

Abstract
Previous studies have suggested that proline endopeptidase (PE) could participate to the catabolism of the β-amyloid peptide (Aβ) or to the physiopathological maturation of the β-amyloid protein precursor (βAPP). We have examined the putative ability of human purified PE to catabolize Aβ40 and Aβ42 and the possible contribution of this enzyme to the generation of Aβ40 and Aβ42 in human HEK293 cells.
We show first that purified human PE does not degrade synthetic Aβ40 and Aβ42, in vitro.
We establish that HEK293 cell homogenates exhibit a Z-Gly-Pro-7AMC-cleaving enzyme, the activity of which is inhibited by Z-Pro-Prolinal and S17092 and S19825, two novel PE inhibitors, with affinities similar to those displayed on the purified human PE. These inhibitors also penetrate cells and achieve a full inhibition of endogenous proline endopeptidase in human cells.
By means of selective antibodies directed towards the C-terminal of Aβ40 and Aβ42, we assessed the effect of PE inhibitors on the recovery of both Aβ species. This was examined in HEK293 cells stably overexpressing the wild-type and the familial Alzheimer' disease-related Swedish mutated β-APP. We establish that none of these inhibitors affected Aβ40 or Aβ42 production in these transfected cells.
Overall, our study indicates that human PE does not degrade Aβ40 and Aβ42. Furthermore, PE does not contribute to Aβ40 and Aβ42 formation in HEK293 cells. Therefore, PE does not appear to contribute to the Aβ-related aetiology of Alzheimer' disease.
Keywords: Proline endopeptidase, inhibitors, secretases, Aβ secretion, Aβ degradation, HEK293 cells, wild-type βAPP, Swedish mutant βAPP, Alzheimer' disease
Introduction
The senile plaques spreading over the cortical areas of brains affected by Alzheimer' disease are typical neuropathological hallmarks of this neurodegenerative disease. The main proteic component of this extracellular deposit is a 39–42/43 amino-acid-long peptide called amyloid β-peptide or Aβ (for review see Selkoe, 1989). This peptide derives from the proteolytic maturation of a precursor, the β-amyloid precursor protein (βAPP) by β- and γ-secretases liberating the N- and C-terminus of Aβ, respectively (for review see Checler, 1995). The overload of Aβ can be envisioned as a cell disturbance of the balance between its production and its catabolism/clearance. Familial early-onset forms of Alzheimer' disease are due to mutations borne by the βAPP itself and two related proteins called presenilins 1 and 2 (for reviews see Van Broeckhoven, 1995; Hutton & Hardy, 1997; Checler, 1999). All but one of these mutations (Ancolio et al., 1999) trigger increased production of total Aβ, and more particularly of its 42-amino-acids long aggregatable form, Aβ42. There is, therefore, a great effort in the scientific community to elucidate the nature of the proteolytic activities responsible for Aβ production or degradation and the mechanisms by which these events are affected in Alzheimer' disease.
Several studies have suggested that proline endopeptidase could participate to the symptomatology and/or aetiology of Alzheimer' disease. Thus, this enzyme could contribute to the degradation of promnesic neuropeptides through its endooligopeptidase activity (Checler, 1993). In relation with the βAPP maturation, it was reported that proline endopeptidase displayed γ-secretase-like activity toward synthetic peptides mimicking the sequence targeted by this enzyme (Ishiura et al., 1990). Furthermore, proline endopeptidase inhibitors were shown to abolish the formation of Aβ in NG108-15 cells (Shinoda et al., 1997). Proline endopeptidase inhibitors also prevented the amyloid deposition in the brain of mouse with accelerated senescence (Kato et al., 1997). Finally, it was reported that anti-proline endopeptidase agents elicit an antiamnesic effect in the rat in the scopolamine-induced amnesia paradigm (Yoshimoto et al., 1987; De Nanteuil et al., 1998).
We have recently reported on the design of S17092-1, a highly potent, specific and bioavailable human PE inhibitor (Barelli et al., 1999). Here we examine the putative effect of S17092-1 and of another PE inhibitor, S19825 on the recovery of Aβ40 and Aβ42 generated by human cells overexpressing wild-type and familial Alzheimer disease (FAD)-linked Swedish mutated βAPP. Furthermore, we examine the ability of purified human PE to hydrolyse synthetic Aβ40 and Aβ42. Our study indicates that PE does not contribute to the degradation of Aβ40/42 and does not participate in their formation in HEK293 cells.
