

Abstract
In common with human bronchial epithelial cells, pulmonary A549 cells release prostaglandin (PG) E2 in response to pro-inflammatory cytokines. We have therefore used these cells to examine the effect of the selective mitogen activated protein (MAP) kinase inhibitors; PD098059, a mitogen activated and extracellular regulated kinase kinase (MEK) 1 inhibitor, UO126, a dual MEK1 & MEK2 inhibitor, and SB203580, a p38 MAP kinase inhibitor in the IL-1β-dependent release of PGE2.
Following IL-1β treatment the extracellular regulated kinases (ERKs) and the p38 MAP kinases were rapidly phosphorylated.
PD09059, UO126 and SB203580 prevented IL-1β-induced PGE2 release at doses that correlated closely with published IC50 values. Small or partial effects at the relevant doses were observed on induction of cyclo-oxygenase (COX) activity or COX-2 protein suggesting that the primary effects were at the level of arachidonate availability.
Neither PD098059 nor SB203580 showed any effect on IL-1β-induced arachidonate release. We therefore speculate that the MEK1/ERK and p38 kinase cascades play a role in the functional coupling of arachidonate release to COX-2.
In contrast, UO126 was highly effective at inhibiting IL-1β-dependent arachidonate release, implicating MEK2 in the activation of the PLA2 that is involved in IL-1β-dependent PGE2 release.
We conclude that the MEK1, MEK2 and p38 MAP kinase inhibitors, PD098059, UO126 and SB203580, are highly potent in respect of inflammatory PG release. Finally, we conclude that these inhibitors act via mechanistically distinct processes, which may have anti-inflammatory benefits.
Keywords: Prostaglandin E2, prostaglandin GH synthase, phospholipase A2, MAP kinase, epithelial cell
Introduction
Prostaglandins (PGs) are lipid mediators that are involved in many normal physiological processes, and are implicated in many pathophysiological processes such as inflammation, edema, bronchoconstriction, platelet aggregation, fever and hyperalgesia (Mitchell et al., 1995). PG synthesis predominantly involves phospholipase A2 (PLA2)-catalysed release of arachidonic acid from the sn-2 position of membrane phospholipids and conversion by the two cyclo-oxygenase (COX) enzymes to PGH2. Subsequently cell type-specific expression of downstream synthases is responsible for the production of biologically relevant PGs.
Numerous PLA2 activities have been identified and many of the genes cloned (Balsinde & Dennis, 1997; Dennis, 1997). Of these, the group II secretory PLA2 (sPLA2) and the group IV Ca2+-dependent cytosolic PLA2 (cPLA2) have long been thought to play the predominant roles in respect of inflammatory prostaglandin synthesis (Balsinde & Dennis, 1997; Dennis, 1997). Similarly, the two COX isoforms are encoded by distinct genes. COX-1 is a constitutively expressed housekeeping gene whilst COX-2 is an acute phase gene that is rapidly induced by inflammatory and mitogenic stimuli (Mitchell et al., 1995). As COX is a target for non-steroidal anti-inflammatory drugs (NSAIDs) this pathway is pharmacologically important (Mitchell et al., 1995). Furthermore the use of isoform selective COX inhibitors has revealed that many anti-inflammatory benefits of NSAIDs derive from COX-2 inhibition whilst many undesirable side effects result from COX-1 inhibition (DeWitt, 1999; Mitchell et al., 1995; Seibert et al., 1994). However, more recent evidence suggests that COX-2-dependent PG release may also play a role in the resolution of inflammation (Gilroy et al., 1999). Despite the clinical usefulness of NSAIDs, currently the most effective drugs in the treatment of chronic inflammatory diseases, such as asthma, are corticosteroids (Barnes, 1999). These down-regulate various inflammatory processes, including prostaglandin synthesis, via repression of pro-inflammatory genes such as COX-2 (Barnes, 1999; Newton et al., 1997a).
Recently the development of small molecule inhibitors of the mitogen activated protein (MAP) kinase pathways has attracted much attention as potential therapeutic agents (Alessi et al., 1995; Cuenda et al., 1995; Dudley et al., 1995; Favata et al., 1998; Lee et al., 1994). Various cellular stresses, as well as pro-inflammatory cytokines such as IL-1β and TNFα, are known to activate multiple MAP kinase signalling pathways, which then play major effector roles in numerous cellular responses (Kyriakis & Avruch, 1996). These pathways comprise of kinase cascades in which small GTP-binding proteins, such as Ras, Rac or Rho, activate MAP kinase kinase kinases (MAP3K), such as Raf. These activate, by phosphorylation, MAP kinase kinases (MAP2K), which in turn activate the MAP kinases (MAPK). Finally, the MAP kinases phosphorylate and activate various effector molecules to elicit cellular responses. In particular, inhibitors of the MAP2K, mitogen activated protein kinase (MAPK)/extracellular regulated kinase (ERK) kinase (MEK), and the p38 MAPK are effective in preventing induction of pro-inflammatory genes (Alessi et al., 1995; Cuenda et al., 1995; Dudley et al., 1995; Lee et al., 1994; Scherle et al., 1998).
Epithelial cells have an active role in inflammation by producing multiple mediators and airway epithelial cells respond to pro-inflammatory cytokines, such as IL-1β, by induction of COX-2 and PGE2 release (Mitchell et al., 1994). As this response is also observed in human A549 cells (Mitchell et al., 1994; Newton et al., 1997a), we have used these cells to examine the effect of selective MAP kinase pathway inhibitors on the induction of COX-2 and PGE2 release by IL-1β.
Methods
Cell culture
A549 cells were grown to confluency as previously described (Newton et al., 1997a). Cells were incubated over-night in serum-free medium before changing to fresh medium containing 1 ng ml−1 IL-1β (2×105 u μg−1) (Genzyme, MA, U.S.A.), 3 μM ionomycin (Sigma, Poole, U.K.), drugs or vehicle. PD098059 (2′-amino-3′methoxyflavone) (Alexis, Bingham, U.K.), UO126 (1,4-diamino-2,3-dicyano-1,4-bis (2-aminophenylthio)butadiene) (Calbiochem, Nottingham, U.K., SB203580 (4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole) (Alexis) and methyl arachidonyl fluorophosphonate (MAFP) (Calbiochem) were dissolved in DMSO and diluted at least 1 : 1000 in culture media prior to use. DMSO at such concentrations had no effect on cell viability, PGE2 release or COX activity (data not shown).
