

Introduction
Protein kinase C (PKC) is a multifunctional, cyclic nucleotide-independent protein kinase that phosphorylates serine and threonine residues in many target proteins. This enzyme was identified in bovine cerebellum by Nishizuka and co-workers (Takai et al., 1977; Inoue et al., 1977) as a protein kinase that phosphorylated histone and protamine. Since its discovery, much interest has been shown in PKC and its role in signal transduction. Development (Otte et al., 1991), memory (Alkon, 1989), differentiation (Cutler et al., 1993), proliferation (Murray et al., 1993) and carcinogenesis (Ashendel, 1985) all are processes for which PKC has been implicated. Once thought to be a single protein, PKC is now known to comprise a large family of enzymes that differ in structure, cofactor requirements and function. Indeed, the PKC family is the largest serine/threonine-specific kinase family known (Parker, 1992) to which many cellular responses have been credited (Nishizuka, 1995). This enzyme multiplicity, together with variation in cellular and tissue distribution, and abundance might explain why so many signal transduction functions have been attributed to this kinase. Here we briefly describe the organization and regulation of PKC and review the current understanding of their role in the regulation of airways smooth muscle (ASM) tone and mitogenesis, which have been investigated in some detail.
Classification and organization of PKC
PKC represents a structurally homologous group of proteins of which at present 10 isoforms have been identified unequivocally. The PKC family can be broadly divided into three groups that differ in their cofactor requirements. These are known as conventional (c)PKC isoforms (α, βI, βII and γ), that require Ca2+ and diacylglycerol (DAG) to become activated; novel (n)PKC isoforms (δ, ε, ζ, θ and μ) that require only DAG; and the atypical (a)PKC isoforms, namely ζ, ι and λ (the mouse homologue of human PKCι), that require neither Ca2+ nor DAG. All PKC family members possess a phosphatidylserine (PS) binding domain for membrane interaction (though the specific lipid activators of aPKC' are not yet known). The general structure of a PKC molecule consists of a catalytic and a regulatory domain found at the C- and N-terminus respectively. Both domain structures are composed of a number of conserved regions (C1–C4) interspersed with regions of lower homology, so-called variable domains, V0–V5 (Figure 1).
Figure 1.

Diagrammatic representation of the domain structure of PKC isoenzymes. The PKC family can be divided into three sub-families consisting of cPKC', namely α, β1, β2 and γ, the nPKC' namely δ, ε, ζ, θ and μ that do not require Ca2+ (as they do not possess the C2 domain) and the aPKC', namely ζ, ι (human) and λ (mouse) that require neither Ca2+ nor DAG for activation. The length and relative sizes of the isoforms are given to demonstrate the variability within this kinase family. V regions represent poorly conserved (variable) sequences between the isoforms and separate the highly conserved functionally-specific domains. The V3 region acts as a link between the regulatory and catalytic domains and also provides a site for proteolytic cleavage by calpains and trypsin, which results in constitutive and activator-dependent kinase activity (Mochly-Rosen et al., 1990). V0 is an N-terminal domain not present in the cPKC'. The C domains are conserved sequences throughout the PKC family. C1 features the DAG and PS binding sites. At the N-terminus of the C1 domain is located a pseudosubstrate site that regulates PKC activity. These ‘so-called' autoinhibitory domains contain a consensus phosphorylation sequence but no phosphorylatable residue, thus controlling kinase activity by blocking access of the substrate through occupation of the catalytic site (Soderling, 1990). C2 is the Ca2+-binding domain present only in cPKC'. The C-terminus of all PKCs features the highly conserved catalytic domain (the C3 and C4 regions), separated by a short variable (V4) insert present only in PKCγ (Parker, 1992). Various sub-domains within the C3-C4 regions have also been identified (Hanks et al., 1988). The C3 region, which is essential for enzyme activity, represents the conserved ATP-binding site sequence found in all isoforms in which an invariant lysine residue (lys-380) is essential for kinase activity (Freisewinkel et al., 1991). The C4 domain represents the catalytic core of PKC'. For further details on the structure of PKC, interested readers should refer to Azzi et al. (1992), Hug & Sarre (1993), Stabel & Parker (1991), Dekker & Parker (1994) and Mellor & Parker (1998).
The most recently discovered members of the ‘PKC-superfamily' are the protein kinase C-related kinases (PRKs), which display sequence homology with PKC' and, like aPKC', are insensitive to Ca2+, DAG and phorbol diesters. These enzymes were discovered independently by Palmer et al. (1994) and Kitagawa et al. (1995) (who named it PKN) utilizing PCR and low-stringency screening, and are regulated by small GTP-binding proteins such as Rho. Indeed, PRK' belong to the Rho-activated kinase family that include rhotekin and rhophilin (see Mellor & Parker, 1998).
Another novel serine/threonine protein kinase that binds phorbol diesters was identified in 1994 by Valverde and colleagues but was not classified into a particular kinase family. This enzyme shows greatest homology (41%) with myosin light chain kinase (MLCK) (Valverde et al., 1994) and was named protein kinase D (PKD), although it may represent the mouse equivalent of human PKCμ (which at 916 amino acids is six residues shorter than PKD) or of an undiscovered human homologue of human PKCμ (Johannes et al., 1994). However, the 54 amino acid difference between PKD and PKCμ exceeds the number seen for other isoforms when inter-species comparisons are made and considerable controversy, thus, surrounds the issue of whether PKD/PKCμ are true PKC family members (see Ron & Kazanietz, 1999).
Finally, protein kinase B (also known as RAC-PK (related to A- & C-kinase) was identified as a serine/threonine protein kinase having a catalytic domain resembling PKC and PKA (hence the nomenclature) (Coffer & Woodgett, 1991; Jones et al., 1991). This kinase has been implicated in a number of signalling pathways, most recently in those controlling ASM proliferation (Walker et al., 1998).
Regulation of PKC
Activation
Membrane association
Activation of cPKC', regardless of the stimulus, seemingly involves translocation from the cytosol to specific binding domains at cell membranes and can involve four basic interactions. First, these isoforms can form catalytically inactive, membrane-associated complexes in a Ca2+-dependent manner; second, they may form two membrane contacts in the C1 and C2 domains (see legend to Figure 1 for description of PKC domain sequence); third, membrane interactions can occur that facilitate conformational changes of the PKC molecule, which are distinct from an activation event; and fourth, additional changes can occur following DAG/phorbol diester binding to the membrane-associated kinase. See Zidovetzki & Lester, 1992; Lester et al., 1990; Bazzi & Nelsestuan 1987 for further details.
The activity of PKC is controlled by its compartmentalization within the cell. Mochly-Rosen (1995) indicated that specific anchoring proteins (immobilized at particular intracellular sites) localize the kinases to their sites of action. These include ‘receptors for activated C-kinase' (RACKS), annexins and other cytoskeletal proteins. For a protein to be considered a RACK, certain criteria must be fulfilled: (i) it should bind PKC in the presence of activators; (ii) PKC binding to recombinant RACK1 should not be inhibited by a pseudosubstrate peptide; (iii) binding of PKC to RACK1 should be saturable and specific; and (iv) RACKS should contain a sequence homologous to a PKC binding motif.
