed the release of this transmitter solely under the conditions of autoinhibition may also be explained by use-dependent effects of riluzole since autoinhibition conditions had 90 pulses whereas ‘autoinhibition-free' conditions only had 4 pulses. When 240 pulses were applied with a current of only 28 mA, however, no effect of riluzole on the release of [3H]-NA was observed, at variance to the strong effects of this drug on the release of [3H]-ACh, [3H]-Glu and [3H]-DA under this constellation of parameters. This comparison indicates that the effects of riluzole on different transmitter systems cannot be similarly explained, e.g. by a similar action on Na+ channels.
In comparison with other studies our IC50 value of the inhibition by riluzole of [3H]-Glu release (19.5 μM) compares with >50 μM of Lingamaneni & Hemmings (1999) who evoked Glu release by 30 mM K+. Martin et al. (1993) found an IC50 value of about 30 μM for evoked Glu release evoked with 50 mM K+. In addition the Imax value of our study was larger. These differences may be due to the fact that in our hands [3H]-Glu was mainly of neuronal origin which may be subject to a more pronounced modulation. For ACh release, evoked by electrical pulses of only 1 ms duration, an IC50 value of about 25 μM (Benavides et al., 1985) is at variance to our value of only 3.3 μM. This discrepancy may be explained by the different stimulation conditions applied, including that pulses with 2 ms duration may allow the generation of more action potentials, and the proposed development of use-dependence (MacIver et al., 1996) than pulses of 1 ms. In the study of Boireau et al. (1998) the ouabain-induced [3H]-DA release was half-maximally inhibited by 0.9 μM riluzole which is less than the IC50 found in the present experiments (6.8 μM). IC50 values between 1.6 and 9.5 μM have been found by Keita et al. (1997) using different stimulating drugs to evoke [3H]-DA release. Veratridine-induced catecholamine release from adrenal chromaffin cells was inhibited by riluzole with an IC50 of about 5 μM in the study of Yokoo et al. (1998). Since in our hands riluzole was hardly effective on [3H]-NA release we cannot provide an IC50 for comparision. As far as we know, in vitro studies investigating the effect of riluzole on 5-HT release do not exist. We found a rather low potency of riluzole (IC50 of 39.8 μM) with respect to this neurotransmitter.
Riluzole inhibits several ion channels that are assumed to be essential for the release of all neurotransmitter (i.e. Benoit & Escande, 1991; Hubert, 1994; Huang et al., 1997; Zona et al., 1998). As outlined above, the mechanism of riluzole to block presynaptic Na+ and Ca2+ channels, however, hardly explains the missing inhibition seen with the transmitter NA, and the weaker effects on 5-HT release we found. Two other studies (MacIver et al., 1996; Martin et al., 1993) also compared the effects of riluzole on different transmitter systems, this time on Glu and GABA, and concluded that Glu neurotransmission is preferentially diminished.
Concerning our results, we propose that inhibition of neurotransmitter release by riluzole may not occur through those ion channels which are nearly equal in most nerve terminals, but is rather mediated through receptors, which might differ between neurotransmitter systems. This hypothesis of receptor-mediated effects of riluzole is supported by earlier findings (Doble et al., 1992; Huang et al., 1997) demonstrating that G-proteins are involved in riluzole effects. However, the receptors influenced by riluzole to depress transmitter release are not known. A specific modulation of glutamatergic transmission rather exists postsynaptically, as shown by Keita et al. (1997) and Hubert et al. (1994), but not presynaptically with respect to transmitter release.
We excluded that inhibition of [3H]-ACh release by riluzole is mediated through blockade of NMDA heteroreceptors on axon terminals or through blockade of other ionotropic Glu receptors. This can be concluded from the finding that the effects of riluzole were maintained in the presence of MK 801 and NBQX, antagonists at NMDA and AMPA/kainate receptors, respectively. Thus, riluzole does obviously not affect ionotropic Glu receptors to inhibit [3H]-ACh release. The release of this transmitter in mouse neocortex was also not modulated by the elevation of extracellular Glu through PDC. Taken together, the terminal release of [3H]-ACh in mouse neocortex seems not affected by the neurotransmitter Glu which is also underlined by the lack of Glu to evoke [3H]-ACh release from nerve terminals (Lupp et al., 1992).
