

Abstract
The successive effects of the angiotensin-converting enzyme inhibitor captopril (CAP, 2 mg kg−1+1 mg kg−1 30 min−1 infusion) and the neutral endopeptidase 24-11 inhibitor retrothiorphan (RT, 25 mg kg−1+12.5 mg kg−1 30 min−1 infusion) were studied on femoral vascular conductance (FVC) in streptozotocin-induced diabetic (STZ-SD) and control Sprague-Dawley (C-SD) rats. The role of the kinin-nitric oxide (NO) pathway was assessed by (1) using pre-treatments: a bradykinin (BK) B2 receptor antagonist (Hoe-140, 300 μg kg−1), a NO-synthase inhibitor (Nω-nitro-L-arginine methyl ester, L-NAME, 10 mg kg−1), a kininase I inhibitor (DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid, MGTA, 10 mg kg−1+20 mg kg−1 20 min−1 infusion) and (2) comparing the effects in STZ-induced diabetic (STZ-BN) and control Brown-Norway kininogen-deficient (C-BN) rats.
In C-SDs, CAP and CAP+RT increased FVC similarly. In STZ-SDs, FVC and FBF were decreased compared to C-SDs. CAP+RT increased them more effectively than CAP alone.
In both C-SDs and STZ-SDs, the femoral bed vasodilatation elicited by CAP was inhibited by Hoe-140 and L-NAME. The FVC increase elicited by CAP+RT was not significantly reduced by Hoe-140 but was inhibited by L-NAME and Hoe-140+MGTA.
In C-BNs, the vasodilatator responses to CAP and CAP+RT were abolished and highly reduced, respectively. In STZ-BNs, these responses were abolished.
These results show that in STZ-SDs, CAP+RT improve FBF and FVC more effectively than CAP alone. These effects are linked to an increased activation of the kinin-NO pathway. BK could lead to NO production by BK B2 receptor activation and another pathway in which kininase I may be involved.
Keywords: Streptozotocin-induced diabetes, femoral vascular conductance, angiotensin-converting enzyme inhibitor, neutral endopeptidase 24-11 inhibitor, MGTA, bradykinin, nitric oxide
Introduction
A decrease in endothelium-dependent vascular relaxation is a common feature in diabetes mellitus and is mainly due to an impairment in nitric oxide (NO) metabolism (for review, see Pieper, 1998). Clinical (O'Driscoll et al., 1997) and experimental (Olbrich et al., 1996) studies have demonstrated that angiotensin converting enzyme (ACE) inhibitors improve endothelium-dependent vasodilatation in type I diabetes. This may be explained, at least partly, by an activation of the kinin-NO pathway (for review, see Gazis et al., 1998).
In non-diabetic situations, ACE is the primary enzyme responsible for the breakdown of bradykinin(1–9) into bradykinin(1–7) in the vascular wall (Bhoola et al., 1992). However, increasing evidence indicates that neutral endopeptidase 24-11 (NEP 24-11) is also involved in this catabolism, particularly during ACE inhibition (Graf et al., 1993). Indeed, in anaesthetized dogs, the combination of captopril and SQ-28603, a NEP 24-11 inhibitor, synergistically enhances the renal vasodilatation induced by exogenous BK (Seymour et al., 1994). In heart failure associated with impaired endothelial NO metabolism (Drexler et al., 1994), combined NEP/ACE inhibition decreases systemic peripheral vascular resistance more effectively than ACE inhibition alone. It has been suggested that bradykinin (BK) could contribute to this synergistic effect (Seymour et al., 1993; Trippodo et al., 1995). Therefore, we tested the hypothesis that, in diabetes, the combined NEP/ACE inhibition may decrease systemic peripheral vascular resistance more effectively than ACE inhibition alone by increased activation of the kinin-NO pathway.
Accordingly, we studied the haemodynamic effects of ACE and combined ACE and NEP 24-11 inhibition in the femoral vascular bed in Sprague-Dawley (SD) rats with diabetes induced by injection of streptozotocin (STZ). We compared these animals to control SD rats. We have chosen this vascular bed since the vasodilatation elicited by BK in the hindquarters vascular bed is particularly dependent on NO and is selectively impaired in STZ-treated rats (Kiff et al., 1991a). As inhibitors, we used retrothiorphan (RT), which is a potent and selective NEP 24-11 inhibitor (Ki=6 nM) with a very low affinity for ACE (IC50>10 μM) (Roques et al., 1983) and captopril (CAP), a classical ACE inhibitor. The role of the kinin-NO pathway was assessed by acute pre-administration of the kinin B2 antagonist Hoe-140 or the NO-synthase inhibitor Nω-nitro-L-arginine methyl ester (L-NAME). In addition, the effects of ACE and combined NEP/ACE inhibition were studied in control and STZ-induced diabetic Brown-Norway Katholiek kininogen-deficient (BN) rats, which have low kinins plasma and tissue levels (Kouyoumadjian & Damas, 1998). Other studies have shown that BK can be cleaved by kininase I (Bhoola et al., 1992, Campbell et al., 1993; Volpe et al., 1996), especially when ACE and NEP activities were reduced (Chodimella et al., 1991), thereby generating the endogenous physiological precursor for NO biosynthesis, L-arginine (Volpe et al., 1996). We therefore assessed the possible involvement of kininase I by the use of the kininase I inhibitor, DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid (MGTA), both alone and in combination with Hoe-140.
Methods
Experimental animals
Male SD rats (6 weeks old) weighing 231±5 g (Janvier, Le Genest St Isle, France) were kept in a conventional animal room. Animals were fed standard rat chow (UAR AO4, Villemoisson, France) and allowed access to water ad libitum. For each experiment, rats were divided at random into two groups. One group was made diabetic by STZ injection (55 mg kg−1 in citrate buffer, pH=4.5, i.m. according to Nakhodo & Wong, 1979). The control group received buffer alone (1 ml kg−1, i.m.). Blood glucose levels were determined from a tail capillary blood sample 7 days later using a glucometer (‘One Touch Profile', Life Scan Co, Paris, France) which allows measurements of blood glucose levels ⩽33.3 mM. Rats with blood glucose levels higher than 17 mM were considered diabetic and included into the experiment. BN rats (139±4 g, Janvier, Le Genest St Isle, France) were used at the same age of 6 weeks and were made diabetic by the same method.