Diacylglycerol and phorbol diester interactions
Following an increase in [Ca2+]i, PKC interacts with the cell membrane in an inactive, but conformationally distinct, form (Bell & Burns, 1991; Huang, 1989). With respect to cPKC' and nPKC', the availability of DAG is essential for their activation through its ability to facilitate the penetration of these isoenzymes into the cell membrane (Lester et al., 1990). Using synthetic lipid bilayers, it has been shown that the region in PKC required for penetration contains cysteines and one or more primary amines. This sequence may have homology with a region in phospholipase A2 that acts as a phospholipid-binding site for the enzyme (Lester et al., 1990).
The effects of different phorbol diesters on the activation of PKC' α, β1, γ, δ and ε have been investigated (Ryves et al., 1991) in an attempt to identify isoenzyme-selective activators without success.
Role of calcium
Activation of cPKC' is critically dependent [Ca2+]i, as well as PS. Indeed, the level of Ca2+ under resting conditions is insufficient to activate PKC. Upon receptor stimulation, and subsequent Ca2+ mobilization, the [Ca2+]i increases transiently and of sufficient magnitude to promote translocation of inactive PKC from the cytosol to the plasma membrane for attachment and activation (Zidovetzki & Lester, 1992). When attached to the membrane, the affinity of PKC for Ca2+ is increased (Nishizuka, 1988) such that activation of the enzyme is achieved.
Fatty acids
Phosphatidylserine has been shown to bind electrostatically to peptides bearing sequence homology to the pseudosubstrate binding site in PKC (Mosier & McLaughlin, 1991). Since unsaturated fatty acid chain length in DAG and PS affect their potential action as PKC cofactors (Lapetina et al., 1985), hydrophobic membrane-interactions must also apply in the activation process (Brumfeld & Lester, 1990).
Other membrane phospholipids may ultimately link extracellular signals to intracellular events through PKC (Nishizuka, 1992). cis-Unsaturated fatty acids such as arachidonic, linoleic and oleic acid can activate PKC in the absence of DAG and PS (Hansson et al., 1986), suggesting an activation mechanism that differs from that effected by DAG. Phosphatidylcholine only activates PKC if a cis-fatty acid is present (Chen et al., 1992), even in the presence of DAG. This is the case for all types of PKC. It has been suggested that fatty acids exert their effect at the regulatory domain of PKC by altering pseudosubstrate binding and unblocking the catalytic site, thereby activating the kinase (Chauhan et al., 1990).
Proteolysis
It is well known that some, if not all, PKC isoforms can be proteolytically cleaved at the V3 region by the calcium-activated protease, calpain (Kishimoto et al., 1983), to generate a cofactor-independent free catalytic subunit known as protein kinase M (PKM). Calpain exists as two distinct isoforms that have a low (μ-calpain) and high (m-calpain) requirement for Ca2+ respectively, where m and μ reflect millimolar and micromolar [Ca2+] respectively (Michetti et al., 1995). The physiological relevance of the ‘calpain-product' was not understood at the time of its discovery but it should not be regarded as an ‘unregulated' enzyme since its generation is, in fact, regulated by proteolysis (Shea et al., 1994). A Ca2+/phospholipid-independent kinase similar to PKM has been observed in several tissues such as the neutrophil (Melloni et al., 1986). In that cell, the rate of disappearance of Ca2+/phospholipid-dependent PKC activity correlated with the appearance of a 65 kDa Ca2+/phospholipid-independent kinase that was identical to that reported for proteolytically modified PKC. Thus, the proteolytic cleavage of the membrane-bound protein can be considered an alternative mechanism of activation.
Down-regulation
Prolonged exposure of mammalian cells and tissues to phorbol diesters and bryostatins has been shown to desensitize agonist- and phorbol-diester-induced responses by a mechanism that can involve down-regulation of conventional and novel PKC isoforms (Newton, 1995; Smith et al., 1985; Huwiler et al., 1994). Although calpains have been implicated in this down-regulation (Eto et al., 1995a; Kishimoto et al., 1989), the evidence is not convincing as it derives from studies with small molecule inhibitors of limited specificity (see Lee et al., 1997). It seems likely that increased vesicle trafficking including lysosomal endocytosis may account for down-regulation of some PKC species (Goode et al., 1994; 1995; Junco et al., 1994). Indeed, isoform-specific PKC down-regulation has been observed in Schizosaccharomyces pombe in response to phorbol diesters (Goode et al., 1995). Alternatively, activation of the ubiquitin/proteasome pathway may play a dominant role in the down-regulation of certain PKC isoforms including PKCα and PKCε (Lee et al., 1996; 1997). All PKCs except the δ isoform express, so-called, PEST sequences (hydrophilic polypeptide segments enriched in proline (P), glutamic acid (E), serine (S) and threonine (T)), that target proteins for degradation by the proteasome (Rechsteiner & Rogers, 1996). An additional level of complexity has been the finding that dephosphorylation of activated PKCs predisposes them to ubiquitination (Lee et al., 1997).
Oxidation
PKC isolated from many tissues is subject to oxidation by a number of agents including peroxide, N-chlorosuccinimide and periodate. In all cases, oxidation is dependent on the presence of Fe2+ and yields a modified form of the enzyme that is constitutively active. Thus, in the case of cPKC', Ca2+ and phospholipid are not required for activity (Palumbo et al., 1992; Gopalakrishna & Anderson, 1987; 1989; Allen et al., 1994; Webb et al., 1997a). Oxidative inactivation of PKC by hydrogen peroxide (H2O2) is enhanced by Ca2+ or by phorbol diesters in the absence of Ca2+. Gopalakrishna et al. (1989) demonstrated that ATP protects the catalytic site from oxidation indicating that the ATP-binding domain features redox-sensitive residues. Under this condition, oxidants increase cofactor-independent PKC activity by exerting an affect at the regulatory site. Using rat brain PKC, it was also demonstrated that mild oxidation of PKC increases Ca2+/phospholipid-independent activity whereas further oxidation inactivates the enzyme (Allen et al., 1994).
N-Chlorosuccinimide as well as H2O2 down-regulate phorbol diester binding to PKC (Gopalakrishna et al., 1987) indicating that oxidation of the DAG-binding domain markedly reduces the affinity of phorbol diesters and DAG for oxidized PKC. This inactivation was shown to be more rapid for membrane-associated PKC than the cytosolic form. At micromolar concentrations, periodate activates PKC through oxidation at the regulatory site, whereas at higher concentrations of the oxidant the enzyme is inactivated through an action at the catalytic domain (Gopalakrishna et al., 1991). Thus, selective regulatory site oxidation constitutively activates PKC whereas oxidation at the regulatory and/or catalytic domain, inactivates the enzyme irreversibly. We have shown (Webb et al., 1997a) that constitutively active cPKC isoforms exist in bovine ASM and behave as ‘unregulated' kinases due to selective oxidation of the regulatory domain. In this respect, it is interesting that treatment of smooth muscle cells with phorbol diesters increases the concentration of intracellular reactive oxygen species (MietusSnyder et al., 1997) raising the potential for exaggerated PKC-driven responses.
Mechanistically, oxidation of PKC may represent a unique method of regulation. Just as the apparently ‘unregulated' activity of PKM is, in fact, controlled by proteolysis, so is the ‘unregulated' activity of oxidized PKC at the regulatory site. Indeed, the constitutive activity is abolished by further oxidation through inactivation of the enzyme at the catalytic core. It is possible that activation of PKC may occur under conditions of oxidative stress, typified by inflammatory disorders such as chronic obstructive pulmonary disease, asthma and arthritis.