PGE2 release and COX activity determination
Culture medium was removed and released PGE2 measured by radioimmunoassay (RIA) using an anti-PGE2 antibody (Sigma) essentially according to the manufacturers instructions (Mitchell et al., 1994). After rinsing cells twice, COX activity assays were performed as described (Mitchell et al., 1994). Briefly, fresh medium containing 30 μM arachidonic acid (Sigma) was added and the plates incubated for 10 min at 37°C. PGE2 produced was measured by RIA and taken as an index of COX activity.
Immunoblot analysis
Cells were harvested and lysed as previously described (Newton et al., 1997c). Total cellular protein was separated by 10% SDS–PAGE (or as indicated) and electroblotted to hybond-ECL membranes (Amersham, Buckinghamshire, U.K.). Membranes were probed with rabbit anti-human antibodies directed to either phosphorylated or total ERK and p38 MAP kinases according to the manufacturers specification (New England Biolabs, Hitchin, U.K.). After washing, membranes were incubated with horseradish peroxidase-linked anti-rabbit immunoglobulin (Dakko) and immune complexes detected by enhanced chemiluminescence (ECL) (Amersham). For COX-2 immunodetection, goat anti-rabbit primary (Santa Cruz) and rabbit-anti-goat secondary (Dakko) antibodies were used with ECL.
Release of [3H]-arachidonate
Confluent cells, in 24-well plates, were incubated over-night in 0.5 ml serum free media supplemented with 0.125 μCi [5,6,8,9,11,12,14,15-3H] arachidonic acid (Amersham). Cells were washed three times with media and treated in media containing 2 mg ml−1 bovine serum albumin (Sigma) to absorb arachidonate metabolites. Following stimulation, supernatants were collected as indicated for liquid scintillation counting. In all cases, [3H]-arachidonate incorporation into cells was measured by harvesting cells in 1% SDS and liquid scintillation counting. Using this methodology [3H]-arachidonate incorporation into cells was typically 75–80%. In each case, release of [3H]-arachidonate and its metabolites was expressed as a percentage of the total incorporated into cells.
Statistical analysis
Data were expressed as means±s.e.mean and statistical analysis performed by ANOVA using a Bonferoni correction for multiple comparisions. Significance is indicated where: *P<0.05, **P<0.01, ***P<0.001.
Results
IL-1β induces ERK and p38 MAP kinase phosphorylation
In A549 cells, IL-1β has previously been shown to activate the p54 and p46 Jun N-terminal kinases (JNK) (Newton et al., 1997c). The effect of IL-1β was therefore examined on the ERK and p38 MAP kinase pathways. As threonine/tyrosine phosphorylation of residues 202 and 204 in ERK1 (p44) or the equivalent residues in ERK2 (p42) and residues 180 and 182 in p38 result in enzymatic activation, we used Western blot analysis to monitor phosphorylation of these sites as an index of kinase activation (Boulton et al., 1991; Payne et al., 1991; Raingeaud et al., 1995). In resting cells, basal ERK phosphorylation was detected whereas no basal level of p38 phosphorylation was observed. Both ERK, p44 and p42, and p38 MAP kinases were rapidly (<5 min) phosphorylated following IL-1β treatment (Figure 1). In each case this was maximal at around 30 min and was declining by 1 h after stimulation.
Figure 1.

Activation of ERK and p38 MAP kinases by IL-1β. Cells were treated with IL-1β (1 ng/ml) for the times indicated and harvested for Western blotting. Immunodetection was performed with antibodies directed to; (A) phosphorylated ERK (upper panel) and pan ERK (lower panel) or, (B) phosphorylated p38 (upper panel) or pan p38 (lower panel). Data presented are representative of at least three similar experiments.
Effect of PD098059 and UO126 on IL-1β-induced PGE2 release
As previously observed, 24 h IL-1β resulted in a substantial increase in PGE2 release, COX activity and COX-2 immunoreactivity (Figure 2) (Mitchell et al., 1994; Newton et al., 1997a, 1998). Since COX-1 message is 100 fold less abundant than COX-2 and COX-1 protein is undetectable in both cytokine stimulated and untreated cells, the increase in COX activity and PGE2 release can be attributed to de novo COX-2 synthesis (Mitchell et al., 1994; Newton et al., 1997a). The increase in PGE2 release was dose-dependently inhibited by PD098059 (EC50 3.0 μM) (Figure 2) and corresponds well with the published values for MEK1 inhibition (IC50 2–10 μM) (Alessi et al., 1995; Dudley et al., 1995). However, 10 μM PD098059 showed little effect on induction of COX activity and protein, indicating that the inhibition of PGE2 release must occur upstream of COX-2, presumably at the level of PLA2 and arachidonic acid release. The partial inhibition of 50 μM PD098059 on COX activity and COX-2 protein suggest an effect on COX-2 synthesis (Figure 2A,C). However, this effect failed to reach statistical significance.
Figure 2.
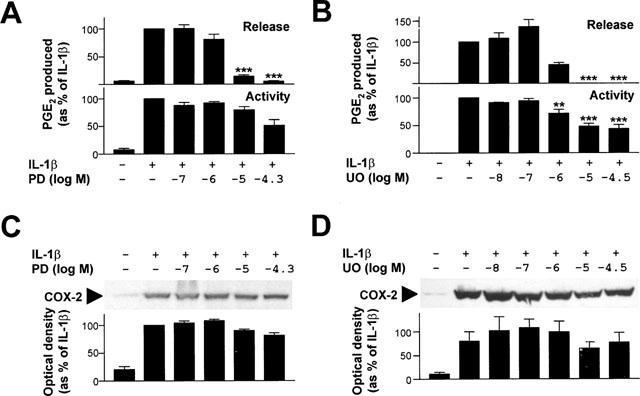
Effect of PD098059 and UO126 on PGE2 release, COX activity and COX-2 protein expression. Cells were treated with various concentrations of PD098059 (PD) or UO126 (UO) as indicated and then stimulated with IL-1β (1 ng ml−1) for 24 h. (A, B) Supernatants were collected for PGE2 release measurement (upper panels) and COX activity (lower panels) determination performed. Data (n=7 and 6 for PD098059 and UO126 respectively) are expressed as a percentage of IL-1β treated as means±s.e.mean. (C, D) In addition cells were harvested for Western blot analysis of COX-2 protein. Representative blots are shown (upper panels). After densitometric analysis, data from four or five (PD098059 and UO126 respectively) experiments were expressed as a percentage of IL-1β treated and are shown as means±s.e.mean (lower panels).