Animal preparation and regional haemodynamic measurements
Experiments began 4 weeks later. Since STZ or buffer had been injected to the rats at the same time, duration of diabetes was variable within an experiment (from 28 days for the first rat to 35–42 days for the last one) but reproducible between experiments.
Animals were anaesthetized with a mixture of chloralose (100 mg kg−1) and urethan (450 mg kg−1) administered i.p. Anaesthesia was maintained throughout the experiment by subcutaneous injections of one eighth of the initial dose every 30–40 min. Animals breathed room air spontaneously via a tracheal cannula. Arterial blood pressure was monitored by a BP-T transducer (Emka technologies, Paris, France) via a cannula containing heparinized saline inserted in the right common artery. The pudendal vein was cannulated to allow i.v. injections and infusions. A small incision was made in the skin overlaying the femoral vessels of the right leg. The femoral artery was separated from the femoral vein and the saphenous nerve. A flow probe (VB series 0.5 mm internal diameter, Transonic Systems Inc., Emka technologies, Paris, France) was positioned around the femoral artery just distal to the rectus abdominis muscle. The cavity containing the probe was filled with acoustic lubricating jelly (Mohawk Medical Supply, Utica, U.S.A.). The probe was connected to a transit time flowmeter (Model T106 Ultrasonic Volume Flowmeter, Transonic Systems Inc., Emka technologies, Paris, France). Mean flow and arterial blood pressure signals were recorded continuously on a potentiometric recorder (Linear, Bioblock Scientific, Illkirch, France). Mean arterial blood pressure (MAP) and mean femoral blood flow (FBF) were used to calculate femoral vascular conductance (FVC) : FBF (μl min−1)/MAP (mmHg). Body temperature was monitored and maintained at 37°C by a heating pad (Ellab instruments, Carrieri, Paris, France). Blood glucose levels were determined as described above at the end of each experiment. Rats were sacrificed by an i.v. overdose of sodium pentobarbitone (160 mg kg−1, i.v.). All procedures adopted for the care and euthanasia of the rats were in compliance with the European Community Standards on the care and use of laboratory animals (Ministère de l'Agriculture, France : authorisation n° 00.860).
Experimental protocols
Six types of experiments were carried out.
Common protocol
For each type of experiment, the rats were allowed to stabilize for approximately 30 min after surgery. The inhibitors were administered as follows: CAP was administered as a priming bolus (2 mg kg−1, i.v.) followed by a sustaining infusion (2 mg kg−1 h−1) for a 30 min period. RT was then immediately administered as a priming bolus (25 mg kg−1, i.v., 1 ml kg−1) followed by a sustaining infusion (25 mg kg−1 h−1) for a 30 min period (Pham et al., 1992). Angiotensin I (0.2 μg kg−1, i.v.) was injected before and at the end of CAP infusion and also at the end of RT infusion to confirm ACE inhibition. The infusions of the inhibitors were set at 2 ml h−1. Each experiment lasted about 2 h.
Pretreatment protocols
Five types of experiments were performed on control (C-SD) and STZ-induced diabetic SD (STZ-SD) rats. In the first protocol, the inhibitors were administered as described above without any pre-treatment. In the others, the following pre-treatments were administered: (1) the NO-synthase inhibitor L-NAME (10 mg kg−1, i.v., Kiff et al., 1991a), injected 15 min before the inhibitors; (2) the BK B2 receptor antagonist Hoe-140 (300 μg kg−1, i.v), injected 10 min before the inhibitors; (3) the kininase I inhibitor MGTA, administered as a priming bolus (10 mg kg−1, i.v., 1 ml kg−1), followed by a sustaining infusion (20 mg kg−1 20 min−1, Salgado et al., 1986) just before the inhibitors; (4) Hoe-140 (300 μg kg−1, i.v.) followed, 10 min later, by MGTA (10 mg kg−1 i.v.+20 mg kg−1 20 min−1) just before the inhibitors. BK (2 nmol kg−1, i.v.) was tested before and after Hoe-140 and also at the end of each infusion when necessary to confirm the B2 receptor blockade. In the last type of experiment, the inhibitors alone were administered in control (C-BN) and STZ-treated BN (STZ-BN) rats as described above.
Time control rats
Saline was administered as a priming bolus (0.5 ml kg−1) followed by a sustaining infusion (2 ml h−1) for two successive 30 min periods in control SD rats (n=5). BK (2 nmol kg−1, i.v.) was injected before and at the end of the infusions.
Results expression and data analysis
Data are presented as means±s.e.mean. Baseline haemodynamic values were taken just before drug administration. In experiments where animals were pre-treated, baseline haemodynamic values after pre-treatment were taken just before the administration of the inhibitors. The effects of CAP and RT on haemodynamic variables were appreciated during the last 10 min of each infusion when a steady haemodynamic state was achieved. Values taken at 20, 25 and 30 min were averaged. When the inhibitors were given without pre-treatment, results were expressed in absolute value and analysed by one-way analysis of variance (ANOVA) for repeated measures, followed by Newman-Keuls test. To compare the effects of CAP and CAP+RT with or without pre-treatment(s), results were expressed as changes in relation to baseline value (for inhibitors alone) or to baseline value after pre-treatment and analysed by ANOVA followed by one-tail Dunnett's t-test. Responses to exogenous BK and angiotensin I were expressed as changes in values taken just before the injection and at the nadir of the response and analysed by ANOVA followed by one-tail Dunnett's t-test. In other cases, data were analysed by paired or unpaired Student's t-test or Mann and Whitney test as appropriate. A P value <0.05 was considered as significant.
Drugs and peptides
Streptozotocin, bradykinin, angiotensin I, captopril and Nω-nitro-L-arginine methyl ester were purchased from Sigma (Sigma Chemical Co., St Quentin-Fallavier, France), DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid from France Biochem (Meudon, France), D-Arg-(Hyp3,Thi5,D-Tic7,Oic8)-bradykinin (Hoe-140) from Hoechst-Marion Roussel (Frankfurt, Germany), chloralose and urethan from Prolabo (Paris, France). Retrothiorphan was synthesized in our laboratory (Laboratoire de Pharmacochimie moléculaire, INSERM U266, UMR 8600 CNRS). All drugs were dissolved in 0.9% NaCl. RT was dissolved using 1 M CO3Na2 and the pH was adjusted to 7.4 with 1 M HCl. Injections were given as 0.5 ml kg−1 unless otherwise precise and flushed with 0.05 ml of isotonic saline.