Methods
Synthesis of S17092-1 and S19825
The synthesis and structure of S17092-1, (2s,3aS,7aS)-1-{[(R,R)-2-phenyl-cyclopropyl]carbonyl}-2((thiazolidin-1-oxyde-3-yl)carbonyl)perhydroindole, has been previously reported (compound 54 in Portevin et al., 1996). S19825 is the sulphoxide derivative of S17092-1 (see formula in Figure 2).
Figure 2.
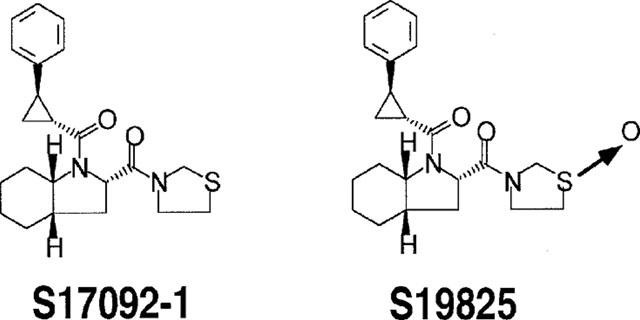
Chemical structures of S17092-1 and S19825.
Purification of human proline endopeptidase and fluorimetric assay
The purification of human cortex proline endopeptidase has been previously reported (Barelli et al., 1999). PE was fluorimetrically assayed by means of N-benzyloxycarbonyl-glycyl-prolyl-7-amino-4-methylcoumarin (Z-Gly-Pro-7AMC) (0.2 mM), in the absence or in the presence of various concentrations of S17092-1, S19825 or N-benzyloxycarbonyl-prolyl-prolinal (Z-Pro-Prolinal) in a final volume of 100 μl of 50 mM Tris-HCl buffer, pH 7.5 containing 0.5 mM dithiothreitol. The release of the 7AMC group was monitored with a fluorimeter setting of 380 and 460 nm as excitation and emission wavelengths, respectively.
Degradation of angiotensin II by human proline endopeptidase and HPLC analysis
Angiotensin II (2 nmol) was incubated for various times at 37°C with human PE in a final volume of 200 μl, in the absence or presence of 1 μM Z-Pro-Prolinal. After acidification, samples are spun and supernatants are HPLC-analysed in the conditions previously detailed (Ichai et al., 1994). Briefly, chromatographies were performed at room temperature, at a 1 ml min−1 flow rate onto a reverse-phase column (RP18 Lichrosorb, Merck, Darmstadt, Germany). Elutions were carried out by means of a 42 min linear gradient between 90 : 10 (vol vol−1) to 60 : 40 (vol vol−1) of 0.1% trifluoroacetic acid, 0.05% triethylamine/0.1% trifluoroacetic acid, 0.05% triethylamine in acetonitrile. Absorbances were monitored at 230 nm.
Degradation of Aβ40 and Aβ42, in vitro
Synthetic Aβ40 and Aβ42 (5 μg) were incubated for various times at 37°C in the presence of human proline endopeptidase. At the end of incubations, proteins were separated on a 16.5% tris-tricine gel, Western blotted and revealed with WO2 antibody as detailed below.
Effect of S17092-1 and S19825 on intracellular proline endopeptidase activity
Mock-transfected HEK293 cells were treated for 5 h as above in the absence or in the presence of inhibitors then cell homogenates preparation (see below) and PE fluorimetric assays were performed as described above.
Cell cultures and transfections
HEK293 cells were grown in 5% CO2 in HAMF12/Dulbecco' modified Eagle' medium (DMEM; vol vol−1) supplemented with 10% foetal calf serum containing penicillin (100 units ml−1), streptomycin (50 μg ml−1) and geneticine (1 mg ml−1). Stable transfectants overexpressing the wild-type and Swedish mutated βAPP751 were obtained with 3N-(N,N′-dimethylaminoethane)-carbamoylcholesterol (DAC30) reagent (Eurogentec) and 2 μg of pcDNA3 encoding the two cDNAs and positive clones were identified by means of a polyclonal antibody (BR188) recognizing the C-terminal end of βAPP751 (Chevallier et al., 1997) or with W02.