Likewise, the inhibition of IL-1β-dependent PGE2 release by UO126 (EC50 0.8 μM) is also consistent with doses previously reported to inhibit MEK1 activity and ERK phosphorylation (Favata et al., 1998). Similarly, whilst a partial inhibition was observed on COX activity (EC50 1.0 μM) and COX-2 (EC50 2.0 μM), these effects were insufficient to account for the total loss of PGE2 release. Thus these data indicate that UO126 inhibits IL-1β-dependent PGE2 release primarily by inhibiting a step (or steps) upstream of COX-2.
To confirm the effects of PD098059 and UO126 on MEK activation, the phosphorylation status of the immediate downstream kinases, ERK1 and 2, was examined (Figure 3). As in Figure 1 above, IL-1β markedly induced phosphorylation of both the p44 and p42 ERK. Pre-incubation with either PD098059 or UO126 totally inhibited this response with similar dose-response characteristics to that for the inhibition of PGE2 release.
Figure 3.

Effect of PD098059 and UO126 on ERK phosphorylation. Cells were treated with IL-1β (1 ng ml−1) and the indicated concentrations of (A) PD098059 (PD) or (B) UO126 (UO). After 30 min cells were harvested for Western blot analysis using antibodies to phosphorylated ERK (upper panel) or pan ERK (lower panel). Blots representative of three such experiments are shown.
Effect of SB203580 on IL-1β-induced PGE2 release
Analysis of the p38 MAP kinase inhibitor, SB203580, again revealed a dose-dependent inhibition of IL-1β-induced PGE2 release (Figure 4). The EC50 for this was 0.18 μM and is consistent with published values for p38 inhibition (IC50 0.6 μM) (Cuenda et al., 1995; Kramer et al., 1996). As little inhibition of COX activity and COX-2 protein was observed at these doses, these data again imply an inhibitory step upstream of COX-2.
Figure 4.

Effect of SB203850 on PGE2 release, COX activity and COX-2 protein expression. Cells were treated with various concentrations of SB203850 (SB) as indicated and then stimulated with IL-1β (1 ng ml−1) for 24 h. (A) Supernatants were collected for PGE2 release measurement (upper panel) and COX activity (lower panel) determination performed. Data (n=5) are expressed as a percentage of IL-1β treated as means±s.e.mean. (B) In addition cells were harvested for Western blot analysis of COX-2 protein. A representative blot is shown (upper panel). After densitometric analysis, data from four such experiments were expressed as a percentage of IL-1β treated and are shown as means±s.e.mean (lower panel).
Effect of PD098059, UO126 and SB203580 on arachidonate release
Following 1 h of IL-1β treatment only modest increases in [3H]-arachidonate metabolite release into the supernatant were observed (untreated 0.90%±0.09, IL-1β 1.60% ±0.11, n=16, P>0.05). Because of this low level of inducibility and release, cells were treated for 6 h with or with out IL-1β and the effects of the PD098059, UO126 and SB203580 examined (Figure 5). At this time point IL-1β caused a 1.5–2 fold increase over basal levels of [3H]-arachidonate release (untreated 3.34%±0.18, IL-1β 5.97%±0.32, n=14, P<0.001). Pretreatment with either PD098059 or SB203580 had little or no effect on [3H]-arachidonate release at or below 10 μM and at higher concentrations a partial inhibition was observed. In marked contrast, UO126 dose-dependently inhibited IL-1β-dependent release of [3H]-arachidonate (EC50 0.3 μM). To confirm that PD098059 and SB203580 were indeed functioning on PGE2 release at this time, measurements of PGE2 release, COX activity and COX-2 protein were carried out as in Figure 2 except that samples were harvested after 6 h. Essentially identical data was obtained to that shown in Figure 2 (data not shown).
Figure 5.

Effect of IL-1β, PD098059, UO126 and SB203580 on arachidonate release. Cellular lipids were loaded with [3H]-arachidonate and then treated with various concentrations of PD098059, UO126 or SB203580, as indicated, prior to stimulation with IL-1β (1 ng ml−1). After 6 h supernatants and cells were harvested for liquid scintillation counting. (A) Data (n=14) are expressed as arachidonate release as a percentage of the total incorporated. (B) The effects of PD098059 (n=7), UO126 (n=4) and SB203580 (n=5) each performed in duplicate are shown as arachidonate release expressed as a percentage of IL-1β treated as means±s.e.mean.
A role for cPLA2 in arachidonate release?
To further examine the mechanisms of arachidonate release in this system, we tested the effect of the highly selective group IV cPLA2 inhibitor, MAFP (Balsinde & Dennis, 1996; Huang et al., 1994). To our surprise there was no inhibitory effect on IL-1β-dependent PGE2 release and at higher doses a significant enhancement of PGE2 release was observed (Figure 6). Little or no effect was observed on COX activity or COX-2 protein expression. Consistent with the elevated levels of PGE2, a dose-dependent increase in arachidonate release was observed, which failed to reach statistical significance.
Figure 6.

Effect of MAFP on PGE2 release, COX activity, COX-2 protein expression and arachidonate release. Cells were treated with various concentrations of MAFP as indicated and then stimulated with IL-1β (1 ng ml−1). (A) After 24 h, supernatants were collected for PGE2 release measurement (upper panel) and COX activity (lower panel) determination performed. Data (n=6) are expressed as a percentage of IL-1β treated as means±s.e.mean. In addition cells were harvested for Western blot analysis of COX-2 protein (B). A blot representative of two such experiments is shown. (C) Cellular lipids were loaded with [3H]-arachidonate and then treated with various concentrations of MAFP, as indicated, prior to stimulation with IL-1β (1 ng ml−1). After 6 h supernatants were removed and cells harvested for liquid scintillation counting. Data (n=4) are shown as arachidonate release expressed as a percentage of IL-1β treated as means±s.e.mean.