Riluzole is used in the treatment of ALS, a disease leading to selective degeneration of the first and second motoneuron (Hugon, 1996; Lacomblez et al., 1996). Glu excitotoxicity is considered as a major pathogenetic factor in ALS (Leigh & Meldrum, 1996) which prompted us to focus on this neurotransmitter in the present study. The marked neuroprotective effects of riluzole in several animal studies of the last years, in vivo and in vitro (see Estevez et al., 1995; Rothstein & Kuncl, 1995; Pratt et al., 1992) seem at variance to the rather short prolongation of life-expectancy and the small improvement in motor function gained by ALS patients receiving riluzole. To test whether riluzole has only minor effects in humans and since, to our knowledge, riluzole has not been tested in human tissue in vitro before, we compared the effects of riluzole in several species, including humans. We found that riluzole also affects the release of [3H]-ACh in human neocortex, but we did not find any relevant difference between the species mouse, rat and human. This indicates that riluzole does affect the cholinergic neurotransmission in patients, and probably also other neurotransmitter systems in man. Among the reasons, which could explain the weak effects of riluzole in ALS, is the fact that it may be given too late to protect motoneurons. In addition, its inhibition of [3H]-ACh release in human tissue suggests that it may also inhibit the cholinergic neurotransmission at muscular endplates, thus possibly diminishing its beneficial effect on motor function.
In conclusion, the neuroprotective agent and major ALS medication, riluzole, is an inhibitor of electrically evoked Glu release. It acts differently on the release of several neurotransmitters, but not selectively on excitatory amino acids. The modulation of ACh release by riluzole is not mediated by glutamate receptors. In human tissue riluzole seems to have the same effects as in animal tissue.
Acknowledgments
The authors thank S. Schmidt, T. Günter, H. Aranda and K. Strasser for excellent technical help. We also thank Dr W. Fischer, Rhône-Poulenc-Rorer, for providing riluzole and financial support. This work was also supported by the Deutsche Forschungsgemeinschaft (SFB 505) and the BMBF (FKZ 01EB9413).
Abbreviations
- ACh
acetylcholine
- ALS
amyotrophic lateral sclerosis
- CI95
95% confidence intervals
- DA
dopamine
- EDTA
ethylenediaminetetraacetic acid
- Gln
glutamine
- Glu
glutamate
- HPLC
high performance liquid chromatography
- 5-HT
serotonin
- Imax
maximum inhibition
- NA
noradrenaline
- PDC
L-trans-pyrrolidine-2,4-dicarboxylic acid
- pIC50
negative logarithm of the concentration at half-maximum inhibition
- TTX
tetrodotoxin
References
- BARNEOUD P., MAZADIER M., MIQUET J.M., PARMENTIER S., DUBEDAT P. Neuroprotective effects of riluzole on a model of Parkinson's disease in the rat. Neuroscience. 1996;74:971–983. doi: 10.1016/0306-4522(96)00249-7. [DOI] [PubMed] [Google Scholar]
- BENAVIDES J., CAMELIN J.C., MITRANI N., FLAMAND F., UZAN A. 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission. Biochemical properties. Neuropharmacology. 1985;24:1085–1092. doi: 10.1016/0028-3908(85)90196-0. [DOI] [PubMed] [Google Scholar]
- BENOIT E., ESCANDE D. Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflügers Arch. 1991;419:603–609. doi: 10.1007/BF00370302. [DOI] [PubMed] [Google Scholar]
- BENSIMON G., LACOMBLEZ L., MEININGER V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- BOIREAU A., MEUNIER M., IMPERATO A. Ouabain-induced increase in dopamine release from mouse striatal slices is antagonized by riluzole. J. Pharm. Pharmacol. 1998;50:1293–1297. doi: 10.1111/j.2042-7158.1998.tb03348.x. [DOI] [PubMed] [Google Scholar]
- BRADFORD H.F., WARD H.K., THOMAS A.J. Glutamine – a major substrate for nerve endings. J. Neurochem. 1978;30:1453–1459. doi: 10.1111/j.1471-4159.1978.tb10477.x. [DOI] [PubMed] [Google Scholar]