Preparation of cell homogenates
Cells (35 mm wells) were rinsed with phosphate buffer saline (PBS), pH 7.4 containing (mM): NaCl, 140; Na2HPO4, 8.5; KCl, 2.7; KH2PO4, 1.5, then scrapped and homogenized with a syringe in 1 ml of 5 mM Tris-HCl, pH 7.5.
Secretion and detection of Aβ40 and Aβ42
Secretion media were treated sequentially overnight with a 200 fold dilution of FCA3542 then with the same dilution of FCA3340 as described (Ancolio et al., 1999). Proteins were immunoprecipitated in the presence of protein A-Sepharose then precipitates were submitted to 16.5% Tris-tricine gels and Western blotted as below. Cell lysates were analysed for their βAPP content by means of WO2 as described (Ancolio et al., 1999).
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE), tris-tricine gels and Western blot analyses
Dried samples and standards were resuspended in 30 μl of 50 mM Tris, pH 6.8 containing 2% sodium dodecyl sulphate (SDS), 10% glycerol and 5% β-mercaptoethanol (Laemmli buffer). Samples were then heated for 5 min at 95°C, electrophoresed for 45 min (at 4°C) at 100 V then proteins were blotted onto nitrocellulose sheets (Hybond-C super, Amersham, Orsay, France) that were heated for 5 min in boiling phosphate buffer saline (PBS), pH 7.5. Membranes were then incubated for 1 h in PBS-0.05% Tween containing 5% skim milk, then exposed overnight to the WO2 monoclonal antibodies in PBS-0.05% Tween containing 1% skim milk. Nitrocellulose sheets were rinsed in PBS (3×5 min) and immunological complexes were revealed as previously described (Ancolio et al., 1999).
Antibodies and materials
FCA3340 and FCA3542 were previously described and are fully selective of the C-terminus of Aβ40 and Aβ42, respectively (Barelli et al., 1997). WO2 recognizes the N-terminal region of Aβ and labels full-length βAPP (Ida et al., 1996). Synthetic Aβ40 and Aβ42 were from Bachem (Voisins-le-Bretonneux, France). Angiotensin II was from Neosystem (Strasbourg, France). Z-Gly-Pro-7AMC was from Cambridge Research Biochemicals (Cleveland, UK). Penicillin, streptomycin, dithiothreitol were from Sigma (Saint Quentin Fallavier, France). Protein-A sepharose was from Zymed (Montrouge, France). Geneticin and HAMF12/DMEM were from Gibco life technologies (Cergy Pontoise, France). Foetal calf serum was from Dutscher (Issy-les-Moulineaux, France).
Results
We have examined the susceptibility of synthetic Aβ40 and Aβ42 to proteolysis by human proline endopeptidase (PE). We previously reported on the partial purification of PE from whole human brain hemisphere (Barelli et al., 1999). The partially purified enzyme hydrolyses Z-Gly-Pro-7AMC and is inhibited by Z-Pro-prolinal with an apparent affinity (4.5 nM, Table 1) that fully matches the Ki value previously reported (Wilk & Orlowski, 1983). Purified human PE also cleaves angiotensin II in a time-dependent- (Figure 1c–e) and Z-Pro-Prolinal-sensitive (Figure 1f) manner. It should be noted that methionine-enkephaline fully resisted degradation by PE (not shown), indicating that the pooled purified proteins were free from contaminating amino and carboxy-exopeptidases. Altogether, our partially purified PE displays the expected properties of the enzyme. Figure 1 shows that Aβ40 (Figure 1a) and Aβ42 (Figure 1b) fully resist degradation by human PE, even after long time incubations.
Table 1.
IC50 values of Z-Pro-prolinal, S17092-1 and S19825 on PE from various sources

Figure 1.