As a number of studies have implicated cPLA2 in the IL-1β-dependent release of PGE2 and/or arachidonate, we decided to further investigate this possibility. The small increases in [3H]-arachidonate release may be explained by the fact that whilst IL-1β is a good inducer of COX-2 synthesis, it is a poor inducer of the PLA2, possibly cPLA2, required for inflammatory PGE2 release (Schalkwijk et al., 1996; Tokumoto et al., 1994). Treatment of cells with Ca2+ mobilizing agents produces substantial increases in cPLA2-dependent arachidonate release (Sa et al., 1995; Schalkwijk et al., 1996). A similar effect is also reported in A549 cells using ionomycin induced Ca2+ mobilisation (Tokumoto et al., 1994). In this case induction of arachidonate release was shown to be Ca2+-dependent as ionomycin had no effect in Ca2+-free media or media containing the Ca2+ chelator, EGTA. We therefore treated cells with combinations of IL-1β and ionomycin (Figure 7). Ionomycin alone produced a significant increase in arachidonate release. In the presence of ionmycin, IL-1β resulted in a consistent doubling of released arachidonate, which was significantly reduced by pre-treatment with 10 and 50 μM PD098059. Consistent with the 6 h data, SB203580, failed to inhibit IL-1β+ionomycin induced arachidonate and at higher concentrations actually enhanced arachidonate release. When PD098059 and SB203580 were tested on IL-1β-induced release of arachidonate at 1 h, again no effect was observed, which is consistent with the data in Figure 5 (data not shown). In contrast to IL-1β stimulation alone, MAFP produced a significant decrease in IL-1β+ionomycin stimulated arachidonate release.
Figure 7.
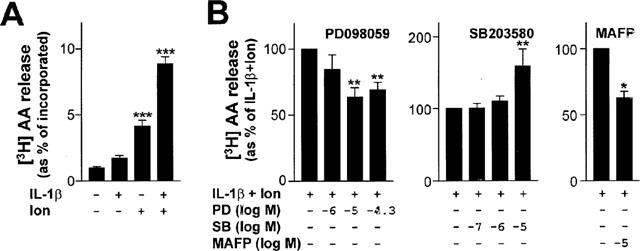
Effect of PD098059, SB203580 and MAFP on IL-1β and ionomycin stimulated arachidonate release. (A) Cells were loaded with [3H]-arachidonate and then treated with various combinations of IL-1β (1 ng ml−1) and ionomycin (3 μM) (as indicated). After 1 h supernatants were removed and cells harvested for liquid scintillation counting. Data (n=6) are expressed as arachidonate release as a percentage of the total incorporated. (B) Cells were loaded with [3H]-arachidonate and then treated with various concentrations of PD098059 (PD), SB203580 (SB) or MAFP, as indicated, prior to stimulation with IL-1β (1 ng ml−1) and ionomycin (3 μM) (Ion). After 1 h, supernatants were removed and cells harvested for liquid scintillation counting. Data (n=4) are shown as arachidonate release expressed as a percentage IL-1β+Ionomycin treated as means±s.e.mean.
Time-dependence of MAP kinase inhibitors on PGE2 release
Confluent cells that had been treated with IL-1β for 20 h (in 12-well plates) were found to produce detectable levels of PGE2 in a 10 min period (0.625±0.04 ng ml−1, n=4), whereas in unstimulated cells any PGE2 release was below the level of detection (Figure 8). This response was totally prevented by both PD098059 and UO126, whereas SB203580 had no effect. Therefore the steps that are inhibited by PD098059 and UO126 are continually required for PGE2 release whereas inhibition by SB203580 is time-dependent.
Figure 8.

Acute effect of PD098059, UO126 and SB203580 on PGE2 release. Cells, in 12-well plates, were treated with IL-1β (1 ng ml−1) for 20 h. Then serum free medium containing PD098059 (50 μM), SB203580 (10 μM) or UO126 (30 μM) was added for 10 min. After this time the media was changed to fresh serum free medium (also containing PD098059, SB203580 or UO126, but not IL-1β). After a further 10 min the medium was harvested for PGE2 release measurements. Data from four experiments are shown as a percentage of IL-1β treated as means±s.e.mean.
Immediate effects of PD098058, UO126 and SB203580 on COX activity
It has been suggested that PD098059 and SB203580 may be capable of acting as direct inhibitors of COX activity (Borsch-Haubold et al., 1998). To examine this possibility, cells were treated with IL-1β for 20 h to induce COX-2. Cells were then pre-treated with each drug before the addition of exogenous arachidonic acid at concentrations of either 1 or 100 μM. After 10 min, PGE2 release was assessed and taken as an index of COX activity (Figure 9). In the absence of inhibitors these doses of arachidonic acid gave rise to 3.07±0.27 and 28.73±3.16 ng ml−1 10 min−1 well−1 of PGE2 respectively. At 100 μM arachidonic acid none of the three inhibitors showed any effect. At 1 μM arachidonic acid, PD098059 and to a lesser extent UO126 showed some degree of inhibition whilst no effect was observed for SB203580.
Figure 9.

Effect of PD098059, UO126 and SB203580 on arachidonic acid induced PGE2 release (COX activity). Cells, in 12-well plates, were treated with IL-1β (1 ng ml−1) for 20 h. Then serum free medium containing PD098059 (50 μM), SB203580 (10 μM) or UO126 (30 μM) was added for 10 min. After this time the media was changed to fresh serum free medium supplemented with either 100 or 1 μM arachidonic acid (also containing PD098059, SB203580 or UO126). After a further 10 min the medium was harvested and PGE2 release taken as an index of COX activity. Data from four experiments are shown as a percentage of IL-1β treated (i.e. no drug) as means±s.e.mean.