Results
Characteristics of the diabetic animals
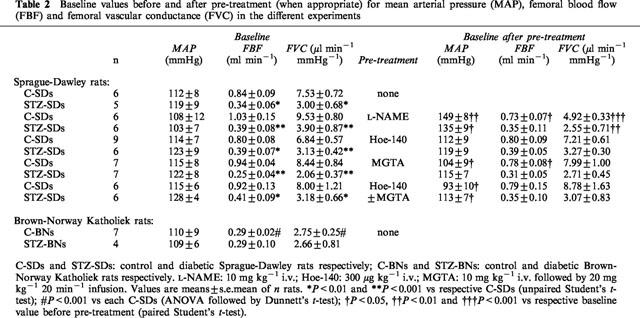
Values of blood glucose levels and body weights of C-SD and STZ-SD rats used for the different experiments were pooled in Table 1. STZ-SDs had higher blood glucose levels and lower body weights than those of age-matched C-SDs. Under basal conditions, FBF were decreased in STZ-SDs when compared to C-SDs, whereas MAP were not significantly modified. Calculated FVC were then lowered (Table 2). This corresponded to a femoral vasoconstriction relative to C-SDs. When compared to C-SDs, the C-BNs had a lower body weight (P<0.001) corresponding to a slower weight curve and slightly but significant higher blood glucose levels (P<0.05, Table 1). They had similar MAP and lower FBF and FVC (Table 2). This strain was very sensititive to STZ (eight deaths out of 12 with 55 mg kg−1 of STZ, i.m.), even when lower doses of STZ were tested (35 and 15 mg kg−1, i.m.). However, the STZ-BNs had a weight gain similar to that of the STZ-SDs (12.2 vs 14%), taking into account that the C-BNs had a slower weight curve than the C-SDs. They had similar MAP, FBF and FVC (Table 2) and higher blood glucose levels (Table 1) when compared to C-BNs.
Table 1.
General characteristics of animals at sacrifice

Table 2.
Baseline values before and after pre-treatment (when appropriate) for mean arterial pressure (MAP), femoral blood flow (FBF) and femoral vascular conductance (FVC) in the different experiments
Time controls
Administration of saline alone had no consistent effect on haemodynamic parameters (MAP was 107±4 vs 100±7 mmHg before and after the infusions respectively, P=0.65; FBF: 0.75±0.10 vs 0.66±0.05 ml min−1, P=0.52; FVC: 6.6±0.5 vs 6.7±0.9 μl min−1 mmHg−1, P=0.68; n=5). Response to exogenous BK was unchanged over the duration of the experiment (at the beginning and at the end of the infusions respectively, for MAP: −31±2 vs −34±2 mmHg, P=0.31; for FVC, +14±2 vs +15±2 μl min−1 mmHg−1, P=0.65; n=5).
Haemodynamic effects of the ACE and NEP inhibitors
In C-SDs, CAP induced a fall in MAP which reached a maximum (−59±8 mmHg) within 2 min. MAP then slightly rose and was stabilized within 10 min until the end of the infusion (Figure 1a). The fall in MAP was associated with an increase in FBF (+0.6±0.1 ml min−1) which returned to baseline levels within 5–10 min (Figure 1b). FVC calculated during the last 10 min of CAP infusion was highly increased (Figure 1c).
Figure 1.
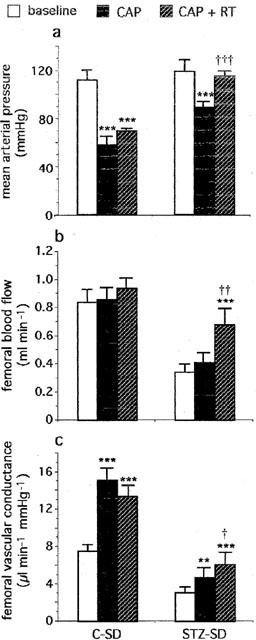
Effects of captopril (CAP, 2 mg kg−1, i.v.+1 mg kg−1 30 min−1 infusion) alone and in combination with retrothiorphan (RT, 25 mg kg−1, i.v.+12.5 mg kg−1 30 min−1 infusion) on (a) mean arterial pressure, (b) femoral blood flow and (c) femoral vascular conductance in control (C-SD, n=6) and diabetic (STZ-SD, n=5) Sprague-Dawley rats. For each rat, values measured at 20, 25 and 30 min after the start of each infusion are averaged. Each column represents the mean value and the T bar the s.e.m. **P<0.01 and ***P<0.001 vs baseline; †P<0.05, ††P<0.01 and †††P<0.001 vs CAP alone (ANOVA for repeated measures followed by Newman-Keuls test).
In the presence of CAP, RT induced a modest fall in MAP (−19±5 mmHg). MAP then increased and was stabilized within 10 min until the end of RT infusion (Figure 1a). The fall in MAP was associated with an increase in FBF (+0.5±0.1 ml min−1) which returned slowly to baseline levels within 10–15 min (Figure 1b). FVC calculated during the last 10 min of RT infusion was significantly increased (Figure 1c). No significant difference was observed between the increases in FVC induced by CAP and CAP+RT (Figure 1c).
In STZ-SDs, haemodynamic changes induced by CAP alone were similar to those observed in C-SDs. CAP induced a fall in MAP which reached a maximum within 2 min. Then MAP rose slightly and became stable within 5–10 min (Figure 1a). No difference was observed in the maximum fall between STZ-SDs and C-SDs (−45±10 vs −59±8 mmHg respectively) but the hypotensive response to CAP during the last 10 min of the infusion was smaller in STZ-SDs (−29±6 vs −54±9 mmHg in C-SDs, P<0.01). No difference was observed in the maximum increase in FBF between STZ-SDs and C-SDs (+0.5±0.1 vs +0.6±0.1 ml min−1 respectively). As in C-SDs, FBF returned to baseline value within 5–10 min (Figure 1b). FVC calculated during the last 10 min of the infusion was slightly but significantly increased (Figure 1c). The vasodilatator response elicited by CAP infusion was smaller in STZ-SDs than in C-SDs (+1.8±0.4 vs +7.6±1.1 μl min−1 mmHg−1 respectively, P<0.01).