Nitrosylation
Protein kinase C contains certain thiol residues that may serve to regulate the enzymes' activity by acting as substrates for S-nitrosylation. Gopalakrishna et al. (1993; 1995) demonstrated (using a number of nitric oxide-generating agents such as sodium nitroprusside) irreversible inactivation of PKC activity and a loss of phorbol diester binding due to disulphide bridge formation following oxidation of the nitrosylated thiols. Inhibition of calpain by S-nitrosylation also may represent an indirect process through which PKC is regulated (Michetti et al., 1995).
Phosphorylation and autophosphorylation
Until recently, there have been few published studies to suggest that PKC is phosphorylated by kinases other than itself. However, it is now appreciated that PKC isoforms are phosphorylated at an ‘activation loop' C-terminal serine/threonine residue in the sequence TFCGTP by phosphatidylinositol-trisphosphate-dependent kinase (PDK1). This is followed by an additional phosphorylation (probably PDK-mediated) or autophosphorylation at a more C-terminal serine/threonine residue in the sequence FSY/FTY. Nineteen amino-acids N-terminal to this FSY/FTY phosphorylation motif is another autophosphorylation site (the ‘so-called' TP site) (Bornancin & Parker, 1996; 1997; Stempka et al., 1997; Li et al., 1997). The order of these final two phosphorylation events is not elucidated, though it is clear that each event induces necessary conformational changes in the PKC molecule that result in altered thermal stability, resistance to phosphatases and catalytic activity (Stempka et al., 1997; Newton & Koshland, 1990; Bornancin & Parker, 1996; 1997).
Autophosphorylation of PKC requires great flexibility of the protein structure, and the region surrounding these sites of phosphorylation contains no common recognition sequence or similarity with substrate phosphorylation sites (Flint et al., 1990). A number of features of PKC autophosphorylation are evident. In particular, it requires higher concentrations of PS than are required for phosphorylation of exogenous substrates (Bazzi & Nelsestuan, 1992; Newton & Koshland, 1990) due to different PS requirements of aggregation by divalent cations.
Tissue expression of PKC isoenzymes
The expression and distribution of PKC isoforms varies markedly between cells and tissues (Nishizuka, 1988). Some isoforms (e.g. PKCα) are ubiquitously expressed whereas others seem to be restricted to certain tissues (see Table 1).
Table 1.
Distribution of PKC isoenzymes between selected mammalian cells and tissues

In ASM, similar patterns of expression of PKC isoforms have been found in the larger central airways of several species (Table 2). In bovine trachealis there is expression of conventional α-, βI-, and βII-PKC isoforms as well as the novel δ-, ε and θ-PKC variants together with trace amounts of atypical PKCζ (Webb et al., 1997a). Canine trachealis shows a similar profile, except that PKCα is apparently absent (Donnelly et al., 1995). With the availability of monoclonal antibodies to additional members of the aPKCs, a recent investigation also identified PKCμ and -ι/λ isoforms in porcine trachealis (Togashi et al., 1997). In human trachealis, there is expression of the conventional α, βI, and βII PKC isoforms as well as novel (δ, ε, ζ, θ) and atypical (ζ) variants (Webb et al., 1997b). Where discrepancies between species have been noted in expression of particular isoforms, it is possible that this involves failure of antibodies raised against human epitopes to cross-react with those of different species, or to the presence of contaminating non-muscle cell types in the tissue preparations. PKCγ, often abundantly expressed in neuronal tissues, has not been detected in ASM from any species thus far investigated.
Table 2.
Protein and mRNA expression of PKC isoforms in airways smooth muscle from different mammalian species

PKC and contraction
Our present understanding of smooth muscle contraction is based on the original studies of Huxley & Niedergerke (1954) and Huxley & Hanson (1954). Using striated muscle these two groups of investigators independently advanced the ‘sliding filament' theory, which proposes that contraction of muscle is the result of a relative interdigitation between two populations of protein filaments, actin and myosin, with the reversible formation of stress-bearing cross-bridges. Many studies have been conducted to elucidate the biochemical basis and regulation of smooth muscle contraction (Allen et al., 1994; Somlyo & Somlyo, 1994) and significant differences have been discovered between the processes that govern force generation and force maintenance (Dillon et al., 1981; Aksoy et al., 1982; Giembycz & Raeburn, 1992). An understanding of smooth muscle contraction and how enzymes, such as PKC, are involved initially requires knowledge of the structure of the contractile proteins themselves and how they interact with each other ultimately to effect force.
Force generation
Myosin is a hexameric protein that forms thick bipolar filaments in muscle cells; it is comprised of a single pair of asymmetric heavy chains and two pairs of light chains. Morphologically, myosin is composed of a head and a tail region. The tail of many myosin molecules combine to form the body of each myosin filament whilst the head, which has a globular appearance, possesses the binding site for attachment to actin and two domains of catalytic (myosin ATPase) activity that provide the energy required for force development. Both pairs of light chains are also located at the globular head of myosin and it is those of 20 kDa (LC20) that are believed to exert important regulatory control over the actin-myosin interaction (see below). Actin is the other major contractile protein. In solution, actin (known as globular- or G-actin) exists as spherical monomers that polymerises in cells into thin filaments, termed filamentous- or F-actin. In force generation, actin performs two important functions. First, each G-actin subunit of the F-actin chain features a binding site for the globular heads of myosin, which permits the formation, breaking and reformation of cross-bridges (force) between the filaments by a process called cross-bridge cycling. Second, actin activates myosin ATPase when LC20 are phosphorylated by a Ca2+- and calmodulin-requiring enzyme known as myosin light chain kinase (MLCK) (Small & Sobieszek, 1977; Sobieszek & Small, 1977; Aksoy et al., 1976). Thus, in the absence of Ca2+, MLCK is catalytically inactive. However, when [Ca2+]i increases some 4–10 fold above the resting level, Ca2+ bind to calmodulin exposing one or more hydrophobic domains for which MLCK has high affinity, and a catalytically-active Ca2+/calmodulin/MLCK complex is formed. LC20 phosphorylation then ensues enabling actin to activate myosin ATPase, which hydrolyses the ATP required for the formation of stress-bearing, actin-myosin cross-bridges (Figure 2).
Figure 2.

Putative model of force generation and maintenance in ASM involving MLCK, PKC and calponin. When a Ca2+-mobilizing agonist, acting through a G-protein-coupled receptor (GPCR), is applied to an ASM cell of the contractile phenotype, there occurs a transient increase in the [Ca2+]i of sufficient magnitude to occupy all four Ca2+-binding sites on calmodulin (CM). In this form Ca2+-bound CM is able to active myosin light chain kinase (MLCK), which phosphorylates the 20 kDa light chains of myosin (LC20) permitting actin activation of myosin ATPase, cross-bridge cycling and, in turn, force generation. When the [Ca2+]i returns towards the resting levels, Ca2+ dissociate from CM and MLCK is dephosphorylated by myosin light chain phosphatase (MLCP). However, in many cases, force is maintained. One explanation for Ca2+-independent force generation is the activation of a novel protein kinase C (PKC) isoform such as PKCε by diacylglycerol (DAG), which is also generated by the contractile agonist and phosphorylates an actin-linked protein, calponin (Cal). Under resting conditions Cal inhibits the ability of actin to activate myosin ATPase and so keeps the muscle relaxed. However, in the phosphorylated state, Cal dissociates from actin thereby relieving its inhibitory effect on myosin ATPase. Ca2+-independent contraction then ensues due to the phosphorylation of LC20 by the resting activity of MLCK. Cal is thought to be regulated by reversible phosphorylation and protein phosphatase 2A (PP2A) has been shown to dephosphorylate Cal to a form that prevents actin-activated myosin ATPase activity (see text for further details).