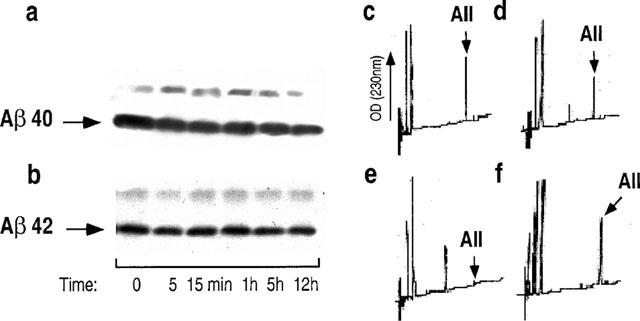
Synthetic Aβ40 and Aβ42 are not degraded by human proline endopeptidase. Synthetic Aβ40 (a) or Aβ42 (b) were incubated for the indicated times with purified human proline endopeptidase then Aβ-related immunoreactivities were analysed after 16.5% Tris-tricine electrophoresis and Western blots as detailed in the Methods. In parallel experiments, angiotensin II was incubated with human proline endopeptidase for 0 (c), 15 min (d) or 60 min (e,f) in absence (c–e) or in the presence (f) of Z-Pro-Prolinal, then incubation were HPLC-analysed as described in the Methods.
Two novel inhibitors of PE (S17092-1 and S19825) have been designed, the structures of which are shown in Figure 2. These two agents inhibit purified human PE in a dose-dependent manner (Figure 3a,b) with Ki in the nanomolar range (1.5 to 3.3 nM). S17092 and S19825 also block the Z-Gly-Pro-7AMC-hydrolyzing activity detectable in the human HEK293 cell line (Figure 3a,b) with similar IC50 affinities (Table 1).
Figure 3.
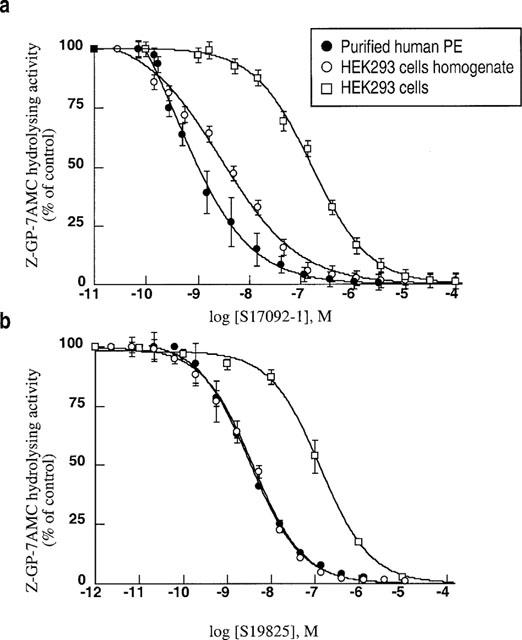
Inhibition curves of S17092-1 and S19825 on proline endopeptidase from various sources. Purified proline endopeptidase, or the Z-Gly-Pro-7AMC-hydrolyzing activities present in HEK293 cells or membrane homogenates were incubated as described in the Methods in the absence (control) or presence of the indicated concentrations of S17092-1 (a) or S19825 (b). Points correspond to the means±s.e.mean of 4–5 independent determinations.
In order to examine the putative contribution of proline endopeptidase to the formation/clearance of endogenous Aβ40 and Aβ42, we first assessed the bioavailability of these inhibitors and their penetrance rate in HEK293 cells. Figure 3 indicates that both S17092 and S19825 inhibit the intracellular endogenous Z-Pro-Prolinal-sensitive Z-Gly-Pro-7AMC cleaving activity, although the apparent affinities were significantly lower than those exhibited on HEK293 cell homogenates (Table 1). This indicates that the inhibitors poorly penetrate in cells. However, the full inhibition observed with inhibitors enabled us to examine the contribution of endogenous human PE activity.
We took advantage of the design of our antibodies able to discriminate between the various Aβ species (Barelli et al., 1997) to assess the influence of S17092-1 on the Aβ40 and Aβ42 production by HEK293 cells overexpressing the wild-type (wt)-βAPP or the FAD-linked βAPP bearing the Swedish mutation. As reported previously (Citron et al., 1992; Cai et al., 1993; Felsenstein et al., 1994), the Swedish mutation triggers an increased recovery of secreted Aβ40 and Aβ42 when compared to wt-βAPP-expressing cells (Figure 4). Clearly, S17092 does not affect βAPP expression and does not modify the secretion of both Aβ species, whatever the transfectants examined (Figure 4), even at a 10 μM concentration that fully blocks the PE activity in HEK293 cells (Figure 3). Figure 5 indicates that S19825 was also ineffective on Aβ40 and Aβ42 recovery in both cell types.
Figure 4.