In the presence of CAP, RT induced a transient fall in MAP in STZ-SDs. There was no difference in the maximum fall between STZ-SDs and C-SDs (−24±7 vs −19±5 mmHg respectively). MAP then rose and returned slowly to baseline levels within 15 min (Figure 1a). Change in MAP was associated with an increase in FBF, which was higher than the corresponding response in C-SDs (+0.7±0.1 vs +0.5±0.1 ml min−1, P<0.01). FBF then slightly decreased and was stabilized within 5 min until the end of RT infusion. After stabilization, FBF was significantly elevated when compared to baseline value or the value observed at the end of CAP infusion (Figure 1b). During the last 10 min of RT infusion, FVC was significantly increased compared to baseline value and significantly higher than that induced by CAP alone (Figure 1c). One can notice that, in STZ-SDs treated with CAP+RT, the FBF and FVC levels have been restored to normal control baseline values (0.68±0.12 ml min−1 and 6.1±1.3 μl min−1 mmHg−1 in the presence of CAP+RT in STZ-SDs vs 0.84±0.09 ml min−1 and 7.5±0.7 μl min−1 mmHg−1 for baseline values in C-SDs, P>0.05).
In the five experiments performed in C-SDs and STZ-SDs, CAP effectively inhibited ACE since the hypertensive response elicited by angiotensin I was markedly reduced throughout the experiment (for C-SDs, +38±3 mmHg before CAP vs +12±2 mmHg and +13±2 mmHg at the end of CAP infusion and RT infusion respectively, P<0.01, n=34; for STZ-SDs, +37±3 mmHg before CAP vs +16±3 mmHg and +11±2 mmHg at the end of CAP infusion and RT infusion respectively, P<0.01, n=30).
Effect of the NO-synthase inhibitor, Nω-nitro-L-arginine methyl ester, on the responses to the ACE and NEP inhibitors
In both C-SDs and STZ-SDs, bolus injection of L-NAME caused an increase in MAP which was associated with a marked and sustained vasoconstriction (Table 2). However, the STZ-SDs showed a smaller decrease in FVC compared to the C-SDs (−1.4±0.3 vs −4.7±0.6 μl min−1 mmHg−1, P<0.01). In both groups, the femoral bed vasodilatations elicited by CAP alone and CAP+RT were markedly reduced (Figures 2c and 3c).
Figure 2.
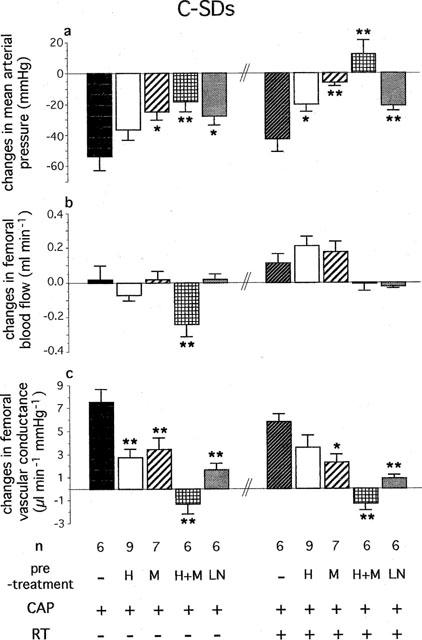
Effects of Hoe-140 (H, 300 μg kg−1, i.v.), MGTA (M, 10 mg kg−1, i.v.+20 mg kg−1 20 min−1 infusion), Hoe-140+MGTA (H+M) and L-NAME (LN, 10 mg kg−1, i.v.) on changes in (a) mean arterial pressure, (b) femoral blood flow and (c) femoral vascular conductance evoked by successive administration of captopril (CAP, 2 mg kg−1, i.v.+1 mg kg−1 30 min−1 infusion) and retrothiorphan (RT, 25 mg kg−1, i.v.+12.5 mg kg−1 30 min−1 infusion) in control Sprague-Dawley rats (C-SDs). For each rat, values measured at 20, 25 and 30 min after the start of each infusion are averaged. Each column represents the mean value of n rats and the vertical T bar the s.e.m. *P<0.05 and **P<0.01 vs CAP without pre-treatment or vs CAP+RT without pre-treatment (ANOVA followed by Dunnett's t-test).
Figure 3.
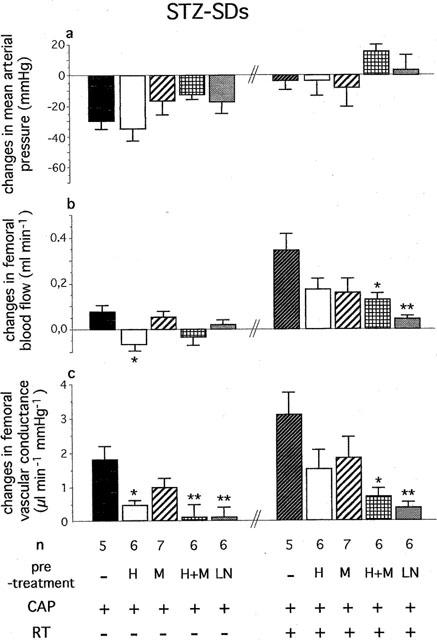
Effects of Hoe-140 (H, 300 μg kg−1, i.v.), MGTA (M, 10 mg kg−1, i.v.+20 mg kg−1 20 min−1 infusion), Hoe-140+MGTA (H+M) and L-NAME (LN, 10 mg kg−1, i.v.) on changes in (a) mean arterial pressure, (b) femoral blood flow and (c) femoral vascular conductance evoked by successive administration of captopril (CAP, 2 mg kg−1, i.v.+1 mg kg−1 30 min−1 infusion) and retrothiorphan (RT, 25 mg kg−1, i.v.+12.5 mg kg−1 30 min−1 infusion) in diabetic Sprague-Dawley rats (STZ-SDs). For each rat, values measured at 20, 25 and 30 min after the start of each infusion are averaged. Each column represents the mean value of n rats and the vertical T bar the s.e.mean. *P<0.05 and **P<0.01 vs CAP without pre-treatment or vs CAP+RT without pre-treatment (ANOVA followed by Dunnett's t-test).