Force maintenance
Although LC20 phosphorylation undoubtedly accounts for force generation in ASM, evidence exists that a different mechanism(s), as well as or in addition to LC20 phosphorylation, may be responsible for steady-state force. This conclusion was based on findings that force is maintained when LC20 are dephosphorylated and myosin ATPase activity is low, and lead to the proposal of a modified stress-bearing, non-cycling, or very slowly cycling, cross-bridge between actin and myosin filaments, which was called the ‘latch-bridge' (Dillon et al., 1981; Chatterjee & Murphy, 1983). However, subsequent studies identified serious inconsistencies with the original ‘latch-bridge' hypothesis (see Giembycz & Raeburn, 1992 for discussion) and led Murphy et al. (1990) to re-define latch as a ‘. . . state characterized by moderate phosphorylation [of LC20] levels when high forces are associated with reduced contraction rates and ATP consumption.' Nevertheless, the general finding that ATP utilization and [Ca2+]i in ASM are low during force maintenance and that LC20 phosphorylation can be clearly dissociated from myosin ATPase activity has led to the proposal that tonic force is controlled and/or regulated, at least in part, by a different mechanism.
One explanation is that cross-bridges, generated during the development of force, are in some way modified to a ‘latch' state, which is responsible for force maintenance. Implicit in this hypothesis is that LC20 phosphorylation is both necessary and sufficient to explain this modified actin-myosin interaction and represents a, so-called, myosin-linked process. Another possibility that has received much attention and is not mutually exclusive with the first, is that latch is due to an undefined actin-linked mechanism(s) that may involve the participation of other proteins including caldesmon, calponin and/or one or more of the actin intermediate filaments. Both postulates are being rigorously investigated, including the potential role played by PKC. These are discussed below.
Myosin-linked regulation – role of LC20 phosphorylation
Phorbol diesters are used widely as activators of conventional and novel PKCs due to their ability to substitute for DAG, and Rembold & Murphy (1988) exploited this pharmacological property to assess if latch is a myosin-linked process that is mediated by PKC through the phosphorylation of LC20. Using porcine carotid arterial smooth muscle strips these investigators reported that the phorbol diester, phorbol 12, 13-dibutyrate (PDBu), evoked a delayed, slow increase in force that was preceded by Ca2+ mobilization and LC20 phosphorylation. Given that phorbol diesters can promote Ca2+ influx into vascular smooth muscle (see Beech, 1997), Rembold & Murphy (1988) reasoned that the PDBu-induced contraction was mediated by Ca2+ through their ability to promote the MLCK-dependent phosphorylation of LC20 and cross-bridge cycling. Consistent with this interpretation is the finding that although PKC catalyses the phosphorylation of LC20 on several residues, this is not associated with an increase in myosin ATPase activity (Ikebe et al., 1987; Bengur et al., 1987). Murphy and co-workers thus concluded that LC20 phosphorylation could account quantitatively for the mechanical effect of PDBu without the need to involve an undefined (actin-linked) PKC-dependent mechanism.
Verification of this conclusion is provided by Singer et al. (1989) who observed that PDBu promoted the phosphorylation of LC20 at S1, S2 and S19 in rabbit aortic smooth muscle, the latter of which was phosphorylated by MLCK but not PKC. The knowledge that S19 phosphorylation is Ca2+-dependent suggests that PDBu evokes this response through the Ca2+/calmodulin-dependent activation of MLCK with much of the Ca2+ being derived extracellularly through voltage-dependent Ca2+ channels (VDCCs).
Of the five main types of VDCCs thus far defined, it is the L-, or long-lasting, type that is most abundant in the plasma membrane of smooth muscle cells including ASM (Small & Foster, 1994). The use of 1,4-dihydropyridine agonists and antagonists as probes has led to the discovery that L-type VDCCs are large oligomeric proteins with dissimilar subunits that, in non-skeletal muscle, combine to form an α1.α2–δ.β complex. In the heart, the α1 subunit is recognized as a target for channel modulation. In particular, it is phosphorylated by PKC that, together with the β subunit (also a substrate for PKC), results in channel activation by relieving an inhibitory constraint exerted by the N-terminus of α1 (Shistik et al., 1998). Whether such regulation is conferred by PKC in smooth muscle is unclear, but some indirect evidence to support this idea is available.
Studies with phorbol diesters
In ASM from the cow (Park & Rasmussen, 1986; Knox et al., 1993), guinea-pig (Menkes et al., 1986), rabbit (Schramm & Grunstein, 1989), rat (Chopra et al., 1994) and humans (Yang & Black, 1996; Rossetti et al., 1995), low concentrations of phorbol diesters elicit slow tonic contractures similar to those evoked in the vasculature (see above). Significantly, force development is antagonized by inhibitors of PKC, including Ro 31-8220 and Ro 31-7549 (Chopra et al., 1994), as well as by staurosporine (Yang & Black, 1995). The rate and magnitude of tension generation is enhanced by Ca2+ ionophores and agonists of L-type VDCCs such as BAY K 8644 (Part & Rasmussen, 1985; Yang & Black, 1996). Conversely, chelation of extracellular Ca2+ or application of nifedipine, diltiazem and verapamil attenuates phorbol diester-induced contractions (Park & Rasmussen, 1985; Knox et al., 1993). Taken together, these data suggest that phorbol diesters promote force generation in ASM, at least in part, by a PKC-dependent process that involves an increase in the open-state probability of L-type VDCCs (possibly by direct phosphorylation of the α1 subunit), Ca2+ influx, LC20 phosphorylation and activation of the contractile machinery. This sequence of events is supported by the finding that phorbol diesters amplify K+-induced contractions of guinea-pig, canine and bovine trachealis (Menkes et al., 1986; Gunst et al., 1994; Yang & Black, 1996). However, more recent data have challenged this hypothesis. For example, phorbol 12,13-diacetate (PDA) and an activating PKC fragment suppressed, rather than enhanced, inward Ca2+ currents through L-type VDCCs in porcine tracheal smooth muscle cells by a mechanism that was antagonized by the PKC inhibitor, calphostin C (Yamakage et al., 1995). Moreover, it has been demonstrated that phorbol diesters can contract smooth muscle by inhibiting, directly or indirectly, the activity of myosin light chain phosphatase (MLCP). This phenomenon, which leads to LC20 phosphorylation at S19 and force development at a constant Ca2+ concentration (Masuo et al., 1994; Peiper et al., 1996; Iizuka et al., 1999), is known as Ca2+ sensitization (Somlyo & Himpens, 1989; Somlyo & Somlyo, 1994; Kitazawa et al., 1989; 1991a) and is well recognized as a mechanism of force generation/maintenance in ASM of several species including the cow (Takuwa et al., 1986, rabbit (Yoshii et al., 1999; Iizuka et al., 1999) and dog (Iizuka et al., 1997).