- CHERAMY A., BARBEITO L., GODEHEU G., GLOWINSKI J. Riluzole inhibits the release of Glu in the caudate nucleus of the cat in vivo. Neurosci. Lett. 1992;147:209–212. doi: 10.1016/0304-3940(92)90597-z. [DOI] [PubMed] [Google Scholar]
- CHERAMY A., ROMO R., GODEHEU G., BARUCH P., GLOWINSKI J. In vivo presynaptic control of DA release in the cat caudate nucleus-II. Facilitatory or inhibitory influence of L-Glu. Neuroscience. 1986;19:1081–1090. doi: 10.1016/0306-4522(86)90124-7. [DOI] [PubMed] [Google Scholar]
- DEBONO M.W., LE G.J., CANTON T., DOBLE A., PRADIER L. Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 1993;235:283–289. doi: 10.1016/0014-2999(93)90147-a. [DOI] [PubMed] [Google Scholar]
- DOBLE A. The pharmacology and mechanism of action of riluzole. Neurology. 1996;47:233–241. doi: 10.1212/wnl.47.6_suppl_4.233s. [DOI] [PubMed] [Google Scholar]
- DOBLE A., HUBERT J.P., BLANCHARD J.C. Pertussis toxin pretreatment abolishes the inhibitory effect of riluzole and carbachol on D-[3H]-aspartate release from cultured cerebellar granule cells. Neurosci. Lett. 1992;140:251–254. doi: 10.1016/0304-3940(92)90114-m. [DOI] [PubMed] [Google Scholar]
- ESTEVEZ A.G., STRUTZMANN J.M., BARBEITO L. Protective effect of riluzole on excitatory amino acid-mediated neurotoxicity in motoneuron-enriched cultures. Eur. J. Pharmacol. 1995;280:47–53. doi: 10.1016/0014-2999(95)00186-o. [DOI] [PubMed] [Google Scholar]
- FEUERSTEIN T.J., ALBRECHT C., WESSLER I., ZENTNER J., JACKISCH R. δ1-Opioid receptor-mediated control of acetylcholine (ACh) release in human neocortex slices. Int. J. Dev. Neurosci. 1998;16:795–802. doi: 10.1016/s0736-5748(98)00086-0. [DOI] [PubMed] [Google Scholar]
- FEUERSTEIN T.J., LIMBERGER N. Mathematical analysis of the control of neurotransmitter release by presynaptic receptors as a supplement to experimental data. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;359:349–359. doi: 10.1007/pl00005361. [DOI] [PubMed] [Google Scholar]
- GODEL H., GRASER T., FOLDI P., PFAENDER P., FURST P. Measurement of free amino acids in human biological fluids by high-performance liquid chromotography. J. Chromatogr. 1984;297:49–61. doi: 10.1016/s0021-9673(01)89028-2. [DOI] [PubMed] [Google Scholar]
- GRASER T.A., GODEL H.G., ALBERS S., FOLDI P., FURST P. An ultra rapid and sensitive high-performance liquid chromatographic method for determination of tissue and plasma free amino acids. Anal. Biochem. 1985;151:142–152. doi: 10.1016/0003-2697(85)90064-8. [DOI] [PubMed] [Google Scholar]
- HUANG C.S., SONG J.H., NAGATA K., YEH J.Z., NARAHASHI T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J. Pharmaco.l. Exp. Ther. 1997;282:1280–1290. [PubMed] [Google Scholar]
- HUBERT J.P., DELUMEAU J.C., GLOWINSKI J., PREMONT J., DOBLE A. Antagonism by riluzole of entry of calcium evoked by NMDA and veratridine in rat cultured granule cells: evidence for a dual mechanism of action. Br. J. Pharmacol. 1994;113:261–267. doi: 10.1111/j.1476-5381.1994.tb16203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGON J. ALS therapy: targets for the future. Neurology. 1996;47:251–254. doi: 10.1212/wnl.47.6_suppl_4.251s. [DOI] [PubMed] [Google Scholar]
- KEITA H., LEPOUSE C., HENZEL D., DESMONTS J.M., MANTZ J. Riluzole blocks DA release evoked by N-methyl-D-aspartate, kainate, and veratridine in the rat striatum. Anesthesiology. 1997;87:1164–1171. doi: 10.1097/00000542-199711000-00021. [DOI] [PubMed] [Google Scholar]
- LACOMBLEZ L., BENSIMON G., LEIGH P.N., GUILLET P., POWE L. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology. 1996;47:242–250. doi: 10.1212/wnl.47.6_suppl_4.242s. [DOI] [PubMed] [Google Scholar]
- LAGREZE W.A., OTTO T., FEUERSTEIN T.J. Neuroprotection in ischemia of the retina in an animal model. Ophthalmology. 1999;96:370–374. doi: 10.1007/s003470050420. [DOI] [PubMed] [Google Scholar]
- LEIGH P.N., MELDRUM B.S. Excitotoxicity in ALS. Neurology. 1996;47:221–227. doi: 10.1212/wnl.47.6_suppl_4.221s. [DOI] [PubMed] [Google Scholar]