Effect of the B2 receptor antagonist Hoe-140 on the responses to the ACE and NEP inhibitors
The administration of Hoe-140 did not modify baseline MAP, FBF and FVC in C-SDs and STZ-SDs (Table 2). In the presence of the B2 receptor antagonist, the FVC increase elicited by CAP alone was significantly reduced in both C-SDs and STZ-SDs. The vasodilatation induced by CAP+RT was also reduced but not significantly (Figures 2c and 3c). The hypotensive and vasodilatator responses elicited by the exogenous BK were markedly reduced after RT infusion (in C-SDs before Hoe-140 administration versus after RT infusion: for MAP, −39±5 vs −5±3 mmHg, P<0.01, for FVC, +18±3 vs +7±3 μl min−1 mmHg−1, P<0.01, n=9; in STZ-SDs: for MAP, −19±4 vs 0 mmHg, P<0.01, for FVC, +7±1 vs +2±1 μl min−1 mmHg−1, P<0.05, n=6). In addition, the vasodilatator response elicited by the exogenous BK was reduced in STZ-SDs compared to C-SDs (+7±1 μl min−1 vs +18±3 mmHg−1 respectively, P<0.01).
Effect of the kininase I inhibitor, DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid, alone or in combination with the B2 receptor antagonist Hoe-140 on the responses to the ACE and NEP inhibitors
The administration of MGTA induced no haemodynamic changes except a slight decrease in MAP and FBF in C-SDs. When MGTA was administered after Hoe-140, baseline MAP was slightly decreased in both groups of rats, whereas FBF and FVC were not modified (Table 1).
In C-SDs, the FVC increases induced by CAP and CAP+RT were reduced by MGTA alone and completely inhibited by MGTA+Hoe-140 (Figure 2c).
In STZ-SDs, MGTA alone reduced but not significantly the vasodilatation induced by CAP and CAP+RT (Figure 3c). When MGTA was administered in combination with Hoe-140, these vasodilatations were strongly and significantly reduced (Figure 3c).
In both C-SDs and STZ-SDs, the depressor and vasodilatator responses elicited by the exogenous BK were markedly reduced at the end of RT infusion (in C-SDs, before Hoe-140 administration versus after RT infusion: for MAP, −34±5 vs −4±3 mmHg, P<0.01, for FVC, +28±2 vs +6±2 μl min−1 mmHg−1, P<0.01, n=6; in STZ-SDs: for MAP, −34±7 vs −6±3 mmHg, P<0.01, for FVC, +13±3 vs +7±2 μl min−1 mmHg−1, P<0.05, n=6). In addition, the vasodilatation elicited by the exogenous BK was reduced in STZ-SDs compared to C-SDs (+13±3 μl min−1 vs +28±2 mmHg−1 respectively, P<0.01).
Haemodynamic effects of the ACE and NEP inhibitors in control and diabetic Brown-Norway Kininogen-deficient rats
In C-BNs, CAP induced a fall in MAP (−45±6 mmHg) which reached a maximum within 1 min. MAP then rose and was stabilized within 10–15 min after the start of CAP infusion (Figure 4a). The depressor response to CAP infusion was reduced when compared to C-SDs (−23±2 vs −54±9 mmHg, P<0.01). No changes in FBF (Figure 4b) and FVC (Figure 4c) were observed.
Figure 4.
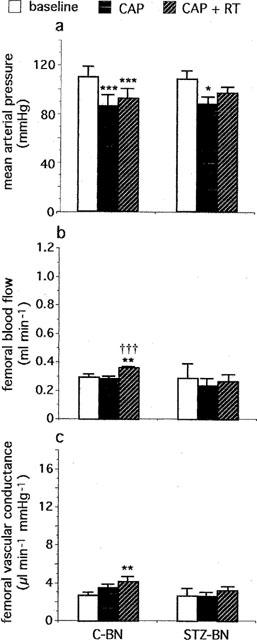
Effects of captopril (CAP, 2 mg kg−1, i.v.+1 mg kg−1 30 min−1 infusion), alone and in combination with retrothiorphan (RT, 25 mg kg−1, i.v.+12.5 mg kg−1 30 min−1 infusion), on (a) mean arterial pressure, (b) femoral blood flow and (c) femoral vascular conductance in control (C-BN, n=7) and diabetic (STZ-BN, n=4) Brown-Norway kininogen-deficient rats. For each rat, values measured at 20, 25 and 30 min after the start of each infusion are averaged. Each column represents the mean value and the T bar the s.e.mean. *P<0.05, **P<0.01 and ***P<0.001 vs baseline; †††P<0.001 vs CAP alone (ANOVA for repeated measures followed by Newman-Keuls test).
In the presence of CAP, RT administration in C-BNs caused a slight decrease in MAP (−12±1 mmHg). MAP then increased and was rapidly stabilized within 5 min (Figure 4a). During the last 10 min of RT infusion, the depressor response to CAP+RT was reduced compared to C-SDs (−17±3 vs −43±9 mmHg, P<0.05). FBF was slightly increased (Figure 4b). FVC was also slightly increased (Figure 4c) but to a much lesser extent than in C-SDs (+1.5±0.4 vs +5.8±0.7 μl min−1 mmHg−1, P<0.001).
In STZ-BNs, haemodynamic changes induced by CAP alone were similar to those observed in C-BNs. CAP induced a fall in MAP (−43±10 mmHg) which reached a maximum within 1 min. MAP then rose and was stabilized within 10–15 min after the start of CAP infusion (Figure 4a). No changes in FBF (Figure 4b) and FVC (Figure 4c) were observed. In the presence of CAP+RT, no changes in MAP, FBF and FVC were observed until the end of the infusion (Figure 4a,b,c).