Recent studies by Bremerich et al. (1998) have confirmed that PDBu increases LC20 phosphorylation in β-escin-permeabilized canine trachealis at a constant Ca2+ concentration. However, these investigators provided two pieces of evidence against the idea that LC20 phosphorylation accounts for force development under these conditions. First, PDBu promotes the phosphorylation of LC20 on residues (S1, S2, T9) that are not thought to contribute to contraction (Ikebe et al., 1987; Bengur et al., 1987; Kamm & Stull, 1989). In fact, phosphorylation of T9 has been shown to suppress the increase in myosin ATPase activity associated with phosphorylation of S19, a major site for MLCK (Kamm & Stull, 1989). In addition, PKC does not affect actin sliding velocity in an in vitro motility assay when LC20 are phosphorylated by MLCK at S19 (Umemoto et al., 1989). Thus, it has been speculated (Bremerich et al., 1998) that phorbol diesters promote the PKC-dependent phosphorylation of LC20 at residues that do not affect force. This conclusion is supported by the additional observation that under certain conditions PDBu can increase LC20 phosphorylation without concomitant force development (Gerthoffer, 1986; Gerthoffer et al., 1989). Second, calphostin C and a pseudosubstrate peptide inhibitor of PKC block PDBu-induced LC20 phosphorylation but not Ca2+ sensitization. Collectively, therefore, it has been proposed that at a constant Ca2+ concentration PDBu increases force in ASM by a PKC-independent mechanism that is unrelated to its ability to phosphorylate LC20 (Bremerich et al., 1998). In this respect PDBu potentiates the effect of endothelin-1 on ovine tracheal smooth muscle (Katoch, 1997) and carbachol on guinea-pig ileum (Sasaguri & Watson, 1989) under conditions where PKC is down-regulated implying that the phorbol diester is acting at a different intracellular locus. A number of potential targets have been identified that can act as receptors for phorbol diesters including Unc-13 (Kazanietz et al., 1995) and n-chimaerin (Ahmed et al., 1993; reviewed in Ron & Kazanietz, 1999). While the role of these proteins in regulating smooth muscle tone is unexplored, it is noteworthy that the C-terminal domain of chimaerins is thought to act as a GTPase-activating protein for members of the Rho subfamily of small GTP-binding proteins (Ahmed et al., 1993). In airways and other smooth muscle, p21rhoA has been implicated in Ca2+ sensitization (Hirata et al., 1992; Fujihara et al., 1997; Gong et al., 1996; 1997; Akopov et al., 1998; Yoshii et al., 1999; Iizuka et al., 1999) and it was recently reported that PDBu-induced contractions of rabbit trachealis at constant Ca2+ concentration were partially reversed by Y-27632, an inhibitor of rho-associated coiled coil-forming protein kinase (ROCK) I and ROCK II (also known as p160ROCK and ROKαrho kinase respectively) (Yoshii et al., 1999). ROCKs lie down-stream of p21rho in smooth muscle and can phosphorylate and, thereby, inactivate, MLCP (Amano et al., 1996).
Given the aforementioned discussion, it is clear that the available evidence is insufficient to formulate a unifying hypothesis that adequately explains phorbol diester-induced contraction of ASM. While certain pharmacological experiments point to the activation of VDCCs, Ca2+ influx and MLCK-driven LC20 phosphorylation as described for vascular smooth muscle, electrophysiological investigations and studies with permeabilized smooth muscles have provided little corroborating support and, in fact, suggest that phorbol diesters may regulate tone independently of PKC. However, as described below, this later idea should be viewed with caution since most of the evidence against a role for PKC is derived from permeabilized preparations from which proteins essential to the contractile process may be lost.
Studies with agonists
Phorbol diesters have the ability to activate at least nine immunologically and, presumably, functionally distinct isoenzymes that comprise the conventional and novel PKC families. As shown in Table 2, multiple cPKCs and nPKCs are co-expressed in ASM from all species thus far examined and it is important to appreciate that phorbol diesters will, theoretically, activate all of these. Therefore, data arising from such studies cannot be considered representative of the effects of receptor agonists which, almost certainly, will selectively activate specific isoforms. Furthermore, phorbol diesters do not permit an evaluation of the role of aPKCs in biological processes since this subfamily of enzymes lacks the diglyceride-binding domain.
Like phorbol diesters, agonism of some G-protein-coupled receptors on ASM also activates L-type VDCCs including the ETA- (Grunstein et al., 1991; Yang et al., 1994), 5-HT2A (Watts et al., 1994) and M3 muscarinic receptor (Tomasic et al., 1992; Kamashima et al., 1992). Therefore, it is conceivable that, as in vascular smooth muscle, this effect is mediated by PKC and leads to contraction through the activation of MLCK. However, although inhibitors of VDCCs, such as nifedipine and verapamil, effectively inhibit L-type Ca2+ currents in ASM cells of some species (Kotlikoff, 1988) they fail, in many cases, to significantly affect tension generation (Coburn, 1979; Farley & Miles, 1978; Cheng & Townley, 1983; Gunst & Pisoni, 1986; Duncan & Douglas, 1984; Ahmed et al., 1985). This discrepancy has led to the belief that most of the Ca2+ necessary for agonist-induced contraction of ASM is derived from the sarcoplasmic reticulum whereas Ca2+ influx through VDCCs is required for refilling the internal Ca2+ stores (Daniel et al., 1992). Exceptions to this general rule include the ability of L-type VDCC antagonists to suppress 5-HT- and carbachol/methacholine-induced tension generation in guinea-pig trachea and rat bronchioles (Watts et al., 1994; Kamishima et al., 1992). The 5-HT results are particularly important since contraction is not associated with inositol phospholipid hydrolysis (Watts et al., 1994) implying that activator Ca2+ are mobilised by an Ins(1,4,5)P3-independent process. Although this might involve the novel second messengers, cyclic ADP ribose or sphingosine 1-phosphate, which can both mobilise intracellular Ca2+ in response to agonists that act through G-protein-coupled receptors (Higashida, 1977; Meyer zu Heringdorf et al., 1998), it is likely that much of the Ca2+ are derived extracellularly and enter the cell through VDCCs. Indeed, the additional finding that 5-HT-induced contractions are abolished by Ro 31-8220, Ro 31-7549 and GF 109203X (Watts et al., 1994; Chopra et al., 1994) suggests that cPKCs and/or nPKCs are involved in the nifedipine-sensitive and insensitive components of contraction. Since 5-HT fails to hydrolyse inositol-containing phospholipids in guinea-pig trachea, it is tempting to speculate that the 5-HT2A receptor subtype couples to PLD, which will indirectly provide the diglyceride necessary to activate nPKCs from phosphatidylcholine.
An interesting, and equally confounding, finding is that ACh reversibly inhibited whole cell L-type Ca2+ currents in guinea-pig tracheal myocytes by a mechanism blocked by the selective M3 muscarinic antagonist 4-DAMP, but not AF-DX 116 (M2-selective) (Yamashita & Kokubun, 1997). Moreover, neither phorbol 12-myristate 13-acetate (PMA) nor the DAG analogue, 1-oleoyl-2-acetyl sn-glycerol (OAG), mimicked this effect suggesting that the inhibition of ACh-induced Ca2+ influx is mediated by a PKC-independent mechanism. Acetylcholine similarly inhibits VDCCs in porcine tracheal smooth muscle cells but, in this tissue, PKC seemingly plays a role since calphostin C and a pseudosubstrate peptide inhibitor antagonized this effect (Yamakage et al., 1995). The reason for these contrary results is unknown. Likewise, the discrepancy between the results of these studies and those of Tomasic et al. (1992) and Kamishima et al. (1992) described above, where M3 muscarinic receptor activation enhanced L-type VDCCs was reported, equally are difficult to reconcile but differences between patch clamp methods have been proposed (Yamakage et al., 1995).