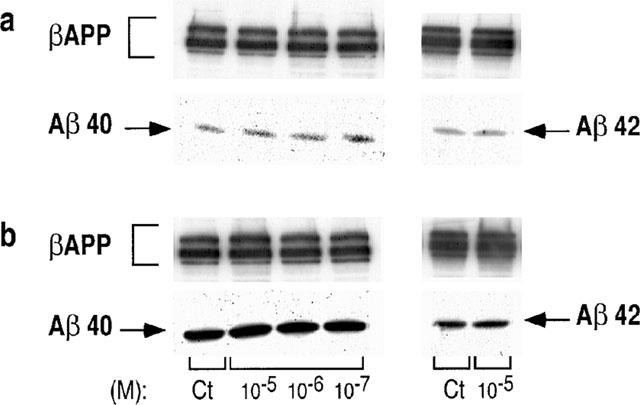
Effect of S17092-1 on the recovery of Aβ40/42 produced by wild-type and Swedish mutated-βAPP-expressing cells. HEK293 cells expressing wild-type (a) or Swedish mutated (b) βAPP were treated for 7 h without (Ct) or with the indicated concentrations of S17092-1. Secretion media were then analysed for their Aβ40 or Aβ42 contents by sequential immunoprecipitation with FCA3542 and FCA3340, respectively, then immunological complexes were revealed as detailed in the Methods.
Figure 5.
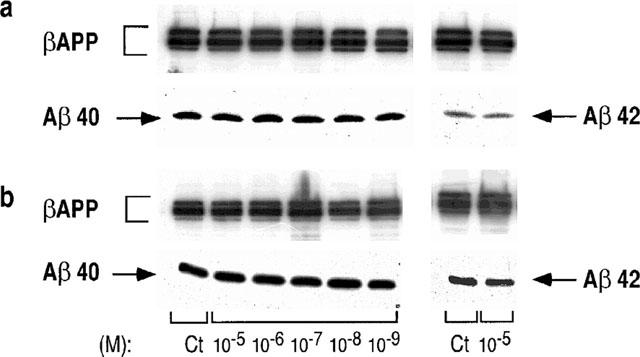
Effect of S19825 on the recovery of Aβ40/42 produced by wild-type and Swedish mutated-βAPP-expressing cells. HEK293 cells expressing wild-type (a) or Swedish mutated (b) βAPP were treated for 7 h without (Ct) or with the indicated concentrations of S19825. Secretion media were then analysed for their Aβ40 or Aβ42 contents by sequential immunoprecipitation with FCA3542 and FCA3340, respectively then immunological complexes were revealed as detailed in the Methods.
Discussion
The mechanisms by which Aβ40 and Aβ42 are generated are likely key events contributing to the neuropathology of Alzheimer' disease. Thus, one of the histopathological hallmarks of both sporadic and genetic forms of the disease is the senile plaque, an extracellular proteinaceous deposit mainly composed of these Aβ species (Selkoe, 1989). A clue of the central role of Aβ came from the observation that familial cases of Alzheimer' disease were due to mutations located on distinct proteins, namely βAPP and presenilins (for reviews see Van Broeckhoven, 1995; Hutton & Hardy, 1997; Checler, 1999). The common phenotypic alteration triggered by these mutations is an exacerbated production of Aβ and particularly its 42-amino-acid-long more aggregatable species (Burdick et al., 1992). The overproduction of Aβ could be due to upregulated concentrations of generating activities, to a misrouting of βAPP to cell compartments permissive for deleterious proteolytic activities, or alternatively could derive from a decrease of Aβ clearance (Checler, 1995).
Several reports suggested that PE could contribute to the physiopathological maturation of βAPP. Thus, PE displays γ-secretase-like activity on synthetic peptides (Ishiura et al., 1990). Furthermore, a PE inhibitor, JTP-4819, (S)-2-[[(S)-2-(hydroxyacetyl)-1-pyrrolidinyl]carbonyl]-N-(phenylmethyl)-1-pyrrolidine-carbox-amide, was reported to prevent the Aβ40 increased production observed in response to serum deprivation of NG108-15 neuroblastoma cells (Shinoda et al., 1997). We have characterized two novel agents, S17092-1 and S19825 that unambiguously display the typical pharmacological inhibitory profile towards human PE. These agents were unable to affect the recovery of Aβ40 and Aβ42 in HEK293 cells overexpressing wild-type βAPP.