Discussion
In these studies, PD098059, a selective MEK1 inhibitor (Alessi et al., 1995; Dudley et al., 1995), UO126, a selective MEK1 & MEK2 inhibitor (Favata et al., 1998), and SB203850, a p38 MAP kinase inhibitor (Cuenda et al., 1995), were highly potent inhibitors of IL-1β-induced PGE2 release. In all three cases the inhibitory effect occurred at the published IC50 values obtained from in vitro kinase assays, indicating that these compounds are readily taken up by the cells and that access to the relevant target enzyme was unhindered. At the concentrations required for almost total inhibition of PGE2 release, 10 μM for PD0998058 and 1 μM for SB203580, little or no effect was observed on IL-1β-induced COX activity or COX-2 protein. These data indicate that neither, activation of MEK1 and the downstream kinases ERK1 and 2, nor the p38 MAP kinase play a major role in the induction of COX-2 by IL-1β. By contrast, inhibition of PG synthesis in zymosan or lipopolysaccharide treated monocytes correlated with inhibition of COX-2 by SB203580 and suggests a major role for p38 kinase in COX-2 synthesis in this system (Dean et al., 1999; Pouliot et al., 1997). Furthermore, a related p38 inhibitor, SC68376, also inhibited PG synthesis seemingly via inhibition of COX-2 mRNA and protein induction in IL-1β-treated rat mesangial cells (Guan et al., 1997). However, our results are consistent with the fact that the NF-κB, C/EBP and JNK signalling pathways, which do not involve p38 MAPK, are primarily thought to mediate COX-2 transcriptional induction (Inoue et al., 1995; Newton et al., 1997b; Xie & Herschman, 1995; Yamamoto et al., 1995). Nevertheless, as higher doses of both PD098059 and SB203850 appeared to inhibit induction of COX activity and COX-2 protein, it is possible that one of these other pathways also becomes partially inhibited. In the case of PD098059 the inhibitory effect on COX activity may be explained by MEK2 inhibition, which occurs with an IC50 of around 50 μM (Alessi et al., 1995). This proposal is supported by the fact that UO126, which is equipotent on both MEK1 and MEK2 (Favata et al., 1998), also results in a partial inhibition of COX-2 protein and COX activity (EC50 1.1 μM) (Figure 2B). Furthermore, a similar inhibitory effect of UO126 was reported at the level of COX-2 mRNA (Scherle et al., 1998). However, whilst providing support for a role of MEKs in the induction of COX-2, this study failed to distinguish between MEK1 and MEK2 inhibition by UO126.
The relative lack of effect of PD098059, UO126 and SB203850 on induction of COX activity and COX-2 protein points to arachidonate release or availability as the primary target of inhibition. However, neither PD098059 nor SB203580 showed any significant effect on IL-1β stimulated arachidonate release at concentrations at or below 10 μM. At 50 μM, PD098059 produced a greater than 50% inhibition of arachidonate release which correlates more closely with inhibition of MEK2 rather than MEK1 (Alessi et al., 1995). This hypothesis is supported by the data using UO126, which dose-dependently inhibits IL-1β-stimulated arachidonate release (Figure 5). The fact that ERK phosphorylation was inhibited over this same range of concentrations shows that the UO126 was gaining access to the cells and was inhibiting MEK1, which is upstream of ERK1 and 2. However, the lack of effect of PD098059 over doses that clearly inhibit MEK1, as measured by ERK1 and 2 phosphorylation, indicate that it is inhibition of MEK2 by UO126 which is responsible for the inhibition of arachidonate release and subsequent PGE2 release.
Previous mRNA and biochemical data point to involvement of group IV cPLA2 and not group II sPLA2 in cytokine mediated PGE2 release in A549 cells (Neagos et al., 1993; Newton et al., 1997a; Tokumoto et al., 1994). However, in the present study the highly selective cPLA2 inhibitor, MAFP, was ineffective at inhibiting IL-1β-dependent PGE2 release suggesting that the group IV cPLA2 may not be involved (Figure 6). As a means of validating this effect, we tested the effect of PD098059 and MAFP in cells that had been co-stimulated with the Ca2+ mobilizing agent, ionomycin, and IL-1β. It is well established that Ca2+ flux and phosphorylation of Ser 505 are both required for full cPLA2 activity (Clark et al., 1991; Lin et al., 1993; Sa et al., 1995; Schalkwijk et al., 1996). The fact that Ca2+ mobilization in the presence of IL-1β gave rise to synergistically increased arachidonate release supports the existence of a role for cPLA2 in A549 cells and this is further substantiated by the inhibitory effect of MAFP (Figure 7). In addition, the inhibition by PD098059 of the synergistic effect of IL-1β on Ca2+-dependent arachidonate release is consistent with previous studies, which indicate that cPLA2 is a target for ERK MAP kinases (Sa et al., 1995; Schalkwijk et al., 1996). Therefore these data indicate that whilst ERK activation of cPLA2 may play a role in IL-1β+Ca2+ stimulated release of arachidonate, the IL-1β-dependent release of PGE2 does not appear to involve cPLA2 and inhibition by PD098059 is not mediated via an effect on arachidonate release. Similarly, the p38 inhibitor, SB203580, resulted in total suppression of IL-1β-dependent release of PGE2 at doses that correlated closely with published IC50 values (Cuenda et al., 1995; Kramer et al., 1996) . However, as no effect of SB203580 was observed on release of arachidonate metabolites at either 1 or 6 h, these data show that p38 MAP kinase is also not directly involved in PLA2 activation per se. This ability of SB203580 to inhibit PGE2 release is time-dependent as cells that have been IL-1β stimulated for 20 h and then treated with SB203580 do not show any inhibition of PGE2 release (Figure 8). Thus either the downstream substrate is fully activated and p38 MAP kinase is no longer required or synthesis of a p38 MAP kinase-dependent protein that is required for PGE2 release has occurred and can no longer be blocked by SB203580.
In A549 cells, epidermal growth factor is reported to cause cPLA2 phosphorylation detectable by mobility shift on SDS–PAGE (Croxtall et al., 1996). However, in our studies we have been unable to reproducibly demonstrate an IL-1β-dependent shift in cPLA2 mobility on SDS–PAGE, and likewise have been unable to show changes in mobility as a result of PD098059 or SB203580 treatment (data not shown). Due to the inherent variability of this technique, these data do not exclude cPLA2 as targets of ERK and/or p38 MAP kinase phosphorylation. However, the lack of inhibition by MAFP suggest that cPLA2 may not actually be involved in the IL-1β-dependent response. Furthermore, a p38 MAP kinase inhibitor inhibited phosphorylation of cPLA2 in platelets, yet no effect on arachidonate release was observed, suggesting that phosphorylation of cPLA2 is not necessarily linked to arachidonate release (Kramer et al., 1996). In addition, numerous new PLA2 activities and genes have been identified (Balsinde & Dennis, 1997). In addition, a 1-O- acylceramide synthase that shows PLA2 activity (Abe & Shayman, 1998), the cPLA2 homologues, cPLA2β and cPLA2γ (Pickard et al., 1999; Underwood et al., 1998), a novel cPLA2 splice variant (Gordon et al., 1996) and a number of novel group II secretory PLA2 (IID, IIE and IIF) have all been cloned (Ishizaki et al., 1999; Valentin et al., 1999). These discoveries raise the possibility that another PLA2 is in fact responsible for IL-1β-dependent PGE2 release in these cells and that this PLA2 activity is downstream of the MEK2-dependent kinase cascade that was inhibited by UO126.