Another mechanism that could account for agonist-induced force maintenance is Ca2+ sensitization. Indeed, there is a consensus that agonists that act through G-protein-coupled receptors sensitize the contractile machinery to Ca2+ primarily by inhibiting MLCP, although exactly how this occurs is controversial (Kitazawa et al., 1991a,1991b; 1999; Li et al., 1998; Shirazi et al., 1994; Parker et al., 1998). Agonist-induced Ca2+ sensitization has been described for canine (Gerthoffer, 1996; Iizuka et al., 1997; Bremerich et al., 1997), human (Yoshii et al., 1999) and rabbit trachealis (Yoshii et al., 1999) and a role for PKC in this response has been sought. Using β-escin-permeabilized canine tracheal smooth muscle cells, Bremerich et al. (1997) demonstrated that at a constant submaximal concentration of Ca2+ and in the presence of GTP, ACh-induced Ca2+ sensitization was neither prevented nor reversed by calphostin C, chelerythrine or a pseudosubstrate peptide inhibitor of PKC. Of particular significance was the result of an additional experiment in which the DAG analogue, OAG, similarly failed to mimic the effect of ACh (Bremerich et al., 1997). In other permeabilized smooth muscle preparations stimulated with GTPγS, PKC inhibitors are also inactive (Akopov et al., 1998; Fujita et al., 1995; Itoh et al., 1994) or poorly active (Iizuka et al., 1999) and there is a wealth of literature implying that neither inhibition nor down-regulation of PKC affect agonist-induced contractions in many intact smooth muscles (Oishi et al., 1992; Sasaguri & Watson, 1989; Yoshida et al., 1994; Fujita et al., 1994). Taken together, these results do not implicate PKC in agonist-induced Ca2+ sensitization and point to other effectors such as the small GTP-binding protein p21rho (Akopov et al., 1998; Yoshida et al., 1994; Otto et al., 1996; Itoh et al., 1994; Fujita et al., 1995; Fujihara et al., 1997; Amano et al., 1996). However, it is conceivable that permeabilization of smooth muscle may alter signal transduction by allowing essential cofactors and integral proteins to be lost through the pores. This possibility was formally investigated in a recent publication by Kitazawa et al. (1999) in which methods of permeabilization of aortic smooth muscle strips were compared. The most salient observation of that study was the absence of Ca2+ sensitization in tissue permeabilized with Triton X-100 due to the depletion of PKC and a novel endogenous smooth muscle-specific inhibitor of MLCP, CPI-17 (Eto et al., 1995b; 1997) from the tissue (Kitazawa et al., 1999). Indeed, reconstitution of tissues with PKCα and CPI-17 increased LC20 phosphorylation and promoted contraction at a constant Ca2+ concentration by a mechanism that was abolished by Go6976, a selective inhibitor of cPKCs (Kitazawa et al., 1999). Significantly, CPI-17 is phosphorylated by PKC on T38 and phospho-CPI-17 inhibits the catalytic subunit and holoenzyme of MLCP with equally high potency resulting in LC20 phosphorylation. These data have culminated in a new, simple model of Ca2+ sensitization in smooth muscle that is independent of regulation at the level of the thin actin filaments (discussed below). In its simplest form, phorbol diesters and agonists evoke Ca2+-independent contractions through the PKC-dependent phosphorylation of CPI-17, which increases basal MLCK activity indirectly through the inhibition of MLCP.
Actin-linked regulation – role of caldesmon, calponin and the actin-intermediate filaments
Although most investigators support the general view that LC20 phosphorylation adequately explains Ca2+-dependent smooth muscle contraction, this concept ignores the observation that under certain conditions force can develop without LC20 phosphorylation (see Gerthoffer, 1991). For example, saponin-permeabilized, single smooth muscle cells isolated from ferret aorta contract in response to phenylephrine and PKM, a constitutively active fragment of PKC, in the absence of Ca2+ and LC20 phosphorylation by a mechanism that is abolished by a pseudosubstrate peptide inhibitor of the α- and β-isoforms of PKC (Collins et al., 1992). Moreover, phorbol diesters and many contractile agonists promote the phosphorylation of a number of, so-called, late phase proteins that may play auxiliary regulatory roles in smooth muscle contraction (Park & Rasmussen, 1986; Rasmussen et al., 1987; Walsh et al., 1996; Pohl et al., 1997). Indeed, exposure of bovine tracheal smooth muscle strips, labelled with inorganic phosphate, to PDBu results in a slow, well sustained phosphorylation of several contractile/cytoskeletal proteins including caldesmon, synemin, filamin and α- and β-desmin that parallels closely the development of force (Park & Rasmussen, 1986; Rasmussen et al., 1987). Significantly, these proteins are also phosphorylated by carbachol with similar kinetics. Another actin-associated protein, calponin, is also phosphorylated in ASM by contractile stimuli (Pohl et al., 1997), and has led to the view that the PKC-dependent phosphorylation of one or more of these proteins may promote, or facilitate, the actin-myosin cross-linking that maintains tonic force.
Caldesmon
Caldesmon is an acidic, actin-binding protein associated with the thin actin filaments that also interacts with calmodulin and myosin (see Walsh, 1990) and is ubiquitously expressed in smooth muscle including the trachealis (Park & Rasmussen, 1986; Gerthoffer & Pohl, 1994; Gerthoffer et al., 1997). The postulate that caldesmon might regulate the actin-myosin interaction stemmed from the in vitro observation that it could reversibly bind to either calmodulin or actin depending on the Ca2+ concentration (Sobue et al., 1981). Although it is now known that caldesmon remains attached to actin during the contraction-relaxation cycle in intact smooth muscle, persuasive evidence is available that it may play a prominent role in regulating smooth muscle contraction. Thus, in a variety of smooth muscles caldesmon inhibits actin-activated myosin ATPase without affecting LC20 phosphorylation (Ngai & Walsh, 1984; Clark et al., 1986). Myosin ATPase activity is also attenuated by caldesmon in the absence of actin (Hemric & Chalovich, 1988; Ikebe & Reardon, 1988). These observations suggest that there must be some intrinsic mechanism by which the inhibition of myosin ATPase activity can be relieved. One possibility is that caldesmon is regulated by reversible phosphorylation. Indeed, Sobue & Sellers (1991) reported that caldesmon is a substrate for several protein kinases, including PKC, and there is evidence that caldesmon is phosphorylated in intact trachealis during agonist-induced contraction (Gerthoffer & Pohl, 1994; Gerthoffer et al., 1997). In vascular smooth muscle, the residues phosphorylated are proline-directed serines (i.e. S789PTKV and S759PAPK), which is characteristic of a mitogen-activated protein (MAP) kinase consensus sequence (Adam et al., 1992; 1993). Thus, in intact smooth muscle extracellular-regulated kinase (ERK)-1 and/or ERK-2, rather than PKC, may be the endogenous caldesmon kinase. In this respect, purified caldesmon is phosphorylated by murine ERK-2 in a cell-free system and addition of ERK-2 to Triton X-100-permeabilized canine ASM fibres potentiates Ca2+-induced tension development (Gerthoffer et al., 1997). Conversely, unphosphorylated caldesmon produces a concentration-dependent inhibition of actin sliding velocity in an in vitro motility assay (Shirinsky et al., 1992), presumably through its ability to inhibit myosin ATPase and prevent actin-myosin interactions. Further support for a MAP kinase/caldesmon cascade in regulating smooth muscle contractility derives from pharmacological studies where phenylephrine-induced contraction of ferret aortic strips in the absence of extracellular Ca2+ was significantly suppressed by PD 098059 (Dessy et al., 1998), a selective inhibitor of MAP kinase kinase-1 (MEK-1).