Several lines of evidence indicate that the Familial Alzheimer Disease (FAD)-related Swedish mutated βAPP is processed in cell compartments distinct from those involved in wt-βAPP maturation, suggesting that secretases involved in the generation of Aβ could be distinct (Haass et al., 1995). However, PE inhibitors did not modify the Aβ40 and Aβ42 production of Swedish βAPP-expressing cells. Altogether, our data indicate that PE does not contribute to the generation of Aβ40 or Aβ42 by wild-type or Swedish mutated βAPP in HEK293 cells and therefore does not display β- or γ-secretase like activity. This agrees well with a very recent study showing that Fmoc-Ala-Pro-CN, (1-(N-(9-fluorenyl-methoxycarbonyl)-alanyl)-2-cyanopyrrolidine), a potent PE inhibitor, does not affect the γ-secretase cleavage of a N-terminally truncated APP fragment corresponding to the last 99 amino-acids of the βAPP C-terminus in SH-SY5Y cells (Johnston et al., 1999).
The fact that PE does not cleave βAPP is not unexpected when considering that this activity belongs to the oligopeptidase family. These enzymes display strong requirements of substrate length and do not cleave peptides longer than 12–15 amino-acids (Checler, 1993). This likely explains why purified PE was unable to cleave synthetic Aβ40 and Aβ42 (our study). This agrees well with the observation that PE inhibitors do not affect Aβ recovery. Any involvement of the enzyme in the clearance of Aβ would have led to an increased recovery of Aβ40 or Aβ42 in HEK293 cells.
It remains possible to envision PE as a contributor of the Alzheimer' disease symptomatology through the degradation of mnemocognitive neuropeptides. Thus, it has been shown that PE inhibitors could display antiamnesic properties in a scopolamine-induced amnesia model in the rat (Yoshimoto et al., 1987). Somatostatin-like immunoreactivity is decreased in Alzheimer' disease-affected brains (Schettini et al., 1988; Bissette & Myers, 1992). Furthermore, drastic memory impairement can be triggered by experimental lowering of endogenous somatostatin content in the rat (Vecsei et al., 1984). Therefore, somatostatin is a likely neuropeptide candidate participating to Alzheimer' symptomatology and therefore, could be envisioned as a possible target of PE. However, it should be noted that somatostatin is only slightly, if at all, susceptible to catabolism by PE (Checler, 1993), ruling out the possible contribution of PE, at least in somatostatin catabolism.
It remains possible to envisage PE contributing to the inactivation of other promnesic neuropeptides. However, our previous study indicated that PE activity was not affected in the frontal cortex and cerebellum, post mortem in Alzheimer' disease-affected brains, and statistically even significantly decreased in the parietal cortex (Ichai et al., 1994). Therefore, such a region-specific decreased activity would have led to an increase in the concentration of any peptide substrate of PE and thereby, to a potentiation of its putative promnesic effect, a feature totally opposed to the cognitive alterations observed in Alzheimer' disease.
Altogether, our study makes any contribution of PE in both symptomatology and etiology of Alzheimer' disease neuropathology unlikely.
Acknowledgments
We are grateful to Drs K. Beyreuther and T. Hartmann (Heidelberg, Germany) for providing us with WO2. We sincerely thank Dr Sherwin Wilk (Mount Sinai School of Medicine, New York, U.S.A.) for the generous gift of Z-Pro-Prolinal. Thanks are due to Dr. M. Goedert for providing BR188. This work was supported by INSERM and the CNRS and by the financial support of Servier.