In both C-BNs and STZ-BNs, CAP effectively inhibited ACE since the hypertensive response elicited by angiotensin I was inhibited throughout the experiment (for C-BNs, +32±2 mmHg before CAP vs 0 and +2±2 mmHg at the end of CAP infusion and RT infusion respectively, P<0.01, n=7; for STZ-BNs, +28±2 mmHg before CAP vs 0 and 0 mmHg at the end of CAP infusion and RT infusion respectively, P<0.01, n=4).
Discussion
The present study shows that the combined NEP/ACE inhibition can correct impaired FBF and FVC in STZ-SD rats more effectively than ACE inhibition alone.
In C-SD rats, CAP did not modify FBF but markedly decreased MAP, leading to an important increase in FVC. These results are consistent with those of Richer et al. (1989) in anaesthetized rats, showing that CAP increases renal and mesenteric blood flows without modifying hindlimb blood flow. Vasodilatator properties of CAP may be due to angiotensin II vasomotor tone suppression but also to the enhancement of vasodilatator peptides such as BK. In our study, pre-treatment with the NO-synthase inhibitor L-NAME and the BK B2 receptor antagonist Hoe-140 strongly reduced the increase in FVC induced by CAP, suggesting that this effect was due to an activation of the kinin-NO pathway. This was supported by the absence of vasodilatation after CAP infusion in C-BN, which have low plasma and tissue kinin levels (Kouyoumadjian & Damas, 1998). These findings are in agreement with studies in vivo showing that the accumulation of endogenous BK and the consequent endothelial NO production are involved in the vascular effects of ACE inhibitors in anaesthetized rats (Tio et al., 1989) and in humans (Hornig et al., 1997).
In the presence of CAP and RT, haemodynamic responses in C-SDs were similar to those induced by CAP alone, with a fall in MAP and no significant change in FBF. It has been previously reported that the combined NEP/ACE inhibition did not significantly modify hindquarters blood flow in conscious rats (Gardiner et al., 1997). This was associated with a fall in MAP and an increase in mesenteric and renal blood flows and conductances. It is possible that the combined NEP/ACE inhibition also leads to an increase in renal and mesenteric blood flows in anaesthetized rats. As cardiac output has been found not to be modified in the presence of CAP+RT (Maxwell et al., 1995), this can explain that the FBF was not consistently modified.
In our study, the femoral vasodilatation induced by CAP+RT in C-SDs was markedly inhibited by L-NAME, suggesting that NO production was involved in this phenomenon. In addition, the femoral vasodilatation induced by CAP+RT was strongly reduced in C-BNs when compared to C-SDs. This suggest that BK play an important role in the vasodilatator response to CAP+RT in C-SDs. However, the femoral vasodilatation was reduced but not significantly by Hoe-140 pre-treatment in C-SDs. The dose of Hoe-140 cannot be questioned since the effects of exogenous BK were still inhibited by Hoe-140 at the end of the experiment. So we must consider that in the presence of CAP+RT, a component of the BK response was independent of the B2 receptor activation. In fact, Volpe et al. (1996) reported in the isolated heart of rat, that NO release induced by exogenous BK was unaffected by Hoe-140 but blocked by the kininase I (KI) carboxypeptidase inhibitor MGTA. The KI can cleave L-arginine from the C terminus of BK(1–9), thus generating des-Arg9-BK and the endogenous physiological precursor for NO biosynthesis, L-arginine (Bhoola et al., 1992, Campbell et al., 1993; Volpe et al., 1996). Indeed, in other vascular territories, the KI has been implicated in the metabolism of BK (Griswold et al., 1998), especially when ACE and NEP activities are reduced (Chodimella et al., 1991). In our study, when the KI inhibitor MGTA was given alone in pre-treatment, the vasodilatation elicited by CAP+RT was 2 fold decreased in C-SDs. When MGTA was given in combination with Hoe-140, the vasodilatation elicited by CAP+RT was abolished. A possible explanation is that CAP+RT could induce a local accumulation of BK to such a level that the KI activity may become significant. In these conditions, BK could yield NO in two ways: BK B2 receptor activation and BK hydrolysis by KI. These results, however, must be interpreted cautiously because the enzyme inhibitor MGTA may have a specificity not strictly restricted to kininase I (Salgado et al., 1986). In C-SDs, MGTA alone did not affect FVC, but it decreased FBF. It also decreased MAP, as previously reported (Salgado et al., 1986).
In STZ-SDs, haemodynamic responses evoked by CAP alone were similar to those observed in C-SDs. CAP did not modify FBF and decreased MAP, leading to a FVC increase. This vasodilatation was blunted by L-NAME and Hoe-140 pretreatment. In addition, the vasodilatator response to CAP was abolished in STZ-BNs. This result clearly demonstrates that the vasodilatation induced by CAP in STZ-SDs was dependent on the activation of the kinin-NO pathway. This is in agreement with a previous study showing that the ACE inhibitor fosinipril enhances the endothelium-dependent relaxations in STZ-induced diabetic rats by increasing the kinin-NO pathway (Olbrich et al., 1996).
A decrease in resting FBF and FVC was observed in STZ-SDs relative to C-SDs. In addition, the femoral vasoconstriction in response to L-NAME and the vasodilatator response to exogenous BK were reduced. These observations are in agreement with those of Kiff et al. (1991a,1991b) who have found that there was a localized impairment of NO production and/or sensitivity to NO in the femoral vascular bed of STZ-treated rats. This may explain why, in our study, the NO-mediated vasodilatator response elicited by CAP was reduced in STZ-SDs compared to C-SDs.
In STZ-SDs, the femoral bed vasodilatation and the FBF increase elicited by CAP+RT were strongly inhibited by L-NAME and reduced but not significantly by Hoe-140 or MGTA alone. On the other hand, they were significantly reduced by MGTA+Hoe-140 and totally abolished in STZ-BNs. Taken all together, these results suggest that, as in C-SDs, CAP+RT could increase endogenous BK concentration to such a level that the peptide can be hydrolysed by KI. Thus, BK should lead to NO production by B2 receptor activation and by supplying the precursor for NO biosynthesis, L-arginine. It has been reported that the reduction in FBF and FVC observed in STZ-SDs was linked to a localized impairment in the production in NO, possibly due to a increase in glutamine synthesis with inhibits the intracellular generation of L-arginine and consequently the synthesis of NO (Kiff et al., 1991b). In addition, plasma arginine concentration is decreased in diabetic rats (for review, see Pieper, 1997). Therefore, it is feasible that a rise in L-arginine concentrations generated by BK hydrolysis induce an increase in NO production, which can restore FBF. This hypothesis is consistent with the observation that in our study, the FBF increase induced by CAP+RT in STZ-SDs was particularly dependent on NO synthesis.