If ERK-1 and/or ERK-2 are direct upstream regulators of caldesmon phosphorylation where does this leave PKC? Khalil & Morgan (1992) have reported that Ca2+-independent, phenylephrine-induced contraction of ferret aortic smooth muscle cells is associated with a sustained translocation of PKCε to the plasma membrane. This effect is accompanied by a transient association of MAP kinase with the membrane followed by a second re-localization towards the vicinity of the contractile filaments prior to active force development (Khalil & Morgan, 1992). The finding that inhibitors of PKC abolished the intracellular redistribution of ERK-1/ERK-2 suggests that members of this MAP kinase family may transmit signals from membrane-bound PKCε to the contractile machinery. Indeed, ERK-1/ERK-2 is a substrate for PKC and is caapable of phosphorylating caldesmon on residues that are phosphorylated in vivo during smooth muscle contraction (Adam et al., 1992).
Calponin
Shortly after the discovery of caldesmon, Takahashi et al. (1986; 1987) identified a basic, heat-stable protein in chicken gizzard and bovine aortic smooth muscle that displayed properties consistent with the regulation of the actin-myosin interaction. This protein was named calponin and has been identified in many tissues including ASM (Gerthoffer & Pohl, 1994; Pohl et al., 1997). In vitro, calponin interacts with actin and calmodulin in a way similar to caldesmon (see above). In addition, calponin inhibits actin-activated myosin ATPase activity without affecting LC20 phosphorylation, an effect that (in a reconstituted cell-free system) is regulated by reversible protein phosphorylation. Indeed, under these conditions calponin is a substrate for PKC (Winder & Walsh, 1990), in particular PKCε (Horowitz et al., 1996b), and displays a dramatic reduction in affinity for actin and is unable to inhibit myosin ATPase activity in the phosphorylated state. An activity capable of dephosphorylating calponin has been found in smooth muscle and identified as a type 2A protein phosphatase (Winder et al., 1992). Moreover, dephosphorylation of calponin by this phosphatase restored actin binding and the inhibition of myosin ATPase (Winder et al., 1992). Functionally, phosphorylation of calponin by PKC results in contraction of porcine coronary artery (Mino et al., 1995) whereas unphosphorylated calponin relaxes single permeabilized vascular smooth muscle cells pre-contracted with phenylephrine and PKCε (Horowitz et al., 1996a,1996b). Similarly, calponin causes a profound inhibition of actin sliding velocity in a cell-free motility assay whereas phosphocalponin is inactive (Pohl et al., 1997). In intact canine tracheal smooth muscle strips, metabolically labelled with inorganic phosphate, carbachol promoted the phosphorylation of calponin and the appearance of an additional, more acidic, isoform over the four variants that were expressed in unstimulated cells (Gerthoffer & Pohl, 1994; Pohl et al., 1997). The subcellular localization of calponin is also affected by PKC-dependent phosphorylation. In unstimulated smooth muscle cells isolated from the ferret portal vein, calponin is primarily cytosolic where it is associated with filamentous structures (presumably F-actin). However, following exposure to phenylephrine, which activates PKC in these cells, calponin redistributes to a region of the cell just underneath the plasma membrane (Parker et al., 1994). The identity of the PKC isoform(s) involved in calponin phosphorylation in ASM has not been explored, although cPKCs or nPKCs are possible candidates given that GF 109203X inhibits the tonic phase of histamine-induced tension generation in human bronchus (Yang & Black, 1995). In the vasculature, a strong case has been made for the novel isoform, PKCε, since it is Ca2+-independent, translocates to the plasma membrane upon stimulation, produces contractile responses in permeabilized muscle strips clamped at low free [Ca2+] and is activated by phorbol diesters (see Walsh et al., 1996). Based upon these data, it has been postulated that agonist-induced, Ca2+-independent contraction of smooth muscle is mediated by PKC through its ability to phosphorylate calponin. This, in turn, facilitates its release from actin thereby relieving the inhibition of myosin ATPase. Slow, sustained contraction then ensues due to the phosphorylation of LC20 by the basal activity of MLCK (Walsh et al., 1996; Figure 2).
More recent studies have shown that calponin binds to both PKCε and ERK-1/ERK-2 in ferret aorta and that it translocates to the cell' cortex with kinetics identical to the association of PKCε and ERK-1/ERK-2 to the plasma membrane (Menice et al., 1997). Of potential significance is that the interaction of ERK with calponin does not result in phosphorylation. Moreover, the amount of calponin in ERK immunoprecipitates decreases at late time-points after agonist (phenylephrine) exposure whereas increased amounts are found at early time-points in PKCε immunoprecipitates. Collectively, these results have led Morgan and co-workers to question the original idea that calponin may directly prevent the actin-myosin interaction (see above). Instead, they propose that calponin is associated with ERK under resting conditions and with PKCε in cells exposed phenylephrine where it acts as an adapter protein to facilitate the migration of ERK and PKCε to the plasma membrane (Menice et al., 1997). Indirect support for this hypothesis is that calponin dissociates from the thin actin filaments after agonist exposure before measurable force is generated (Parker et al., 1994; Menice et al., 1997), which is difficult to reconcile with a direct modulation of the actin-myosin interaction.
Desmin, synemin and filamin
In addition to caldesmon and calponin, four other proteins (filamin, synemin, α-desmin, β-desmin) have been found in airways and other smooth muscles (Park & Rasmussen, 1986) that, together with actin, form the actin-intermediate filaments (Small et al., 1986). It has been proposed that these filaments may constitute a mechanism of force development distinct from the classical actin-myosin system described above (Granger & Lazarides, 1980; Small et al., 1986). Indeed, even though few investigations have considered the functional significance of the actin-intermediate proteins, ultrastructural studies indicate that they may be specifically involved in the genesis of tonic force. This could occur by the formation of actin-actin cross-bridges promoted by proteins such as filamin that, itself, requires the phosphorylation of other proteins including synemin and desmin (Small et al., 1986). Some support for this hypothesis is provided from studies conducted with bovine tracheal smooth muscle in which desmin was found to be the most abundant of the actin-intermediate filaments phosphorylated in response to carbachol and 12-deoxyphorbol 13-isobutyrate (DPB; Park & Rasmussen, 1986). Since the slow contracture evoked by the phorbol diester mimics the maintained phase of the carbachol-induced contraction, a case for phosphorylated desmin catalyzed by PKC has been made (Park & Rasmussen, 1986). Indeed, desmin can be phosphorylated on four residues by PKC (Kitamura et al., 1989).
Subcellular re-distribution of PKC during contraction
In canine and bovine tracheal smooth muscle, methacholine and carbachol promote a rapid translocation of PKC from the cytosol to the membrane implying a functional change from the inactive to the active form (Yamakage, 1992; Langlands & Diamond, 1992). Generally, this effect is transient and less well sustained than phorbol diester-induced responses (Haller et al., 1990). Similar data have been obtained with intestinal smooth muscle, where PKCβII is implicated in methacholine-induced contraction (Xu et al., 1991), and the vasculature where endothelin, histamine and phenylephrine elicit rapid translocation of PKC from the cytosol to the membrane (Haller et al., 1990; Khalil & Morgan, 1992).