One explanation for the effect of PD098059 and SB203580 is that these compounds may directly inhibit COX-2 activity (Borsch-Haubold et al., 1998). In the case of SB203580, this possibility was excluded as incubation of SB203580 with cells in which COX-2 had been induced, in the presence of odifferent exogenous arachidonic acid concentrations, showed no inhibition of COX activity (Figure 9). Furthermore, high dose SB203580 had no acute effect on release of PGE2 from IL-1β-treated cells, again indicating that there is no direct effect on COX-2 enzyme (Figure 8). In the case of PD098059, a possible competitive inhibitory effect on COX (or a downstream enzyme) was apparent (Figure 9). However, as the inhibition of PGE2 release occurred at the same concentrations as that for inhibition of ERK phosphorylation, it is unlikely that this effect was the result of direct COX-2 inhibition. Another explanation for the effect of PD098059 and SB203580 is that the phospholipid pool, which is labelled with [3H]-arachidonate, is not the same pool that gives rise to IL-1β-induced PGE2. However, this hypothesis again seems unlikely as UO126 inhibited both release of PGE2 and [3H]-arachidonate release.
The question therefore remains as to the roles of the MEK1/ERK and p38 MAP kinases in IL-1β-dependent PGE2 release. Whilst MEK1/ERK and p38 inhibition resulted in no change in arachidonate release, our data indicate a functional role for both these MAP kinase pathways in PGE2 generation. Recently the group IV cPLA2 and the group IIA and V sPLA2's, but not the group VI Ca2+-independent PLA2 (iPLA2), have been shown to be functionally linked to COX-2 and PG generation (Murakami et al., 1998). Thus simple release of arachidonic acid in the presence of COX-2 is not sufficient for PG production suggesting that a specific relationship between PLA2 and COX-2 is required (Murakami et al., 1998). It is therefore possible that phosphorylation of the relevant PLA2, by ERK or p38 has no effect on activity, but is important in coupling to or interaction with COX-2. In this context, it has become clear that adaptor or scaffold proteins, which provide a structural support for the various components of signalling pathways, also play an important function in the signal transduction process (Pawson & Scott, 1997). Such proteins may confer a specific spatial arrangement to the various pathway components thus enhancing specificity as well as ensuring that kinase and substrate domains are in close proximity to allow rapid kinetics. Furthermore, it is known that prostaglandin synthetic enzymes are co-localized to the endoplasmic reticulum and the nuclear envelope following cell stimulation (Morita et al., 1995; Schievella et al., 1995). Therefore it is possible that phosphorylation by ERKs or p38 MAP kinase provide an accessory function, which is important in the interaction of the IL-1β-dependent PLA2 with specific anchoring proteins, or even COX-2, such that the correct spatial arrangement can be adopted for transfer of arachidonic acid to COX-2.
In conclusion, this study identifies the MEK1/ERK and MEK2 as well as the p38 MAP kinase pathways as major regulators of IL-1β-dependent PGE2 release in human airway epithelial-like A549 cells. At the relevant doses, inhibitors of these pathways had little or partial effects on induction of COX activity or COX-2 protein yet totally inhibited IL-1β-dependent PGE2 release indicating a primary role upstream of COX-2. Repression of PGE2 release by UO126 was attributed to inhibition of MEK2, which lead to reduced PLA2 activity of a non-cPLA2 phospholipase. Inhibition of MEK1/ERK by PD098059 and of p38 MAP kinase by SB203580 prevented PGE2 release but had no effect on arachidonate release suggesting that these pathways play roles in linking the relevant PLA2 activity to COX-2. In each case, we suggest that inhibitors of the MEK1/ERK, MEK2 and p38 MAP kinase pathways are potent and effective inhibitors of PG production from epithelial cells and may therefore provide useful therapeutic tools.
Acknowledgments
This work was supported by grants from the European Commission (Biomed II), the U.K. Medical Research Council and the Wellcome Trust.
Abbreviations
- COX
cyclo-oxygenase
- cPLA2
cytosolic phospholipase A2
- DMSO
dimethyl sulphoxide
- ERK
extrcellular regulated kinase
- IL
interleukin
- JNK
Jun N-terminal kinase
- MAFP
methyl arachidonyl fluorophosphonate
- MAP
mitogen activated protein
- MAPK
MAP kinase
- MAP2K
mitogen activated protein kinase kinase
- MAP3K
mitogen activated protein kinase kinase kinase
- MEK
mitogen activated protein kinase/extracellular regulated kinase kinase
- PD098059
2′-amino-3′methoxyflavone
- PG
prostaglandin
- PLA
phospholipase
- RIA
radioimmuoassay
- SB203580
4-(4-fluorophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)1H-imidazole
- sPLA2
secretory phospholipase A2
- UO126
1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene
References
- ABE A., SHAYMAN J.A. Purification and characterization of 1-O-acylceramide synthase, a novel phospholipase A2 with transacylase activity. J. Biol. Chem. 1998;273:8467–8474. doi: 10.1074/jbc.273.14.8467. [DOI] [PubMed] [Google Scholar]
- ALESSI D.R., CUENDA A., COHEN P., DUDLEY D.T., SALTIEL A.R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- BALSINDE J., DENNIS E.A. Distinct roles in signal transduction for each of the phospholipase A2 enzymes present in P388D1 macrophages. J. Biol. Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- BALSINDE J., DENNIS E.A. Function and inhibition of intracellular calcium-independent phospholipase A2. J. Biol. Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- BARNES P.J. Therapeutic strategies for allergic diseases. Nature. 1999;402 Suppl.:B31–B38. doi: 10.1038/35037026. [DOI] [PubMed] [Google Scholar]