We observed that RT enhanced the vasodilatator response to CAP in STZ-SDs but not in C-SDs. In the same way, the combined NEP/ACE inhibition has been found to decrease systemic peripheral resistance more effectively than ACE inhibition alone in heart failure (Seymour et al., 1993; Trippodo et al., 1995) but not in healthy rats (Maxwell et al., 1995). Several vasoactive peptides including Atrial Natriuretic Peptide, angiotensin I and II and endothelin are known to be substrates for ACE and NEP (Skidgel et al., 1987). The metabolism of several of these peptides is altered in STZ-treated rats (Todd et al., 1990; Cassis et al., 1992; Makino & Kamata, 1998). The resultant effect could be that the vasodilatator responses to CAP+RT are more dependent on BK in STZ-SDs than in C-SDs. This hypothesis is consistent with the absence of effects of CAP+RT in STZ-BNs.
In conclusion, this study shows that in STZ-SDs, under acute conditions, the combined inhibition of NEP and ACE by retrothiorphan and captopril respectively corrects impaired FBF and FVC more effectively than ACE inhibition alone. The effects are linked to an increase in NO production, which seems to result from a better protection of endogenous BK. BK could lead to NO production by BK B2 receptor activation and by another pathway in which kininase I would be involved. These findings show that the combined NEP/ACE inhibition merits further consideration in the treatment of diabetic vascular complications.
Acknowledgments
We thank Dr K.J. Wirth from Hoechst-Marion Roussel (Frankfurt, Germany) for his kind supply of D-Arg-(Hyp3,Thi5,D-Tic7,Oic8)-bradykinin (Hoe-140).
Abbreviations
- CAP
captopril
- C-BN
control Brown-Norway Katholiek kininogen-deficient rat
- C-SD
control Sprague-Dawley rat
- FBF
femoral blood flow
- FVC
femoral vascular conductance
- MGTA
DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid
- NEP
neutral endopeptidase 24-11
- RT
retrothiorphan
- STZ-BN
streptozotocin-induced diabetic Brown-Norway Katholiek kininogen-deficient rat
- STZ-SD
streptozotocin-induced diabetic Sprague-Dawley rat
References
- BHOOLA K.D., ERGUEROS C.D., WORTHY K. Bioregulation of kinins: kallikreins, kininogens and kininases. Pharmacol. Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- CAMPBELL D.J., KLADIS A., DUNCAN A.M. Bradykinin peptides in kidney, blood and other tissues of the rat. Hypertension. 1993;21:155–165. doi: 10.1161/01.hyp.21.2.155. [DOI] [PubMed] [Google Scholar]
- CASSIS L.A. Downregulation of the renin-angiotensin system in streptozotocin-diabetic rats. Am. J. Physiol. 1992;262:E105–E109. doi: 10.1152/ajpendo.1992.262.1.E105. [DOI] [PubMed] [Google Scholar]
- CHODIMELLA V., SKIDGEL R.A., KROWIAK E.J., MURLAS C.G. Lung peptidases, including carboxypeptidase, modulate airway reactivity to intravenous bradykinin. Am. Rev. Respir. Dis. 1991;144:869–874. doi: 10.1164/ajrccm/144.4.869. [DOI] [PubMed] [Google Scholar]
- DREXLER H., HAYOZ D., MUNZEL T., JUST H., ZELIS R., BRUNNER H.R. Endothelial dysfunction in chronic heart failure: experimental and clinical studies. Arzneim Forsch. 1994;44 Suppl. I:455–458. [PubMed] [Google Scholar]
- GARDINER S.M., KEMP P.A., BRUNNER-FERBER F., BENNETT T. Effects of the dual metallopeptidase inhibitor, MDL 100.240, on regional haemodynamic responses to vasoactive peptides in conscious rats. Br. J. Pharmacol. 1997;122:1687–1693. doi: 10.1038/sj.bjp.0701550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAZIS A., PAGE S.R., COCKROFT J.R. ACE inhibitors, diabetes and cardiovascular disease. Diabetologia. 1998;41:595–597. doi: 10.1007/s001250050952. [DOI] [PubMed] [Google Scholar]
- GRAF K., GRÄFE M., BOSSALLER C., NIEHUS J., SCHULZ K.D., AUCH-SCHWELK W., FLECK E. Degradation of bradykinin by neutral endopeptidase (EC 3.4.24.11) in cultured human endothelial cells. Eur. J. Clin. Chem. Clin. Biochem. 1993;31 5:267–272. doi: 10.1515/cclm.1993.31.5.267. [DOI] [PubMed] [Google Scholar]
- GRISWOLD J.A., BAKER C.R., JR, LITTLE D.T., LITTLE G.H., BEHAL F.J. Bradykinin metabolism in rat hindlimbs. Shock. 1998;10 2:146–152. doi: 10.1097/00024382-199808000-00011. [DOI] [PubMed] [Google Scholar]
- HORNIG B., KOHLER C., DREXLER H. Role of bradykinin in mediating vascular effects of angiotensin-converting enzyme inhibitors in humans. Circulation. 1997;95:1115–1118. doi: 10.1161/01.cir.95.5.1115. [DOI] [PubMed] [Google Scholar]
- KIFF R.J., GARDINER S.M., COMPTON A.M., BENNETT T. Selective impairment of hindquarters vasodilatator responses to bradykinin in conscious Wistar rats with streptozotocin-induced diabetes mellitus. Br. J. Pharmacol. 1991a;103:274–279. doi: 10.1111/j.1476-5381.1991.tb09793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIFF R.J., GARDINER S.M., COMPTON A.M., BENNETT T. The effects of endothelin-l and N-nitro-L-arginine methyl ester on regional haemodynamics in conscious rats with streptozotocin-induced diabetes mellitus. Br. J. Pharmacol. 1991b;103:1321–1326. doi: 10.1111/j.1476-5381.1991.tb09787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOUYOUMADJIAN M., DAMAS J. The fate of plasma kallicrein in normal and kininogen-deficient rats. Arch. Physiol. Biochem. 1998;106 1:25–32. doi: 10.1076/apab.106.1.25.4400. [DOI] [PubMed] [Google Scholar]