- LINGAMANENI R., HEMMINGS H.C., JR Effects of anticonvulsants on veratridine- and KCl-evoked glutamate release from rat cortical synaptosomes. Neurosci. Lett. 1999;276:127–130. doi: 10.1016/s0304-3940(99)00810-1. [DOI] [PubMed] [Google Scholar]
- LUPP A., LÜCKING C.H., KOCH R., JACKISCH R., FEUERSTEIN T.J. Inhibitory effects of the antiparkinsonian drugs memantine and amantadine on N-methyl-D-aspartate-evoked acetylcholine release in rabbit caudate nucleus in vitro. J. Pharmacol. Exp. Ther. 1992;263:717–724. [PubMed] [Google Scholar]
- MACIVER M.B., AMAGASU S.M., MIKULEC A.A., MONROE F.A. Riluzole anesthesia: use-dependent block of presynaptic Glu fibers. Anesthesiology. 1996;85:626–634. doi: 10.1097/00000542-199609000-00023. [DOI] [PubMed] [Google Scholar]
- MALGOURIS C., BARDOT F., DANIEL M., PELLIS F., RATAUD J. Riluzole, a novel antiGlu, prevents memory loss and hippocampal neuronal damage in ischemic gerbils. J. Neurosci. 1989;9:3720–3727. doi: 10.1523/JNEUROSCI.09-11-03720.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN D., THOMPSON M.A., NADLER J.V. The neuroprotective agent riluzole inhibits release of Glu and aspartate from slices of hippocampal area CA1. Eur. J. Pharmacol. 1993;250:473–476. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- MARY V., WAHL F., STUTZMANN J.M. Effect of riluzole on quinolinate-induced neuronal damage in rats: comparison with blockers of Glurgic neurotransmission. Neurosci. Lett. 1995;201:92–96. doi: 10.1016/0304-3940(95)12137-s. [DOI] [PubMed] [Google Scholar]
- OBRENOVITCH T.P., URENJAK J. Glu release inhibitors: a critical assessment of their action mechanism. Amino Acids. 1998;14:143–150. doi: 10.1007/BF01345255. [DOI] [PubMed] [Google Scholar]
- PRATT J., RATAUD J., BARDOT F., ROUX M., BLANCHARD J.C. Neuroprotective actions of riluzole in rodent models of global and focal cerebral ischaemia. Neurosci. Lett. 1992;140:225–230. doi: 10.1016/0304-3940(92)90108-j. [DOI] [PubMed] [Google Scholar]
- REUBI J.C. Comparative study of the release of Glu and GABA, newly synthesized from glutamine, in various regions of the central nervous system. Neuroscience. 1980;5:2145–2150. doi: 10.1016/0306-4522(80)90130-x. [DOI] [PubMed] [Google Scholar]
- RICHARDSON I.W., SZERB J.C. The release of labelled acetylcholine and choline from cerebral cortical slices stimulated electrically. Br. J. Pharmacol. 1974;52:499–507. doi: 10.1111/j.1476-5381.1974.tb09717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTHSTEIN J.D., KUNCL R.W. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J. Neurochem. 1995;65:643–651. doi: 10.1046/j.1471-4159.1995.65020643.x. [DOI] [PubMed] [Google Scholar]
- SINGER E.A. Transmitter release from brain slices elicited by single pulses: a powerful method to study presynaptic mechanisms. Trends Pharmacol. Sci. 1988;8:274–276. doi: 10.1016/0165-6147(88)90004-1. [DOI] [PubMed] [Google Scholar]
- SZATKOWSKI M., BARBOUR B., ATTWELL D. Non-vesicular release of Glu from glial cells by reversed electrogenic Glu uptake. Nature. 1990;348:443–446. doi: 10.1038/348443a0. [DOI] [PubMed] [Google Scholar]
- TIMMERMAN W., WESTERINK B.H. Brain microdialysis of GABA and Glu: what does it signify. Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- WALDMEIER P.C., WICKI P., FELDTRAUER J.J. Release of endogenous Glu from rat cortical slices in presence of the Glu uptake inhibitor L-trans-pyrrolidine-2,4-dicarboxylic acid. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;348:478–485. doi: 10.1007/BF00173206. [DOI] [PubMed] [Google Scholar]
- YOKOO H., SHIRAISHI S., KOBAYASHI H., YANAGITA T., YAMAMOTO R., WADA A. Selective inhibition by riluzole of voltage-dependent sodium channels and catecholamine secretion in adrenal chromaffin cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;357:526–531. doi: 10.1007/pl00005203. [DOI] [PubMed] [Google Scholar]
- ZONA C., SINISCALCHI A., MERCURI N.B., BERNARDI G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience. 1998;85:931–938. doi: 10.1016/s0306-4522(97)00604-0. [DOI] [PubMed] [Google Scholar]