Abbreviations
- Aβ
amyloid β-peptide
- AD
Alzheimer' disease
- βAPP
β-amyloid precursor protein
- FAD
familial Alzheimer' disease
- Fmoc
9-fluorenyl-methoxycarbonyl
- HEK
human embryonic kidney
- PE
proline endopeptidase
- Z
N-benzyloxycarbonyl
References
- ANCOLIO K., DUMANCHIN C., BARELLI H., WARTER J.M., BRICE A., CAMPION D., FRÉBOURG T., CHECLER F. Unusual phenotypic alteration of β amyloid precursor protein (βAPP) maturation by a new Val->Met βAPP-770 mutation responsible for probable early-onset Alzheimer' disease. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4119–4124. doi: 10.1073/pnas.96.7.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARELLI H., LEBEAU A., VIZZAVONA J., DELAERE P., CHEVALLIER N., DROUOT C., MARAMBAUD P., ANCOLIO K., BUXBAUM J.D., KHORKOVA O., HEROUX J., SAHASRABUDHE S., MARTINEZ J., WARTER J.-M., MOHR M., CHECLER F. Characterization of new polyclonal antibodies specific for 40 and 42 aminoacid-long amyloid β peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer' disease and cerebral amyloid angiopathy cases. Mol. Med. 1997;3:695–707. [PMC free article] [PubMed] [Google Scholar]
- BARELLI H., PETIT A., HIRSCH E., WILK S., DENANTEUIL G., MORAIN P., CHECLER F. S17092-1, a highly potent, specific and cell permeant inhibitor of human proline endopeptidase. Biochem. Biophys. Res. Commun. 1999;257:657–661. doi: 10.1006/bbrc.1999.0366. [DOI] [PubMed] [Google Scholar]
- BISSETTE G., MYERS B. Somatostatin in Alzheimer' disease and depression. Life Sci. 1992;51:1389–1410. doi: 10.1016/0024-3205(92)90534-v. [DOI] [PubMed] [Google Scholar]
- BURDICK D., SOREGHAN B., KWON M., KOSMOSKI J., KNAUER M., HENSCHEN A., YATES J., COTMAN C., GLABE C. Assembly and aggregation properties of synthetic Alzheimer' A4/β amyloid peptide analogs. J. Biol. Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- CAI X.-D., GOLDE T.E., YOUNKIN S.G. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- CHECLER F.Neuropeptide-degrading peptidases Methods in Neurotransmitters and Neuropeptides Research. Part 2 1993Amsterdam: Elsevier Science Publishers; 375–418.ed. Nagatsu, T., Parvez, H., Naoi, M. & Parvez, S. pp [Google Scholar]
- CHECLER F. Processing of the β-amyloid precursor protein and its regulation in Alzheimer' disease. J. Neurochem. 1995;65:1431–1444. doi: 10.1046/j.1471-4159.1995.65041431.x. [DOI] [PubMed] [Google Scholar]
- CHECLER F. Presenilins: multifunctional proteins involved in Alzheimer' disease pathology. Iubmb Life. 1999;48:33–39. doi: 10.1080/713803480. [DOI] [PubMed] [Google Scholar]
- CHEVALLIER N., JIRACEK J., VINCENT B., BAUR C.P., SPILLANTINI M.G., GOEDERT M., DIVE V., CHECLER F. Examination of the role of endopeptidase 3.4.24.15 in Aβ secretion by human transfected cells. Br. J. Pharmacol. 1997;121:556–562. doi: 10.1038/sj.bjp.0701151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CITRON M., OLTERSDORF T., HAASS C., MCCONLOGUE L., HUNG A.Y., SEUBERT P., VIGO-PELFREY C., LIEBERBURG I., SELKOE D.J. Mutation of the β-amyloid precursor protein in familial Alzheimer' disease increases β-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- DENANTEUIL G., PORTEVIN B., LEPAGNOL J. Prolyl endopeptidase inhibitors: a new class of memory enhancing drugs. Drugs of the Future. 1998;23:167–179. [Google Scholar]
- FELSENSTEIN K.M., HUNIHAN L.W., ROBERTS S.B. Altered cleavage and secretion of a recombinant β-APP bearing the Swedish familial Alzheimer' disease mutation. Nature Genetics. 1994;6:251–256. doi: 10.1038/ng0394-251. [DOI] [PubMed] [Google Scholar]