- BORSCH-HAUBOLD A.G., PASQUET S., WATSON S.P. Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthase. J. Biol. Chem. 1998;273:28766–28772. doi: 10.1074/jbc.273.44.28766. [DOI] [PubMed] [Google Scholar]
- BOULTON T.G., NYE S.H., ROBBINS D.J., IP N.Y., RADZIEJEWSKA E., MORGENBESSER S.D., DEPINHO R.A., PANAYOTATOS N., COBB M.H., YANCOPOULOS G.D. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- CLARK J.D., LIN L.L., KRIZ R.W., RAMESHA C.S., SULTZMAN L.A., LIN A.Y., MILONA N., KNOPF J.L. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., NEWMAN S., FLOWER R.J. Lipocortin 1 and the control of cPLA2 activity in A549 cells. Glucocorticoids block EGF stimulation of cPLA2 phosphorylation. Biochem. Pharmacol. 1996;52:351–356. doi: 10.1016/0006-2952(95)02442-5. [DOI] [PubMed] [Google Scholar]
- CUENDA A., ROUSE J., DOZA Y.N., MEIER R., COHEN P., GALLAGHER T.F., YOUNG P.R., LEE J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- DEAN J.L., BROOK M., CLARK A.R., SAKLATVALA J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J. Biol. Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- DENNIS E.A. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends. Biochem. Sci. 1997;22:1–2. doi: 10.1016/s0968-0004(96)20031-3. [DOI] [PubMed] [Google Scholar]
- DEWITT D.L. Cox-2-selective inhibitors: the new super aspirins. Mol. Pharmacol. 1999;55:625–631. [PubMed] [Google Scholar]
- DUDLEY D.T., PANG L., DECKER S.J., BRIDGES A.J., SALTIEL A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAVATA M.F., HORIUCHI K.Y., MANOS E.J., DAULERIO A.J., STRADLEY D.A., FEESER W.S., VAN DYK D.E., PITTS W.J., EARL R.A., HOBBS F., COPELAND R.A., MAGOLDA R.L., SCHERLE P.A., TRZASKOS J.M. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- GILROY D.W., COLVILLE-NASH P.R., WILLIS D., CHIVERS J., PAUL-CLARK M.J., WILLOUGHBY D.A. Inducible cyclooxygenase may have anti-inflammatory properties. Nat. Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- GORDON R.D., LEIGHTON I.A., CAMPBELL D.G., COHEN P., CREANEY A., WILTON D.C., MASTERS D.J., RITCHIE G.A., MOTT R., TAYLOR I.W., BUNDELL K.R., DOUGLAS L., MORTEN J., NEEDHAM M. Cloning and expression of cystolic phospholipase A2 (cPLA2) and a naturally occurring variant. Phosphorylation of Ser505 of recombinant cPLA2 by p42 mitogen-activated protein kinase results in an increase in specific activity. Eur. J. Biochem. 1996;238:690–697. doi: 10.1111/j.1432-1033.1996.0690w.x. [DOI] [PubMed] [Google Scholar]
- GUAN Z., BAIER L.D., MORRISON A.R. p38 mitogen-activated protein kinase down-regulates nitric oxide and up-regulates prostaglandin E2 biosynthesis stimulated by interleukin-1beta. J. Biol. Chem. 1997;272:8083–8089. doi: 10.1074/jbc.272.12.8083. [DOI] [PubMed] [Google Scholar]
- HUANG Z., LIU S., STREET I., LALIBERTE F., ABDULLAH K., DESMARAIS S., WANG Z., KENNEDY B., PAYETTE P., REINDEAU D., WEECH P., GRESSER M. Methyl arachidonyl fluorophosphonate, a potent irreversible cPLA2 inhibitor, blocks the mobilization of arachidonic acid in human platelets and neutrophils. Mediat. Inflamm. 1994;3:307–308. [Google Scholar]
- INOUE H., YOKOYAMA C., HARA S., TONE Y., TANABE T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J. Biol. Chem. 1995;270:24965–24971. doi: 10.1074/jbc.270.42.24965. [DOI] [PubMed] [Google Scholar]
- ISHIZAKI J., SUZUKI N., HIGASHINO K., YOKOTA Y., ONO T., KAWAMOTO K., FUJII N., ARITA H., HANASAKI K. Cloning and characterization of novel mouse and human secretory phospholipase A(2)s. J. Biol. Chem. 1999;274:24973–24979. doi: 10.1074/jbc.274.35.24973. [DOI] [PubMed] [Google Scholar]
- KRAMER R.M., ROBERTS E.F., UM S.L., BORSCH, HAUBOLD A.G., WATSON S.P., FISHER M.J., JAKUBOWSKI J.A. p38 mitogen-activated protein kinase phosphorylates cytosolic phospholipase A2 (cPLA2) in thrombin-stimulated platelets. Evidence that proline-directed phosphorylation is not required for mobilization of arachidonic acid by cPLA2. J. Biol. Chem. 1996;271:27723–27729. doi: 10.1074/jbc.271.44.27723. [DOI] [PubMed] [Google Scholar]
- KYRIAKIS J.M., AVRUCH J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J. Biol. Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- LEE J.C., LAYDON J.T., MCDONNELL P.C., GALLAGHER T.F., KUMAR S., GREEN D., MCNULTY D., BLUMENTHAL M.J., HEYS J.R., LANDVATTER S.W., STRICKLER J.E., MCLAUGHLIN M.M., SIEMENS I.R., FISHER S.M., LIVI G.P., WHITE J.R., ADAMS J.L., YOUNG P.R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- LIN L.L., WARTMANN M., LIN A.Y., KNOPF J.L., SETH A., DAVIS R.J. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- MITCHELL J.A., BELVISI M.G., AKARASEREENONT P., ROBBINS R.A., KWON O.J., CROXTALL J., BARNES P.J., VANE J.R. Induction of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. Br. J. Pharmacol. 1994;113:1008–1014. doi: 10.1111/j.1476-5381.1994.tb17093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHELL J.A., LARKIN S., WILLIAMS T.J. Cyclooxygenase-2: regulation and relevance in inflammation. Biochem. Pharmacol. 1995;50:1535–1542. doi: 10.1016/0006-2952(95)00212-x. [DOI] [PubMed] [Google Scholar]