- MAKINO A., KAMATA K. Elevated plasma endothelin-1 level in streptozotocin-induced diabetic rats and responsiveness of the mesenteric arterial bed to endothelin-l. Br. J. Pharmacol. 1998;123 6:1065–1072. doi: 10.1038/sj.bjp.0701704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAXWELL A.J., HUSSEINI W.K., PIEDIMONTE G., HOFFMAN J.I. Effects of inhibiting neutral endopeptidase and kininase II on coronary and systemic hemodynamics in rats. Am. J. Physiol. 1995;269:H1016–H1029. doi: 10.1152/ajpheart.1995.269.3.H1016. [DOI] [PubMed] [Google Scholar]
- NAKHODO A., WONG H.A. The induction of diabetes in rats by intramuscular administration of streptozotocin. Experientia. 1979;35:1679–1680. doi: 10.1007/BF01953269. [DOI] [PubMed] [Google Scholar]
- O'DRISCOLL G., GREEN D., RANKIN J., STANTON K., TAYLOR R. Improvement in endothelial function by angiotensin converting enzyme inhibition in insulin-dependent diabetes mellitus. J. Clin. Invest. 1997;100 3:678–684. doi: 10.1172/JCI119580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLBRICH A., RÖSEN P., HILGERS K.D., DHEIN S. Fosinopril improves regulation of vascular tone in mesenteric bed of diabetic rats. J. Cardiovasc. Pharmacol. 1996;27:187–194. doi: 10.1097/00005344-199602000-00003. [DOI] [PubMed] [Google Scholar]
- PHAM I., El AMRANI A-I, K., FOURNIÉ-ZALUSKI M.C., CORVOL P., ROQUES B.P., MICHEL J.B. Effects of the selective NEP inhibitor, retrothiorphan, on renal function and blood pressure in conscious normotensive Wistar and hypertensive DOCA-salt rats. J. Cardiovasc. Pharmacol. 1992;20:847–857. doi: 10.1097/00005344-199212000-00001. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M. Review of alterations in endothelial nitric oxide production in diabetes. Protective role of arginine on endothelial dysfunction. Hypertension. 1998;31:1047–1060. doi: 10.1161/01.hyp.31.5.1047. [DOI] [PubMed] [Google Scholar]
- RICHER C., DOUSSAU M.P., GIUDICELLI J.F. Differential systemic and regional haemodynamic profiles of four angiotensin-I converting-enzyme inhibitors in the rat. Cardiovasc. Drugs Therap. 1989;3 6:865–872. doi: 10.1007/BF01869574. [DOI] [PubMed] [Google Scholar]
- ROQUES B.P., LUCAS-SORACA E., CHAILLET P., COSTENTIN J., FOURNIÉ-ZALUSKI M.C. Complete differentiation between enkephalinase and angiotensin-converting enzyme inhibition by retrothiorphan. Proc. Natl. Acad. Sci. U.S.A. 1983;80:3178–3182. doi: 10.1073/pnas.80.11.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALGADO H.C., CARRETERO O.A., SCICLI G., MURRAY R.D. Effect of DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid on the blood pressure response to vasoactive substances. J. Pharmacol. Exp. Ther. 1986;237 1:204–208. [PubMed] [Google Scholar]
- SEYMOUR A.A., ASAAD M.M., LANOCE V.M., LANGENBACHER K.M., FENNELL S.A., ROGERS W.L. Systemic hemodynamics, renal function and hormonal levels during inhibition of neutral endopeptidase 3.4.24.11 and angiotensin-converting enzyme in conscious dogs with pacing-induced heart failure. J. Pharmacol. Exp. Ther. 1993;266 2:872–883. [PubMed] [Google Scholar]
- SEYMOUR A.A., SHELDON J.H., SMITH P.L., ASAAD M., ROGERS W.L. Potentiation of the renal responses to bradykinin by inhibition of neutral endopeptidase 3.4.24.11 and angiotensin-converting enzyme in anaesthetized dogs. J. Pharmacol. Exp. Ther. 1994;269 1:263–270. [PubMed] [Google Scholar]
- SKIDGEL R.A., SCHULZ W.W., TAM L.-T., ERDÖS E.G. Human renal angiotensin I converting enzyme and neutral endopeptidase. Kidney Int. Suppl. 1987;20:S45–S48. [PubMed] [Google Scholar]
- TIO R.A., HEILIGERS J.P., DE LANGEN C.D., VAN GILST W.H., KONIG W., SAXENA P.R., WESSELING H. The haemodynamic effects of two angiotensin converting enzyme inhibitors, enalaprilat and zofenoprilat, in the rat : evidence for the involvement of bradykinin. J. Hypertens. Suppl. 1989;7 6:S296–S297. doi: 10.1097/00004872-198900076-00144. [DOI] [PubMed] [Google Scholar]
- TODD M.E., HEBDEN R.A., GOWEN B., TANG C., MCNEILL J.H. Atrial structure and plasma ANF levels in rats with chronic diabetes mellitus. Diabetes. 1990;39 4:483–489. doi: 10.2337/diab.39.4.483. [DOI] [PubMed] [Google Scholar]
- TRIPPODO N.C., PANCHAL B.C., FOX M. Repression of angiotensin II and potentiation of bradykinin contribute to the synergistic effects of dual metalloprotease inhibition in heart failure. J. Pharmacol. Exp. Ther. 1995;272 2:619–627. [PubMed] [Google Scholar]
- VOLPE A.R., GIARDINA B., PREZIOSI P., CARMIGNANI M. Biosynthesis of endothelium-derived nitric oxide by bradykinin as endogenous precursor. Immunopharmacology. 1996;33:287–290. doi: 10.1016/0162-3109(96)00043-4. [DOI] [PubMed] [Google Scholar]