- HAASS C., LEMERE C.A., CAPELL A., CITRON M., SEUBERT P., SCHENK D., LANNFELT L., SELKOE D. The Swedish mutation causes early-onset Alzheimer' disease by β-secretase cleavage within the secretory pathway. Nature Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- HUTTON M., HARDY J. The presenilins and Alzheimer' disease. Human Mol. Gen. 1997;6:1639–1646. doi: 10.1093/hmg/6.10.1639. [DOI] [PubMed] [Google Scholar]
- ICHAI C., CHEVALLIER N., DELAERE P., DOURNAUD P., EPELBAUM J., HAUW J.J., VINCENT J.P., CHECLER F. Influence of region-specific alterations of neuropeptidases content on the catabolic fates of neuropeptides in Alzheimer' disease. J. Neurochem. 1994;62:645–655. doi: 10.1046/j.1471-4159.1994.62020645.x. [DOI] [PubMed] [Google Scholar]
- IDA N., JOHANNES H., PANTEL J., SCHRÖDER J., ZERFASS R., FÖRSTL H., SANDBRINK R., MASTERS C.L., BEYREUTHER K. Analysis of heterogeneous βA4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J. Biol. Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- ISHIURA S., TSUKAHARA T., TABIRA T., SHIMIZU T., ARAHATA K., SUGITA H. Identification of a putative amyloid A4-generating enzyme as a prolyl endopeptidase. FEBS Lett. 1990;260:131–134. [Google Scholar]
- JOHNSTON J.A., JENSEN M., LANNFELT L., WALKER B., WILIAMS C.H. Inhibition of prolylendopeptidase does not affect γ-secretase processing of amyloid precursor protein in a human neuroblastoma cell line. Neurosci. Lett. 1999;277:33–36. doi: 10.1016/s0304-3940(99)00834-4. [DOI] [PubMed] [Google Scholar]
- KATO A., FUKUNARI A., SAKAI Y., NAKAJIMA T. Prevention of amyloid-like deposition by a selective prolyl endopeptidase inhibitor, Y-29794, in senescence-accelerated mouse. J. Pharm. Exp. Ther. 1997;283:328–335. [PubMed] [Google Scholar]
- PORTEVIN B., BENOIST A., RÉMOND G., HERVÉ Y., VINCENT M., LEPAGNOL J., DENANTEUIL G. New prolyl endopeptidase inhibitors: in vitro and in vivo activities of Azabicyclo[2.2.2.]octane, Azabicyclo[2.2.1]heptane, and perhydroindole derivatives. J. Med. Chem. 1996;39:2379–2391. doi: 10.1021/jm950858c. [DOI] [PubMed] [Google Scholar]
- SCHETTINI G., FLORIO T., MAGRI G., GRIMALDI M., MENCCI O., LANDOLFI E., MANICRO A. Somatostatin and SMS 201-995 reverse the impairment of cognitive functions induced by cysteamine depletion of brain somatostatin. Eur. J. Pharmacol. 1988;151:399–407. doi: 10.1016/0014-2999(88)90536-5. [DOI] [PubMed] [Google Scholar]
- SELKOE D.J. Amyloid beta protein precursor and the pathogenesis of Alzheimer' disease. Cell. 1989;58:611–612. doi: 10.1016/0092-8674(89)90093-7. [DOI] [PubMed] [Google Scholar]
- SHINODA M., TOIDE K., OHSAWA I., KOHSAKA S. Specific inhibitor for prolyl endopeptidase suppresses the genertion of amyloid β protein in NG108-15 cells. Biochem. Biophys. Res. Commun. 1997;235:641–645. doi: 10.1006/bbrc.1997.6730. [DOI] [PubMed] [Google Scholar]
- VANBROECKHOVEN C. Presenilins and Alzheimer disease. Nature Genet. 1995;11:230–232. doi: 10.1038/ng1195-230. [DOI] [PubMed] [Google Scholar]
- VECSEI L., BOLLOK I., TELEGDY G. Phenoxybenzamine antagonizes somatostatin-induced anti-amnesia in rats. Eur. J. Pharmacol. 1984;99:325–328. doi: 10.1016/0014-2999(84)90139-0. [DOI] [PubMed] [Google Scholar]
- WILK S., ORLOWSKI M. Inhibition of rabbit brain prolyl endopeptidase by N-benzyloxycarbonyl-prolyl-prolinal, a transition state aldehyde inhibitor. J. Neurochem. 1983;41:67–75. doi: 10.1111/j.1471-4159.1983.tb11815.x. [DOI] [PubMed] [Google Scholar]
- YOSHIMOTO T., KADO K., MATSUBARA F., KORIYAMA N., KANETO H., TSURU D. Specific inhibitors for prolyl endopeptidase and their anti-amnesic effect. J. Pharmacobiol. Dyn. 1987;10:730–735. doi: 10.1248/bpb1978.10.730. [DOI] [PubMed] [Google Scholar]