- MORITA I., SCHINDLER M., REGIER M.K., OTTO J.C., HORI T., DEWITT D.L., SMITH W.L. Different intracellular locations for prostaglandin endoperoxide H synthase-1 and -2. J. Biol. Chem. 1995;270:10902–10908. doi: 10.1074/jbc.270.18.10902. [DOI] [PubMed] [Google Scholar]
- MURAKAMI M., SHIMBARA S., KAMBE T., KUWATA H., WINSTEAD M.V., TISCHFIELD J.A., KUDO I. The functions of five distinct mammalian phospholipase A2S in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2S are functionally redundant and act in concert with cytosolic phospholipase A2. J. Biol. Chem. 1998;273:14411–14423. doi: 10.1074/jbc.273.23.14411. [DOI] [PubMed] [Google Scholar]
- NEAGOS G.R., FEYSSA A., PETERS GOLDEN M. Phospholipase A2 in alveolar type II epithelial cells: biochemical and immunologic characterization. Am. J. Physiol. 1993;264:L261–L268. doi: 10.1152/ajplung.1993.264.3.L261. [DOI] [PubMed] [Google Scholar]
- NEWTON R., KUITERT L.M., BERGMANN M., ADCOCK I.M., BARNES P.J. Evidence for involvement of NF-kB in the transcriptional control of COX-2 gene expression by IL-1β. Biochem. Biophys. Res. Commun. 1997b;237:28–32. doi: 10.1006/bbrc.1997.7064. [DOI] [PubMed] [Google Scholar]
- NEWTON R., KUITERT L.M., SLATER D.M., ADCOCK I.M., BARNES P.J. Cytokine induction of cytosolic phospholipase A2 and cyclooxygenase-2 mRNA is suppressed by glucocorticoids in human epithelial cells. Life Sci. 1997a;60:67–78. doi: 10.1016/s0024-3205(96)00590-5. [DOI] [PubMed] [Google Scholar]
- NEWTON R., SEYBOLD J., KUITERT L.M.E., BERGMANN M., BARNES P.J. Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J. Biol. Chem. 1998;273:32312–32321. doi: 10.1074/jbc.273.48.32312. [DOI] [PubMed] [Google Scholar]
- NEWTON R., STEVENS D.A., HART L.A., LINDSAY M., ADCOCK I.M., BARNES P.J. Superinduction of COX-2 mRNA by cycloheximide and interleukin-1β involves increased transcription and correlates with increased NF-kappa B and JNK activation. FEBS Lett. 1997c;418:135–138. doi: 10.1016/s0014-5793(97)01362-8. [DOI] [PubMed] [Google Scholar]
- PAWSON T., SCOTT J.D. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- PAYNE D.M., ROSSOMANDO A.J., MARTINO P., ERICKSON A.K., HER J.H., SHABANOWITZ J., HUNT D.F., WEBER M.J., STURGILL T.W. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase) EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PICKARD R.T., STRIFLER B.A., KRAMER R.M., SHARP J.D. Molecular cloning of two new human paralogs of 85-kDa cytosolic phospholipase A2. J. Biol. Chem. 1999;274:8823–8831. doi: 10.1074/jbc.274.13.8823. [DOI] [PubMed] [Google Scholar]
- POULIOT M., BAILLARGEON J., LEE J.C., CLELAND L.G., JAMES M.J. Inhibition of prostaglandin endoperoxide synthase-2 expression in stimulated human monocytes by inhibitors of p38 mitogen- activated protein kinase. J. Immunol. 1997;158:4930–4937. [PubMed] [Google Scholar]
- RAINGEAUD J., GUPTA S., ROGERS J.S., DICKENS M., HAN J., ULEVITCH R.J., DAVIS R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- SA G., MURUGESAN G., JAYE M., IVASHCHENKO Y., FOX P.L. Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase- dependent phosphorylation pathway in endothelial cells. J. Biol. Chem. 1995;270:2360–2366. doi: 10.1074/jbc.270.5.2360. [DOI] [PubMed] [Google Scholar]
- SCHALKWIJK C.G., VAN DER HEIJDEN M.A., BUNT G., MAAS R., TERTOOLEN L.G., VAN BERGEN EN HENEGOUWEN P.M., VERKLEIJ A.J., VAN DEN BOSCH H., BOONSTRA J. Maximal epidermal growth-factor-induced cytosolic phospholipase A2 activation in vivo requires phosphorylation followed by an increased intracellular calcium concentration. Biochem. J. 1996;313:91–96. doi: 10.1042/bj3130091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHERLE P.A., JONES E.A., FAVATA M.F., DAULERIO A.J., COVINGTON M.B., NURNBERG S.A., MAGOLDA R.L., TRZASKOS J.M. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J. Immunol. 1998;161:5681–5686. [PubMed] [Google Scholar]
- SCHIEVELLA A.R., REGIER M.K., SMITH W.L., LIN L.L. Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J. Biol. Chem. 1995;270:30749–30754. doi: 10.1074/jbc.270.51.30749. [DOI] [PubMed] [Google Scholar]
- SEIBERT K., ZHANG Y., LEAHY K., HAUSER S., MASFERRER J., PERKINS W., LEE L., ISAKSON P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOKUMOTO H., CROXTALL J.D., FLOWER R.J. Differential role of extra- and intracellular calcium in bradykinin and interleukin 1 alpha stimulation of arachidonic acid release from A549 cells. Biochim. Biophys. Acta. 1994;1211:301–309. doi: 10.1016/0005-2760(94)90154-6. [DOI] [PubMed] [Google Scholar]
- UNDERWOOD K.W., SONG C., KRIZ R.W., CHANG X.J., KNOPF J.L., LIN L.L. A novel calcium-independent phospholipase A2, cPLA2-gamma, that is prenylated and contains homology to cPLA2. J. Biol. Chem. 1998;273:21926–21932. doi: 10.1074/jbc.273.34.21926. [DOI] [PubMed] [Google Scholar]
- VALENTIN E., GHOMASHCHI F., GELB M.H., LAZDUNSKI M., LAMBEAU G. On the diversity of secreted phospholipases A(2). Cloning, tissue distribution, and functional expression of two novel mouse group II enzymes [published erratum appears in J Biol Chem 2000 Jan 21;275(3):2246] J. Biol. Chem. 1999;274:31195–31202. doi: 10.1074/jbc.274.44.31195. [DOI] [PubMed] [Google Scholar]
- XIE W., HERSCHMAN H.R. v-src induces prostaglandin synthase 2 gene expression by activation of the c-Jun N-terminal kinase and the c-Jun transcription factor. J. Biol. Chem. 1995;270:27622–27628. doi: 10.1074/jbc.270.46.27622. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO K., ARAKAWA T., UEDA N., YAMAMOTO S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J. Biol. Chem. 1995;270:31315–31320. doi: 10.1074/jbc.270.52.31315. [DOI] [PubMed] [Google Scholar]