The identity of the PKC isoforms involved in agonist-induced contraction of ASM is unknown. However, in vascular smooth muscles, data are emerging that vessel type, even within the same species and in response to the same agonist, dictates the complement of isoforms activated. This phenomenon is elegantly illustrated in studies performed by Morgan and colleagues with permeabilized vascular smooth muscle from the ferret. Thus, at a constant Ca2+ concentration, Lee et al. (1999) found that a pseudosubstrate inhibitor of cPKCs inhibited phenylephrine-induced contraction of the portal vein but not aorta. Conversely, a peptide targeted against the N-terminus (translocation domain) of PKCε blocked contractions of the aorta elicited by phenylephrine but not those of the portal vein under identical experimental conditions. These functional data were supported by Western blotting where PKCα and PKCε were strongly expressed in portal vein and aorta respectively (Lee et al., 1999). Moreover, digital imaging of the distribution of PKCs in these two muscles before and after exposure to phenylephrine found that PKCα translocated from the cytosol to membrane in cells of the portal vein but not aorta (Khalil et al., 1994; Lee et al., 1999). In contrast, PKCε migrated to the periphery of aortic smooth muscle cells in response to phenylephrine whereas no such redistribution was detected in the portal vein (Khalil et al., 1992; Lee et al., 1999). This selective effect on specific PKC isoenzymes may reflect discrete activation in a subcellular compartment or differences in the expression of, so-called, scaffold or anchoring proteins such as RACKs to which these enzymes interact.
PLD and PKC-mediated, Ca2+-independent contraction
Several agonists that act through G-protein-coupled receptors including ACh and bradykinin activate PLD in [3H]-palmitic acid-labelled porcine and guinea-pig tracheal smooth muscle respectively in the presence of a primary alcohol as indicated by a significant increase in the formation of [3H]-phosphatidylalcohol (Pyne & Pyne, 1993a,1993b; Mamoon et al., 1999). Such an effect leads to the formation of phosphatidic acid and, ultimately, DAG by the action of phosphatidic acid phosphohydrolase. Since agonist-induced PLD activation is not associated with Ca2+ mobilization, this signal transduction pathway could account for Ca2+-independent contraction of smooth muscles elicited by phorbol diesters and receptor-mediated agonists through the selective activation of nPKC isoforms such as PKCε (see above). However, ACh and bradykinin also stimulate PLC in ASM (Takuwa et al., 1986; Pyne & Pyne, 1993b; see Farmer, 1995) liberating DAG for the activation of cPKC and nPKC isoforms. Although the functional significance of this effect is obscure, it is clear that multiple PKC isoforms could be activated in response to a single stimulus that may act in a complex fashion to fine-tune various functional responses including contraction and proliferation.
PKC and relaxation
Na+/K+-dependent ATPase, Na+-H+ exchanger and Na+/K+/Cl−-co-transporter as targets for PKC
High concentrations of phorbol diesters including DPB cause relaxation of methacholine-contracted ASM (Schramm & Grunstein, 1989; Menkes et al., 1986), and evidence is available that this may be due to the activation of the Na+/K+-ATPase. Mechanistically, an increase in extracellular Na+ would lower the [Ca2+]i through enhanced Na+/Ca2+ exchange and also hyperpolarize the membrane. Indirect evidence for such a mechanism is the greater relaxation produced in DPB-treated rabbit tracheal smooth muscle when the ATPase is activated by loading the muscle with Na+ than that produced in the absence of DPB. In addition, an inhibitory effect of DPB on agonist-induced contractions is prevented by inhibiting the ATPase with ouabain or by incubating the muscle in K+-free media. This effect is independent of the activation of the Na+-H+ exchanger and of Ca2+-influx through VDCCs (Schramm & Grunstein, 1989). Human ASM also relaxes in response to a high concentration of PDBu but this is then followed by contraction (Yang & Black, 1996). Consistent with the results obtained in rabbit trachea, PDBu-induced relaxation is inhibited by ouabain whereas the contractile component is augmented (Yang & Black, 1996).
In guinea-pig trachea, phorbol diesters promote either relaxation (Morrison & Vanhoutte, 1991) or contraction followed by a relaxation (Souhrada & Souhrada, 1989a,1989b). Moreover, in a study where electrical and mechanical measurements were made simultaneously, phorbol diesters produce a triphasic electrical response: an initial rapid depolarization of the membrane and associated muscle contraction, hyperpolarization of the membrane and coincident relaxation followed by a second slow depolarization and a gradually developing contracture (Souhrada & Souhrada, 1989a). The biochemical basis for this triphasic effect is complex. All phases of the response were suppressed, to some extent, by verapamil indicating that the phorbol diester promotes Ca2+ influx through L-type VDCCs. In addition, the first and second phase were inhibited by amiloride (c.f. rabbit trachea) implicating an alteration in Na+-H+ exchange that ultimately modifies the [Ca2+]i. The second (relaxation) component was also inhibited by ouabain suggesting that it is due, in part, to the activation of the Na+/K+ ATPase which would lower [Ca2+]i through enhanced Na+/Ca2+ exchange. The final slow contraction was attenuated by the diuretic frusemide, although whether this is through the inhibition of the Na+/K+/Cl−-co-transporter is unknown. In bovine trachealis, 5-N,N-hexamethylene amiloride and frusemide, inhibitors of the Na+-H+ exchanger and Na+/K+/Cl−-co-transporter respectively, have no effect on PDBu-induced contractions which might point to differences in regulation between species.
ATP-sensitive K+ channels as a target for PKC
Path clamp studies using porcine tracheal smooth muscle cells have established that ACh and PMA inhibit the activation of ATP-sensitive K+ channels (KATP) in response to levcromakalim (Nuttle & Farley, 1997) implying that a cPKC or nPKC is responsible. Staurosporine antagonized the effect of both ACh and PMA from which the authors suggest that activation of the muscarinic receptor on porcine trachealis inhibits KATP by a PKC-dependent mechanism. By reducing the open state probability of KATP, PMA and ACh would promote hyperpolarization, prevent further Ca2+ entry through VDCCs and so facilitate relaxation (Nuttle & Farley, 1997). However, given the pharmacology of staurosporine, this interpretation must be viewed cautiously.
Subcellular re-distribution of PKC during relaxation
In methacholine-contracted bovine trachealis, verapamil evokes relaxation and dissociation of PKC from the membrane presumably through an effect on L-type VDCCs (Langlands & Diamond, 1994). Sodium nitroprusside and the β-adrenoceptor agonist, isoprenaline, which are believed to act via cyclic GMP and cyclic AMP respectively, also release PKC from the membrane fraction and relax the tissue (Langlands & Diamond, 1992; 1994).
PKC and mitogenesis
Mitogenesis is believed to play an important role not only in normal homeostasis but also in the development of increased smooth muscle content in the airway wall in diseases such as chronic severe asthma. The mitogens involved in this process in vivo are undefined, but cell culture-based studies conducted over the last 10 years have identified several ASM mitogens, which are also contractile agonists. These include α-thrombin (Panettieri et al., 1995), histamine (Panettieri et al., 1990), leukotriene D4 (Cohen et al., 1995), thromboxane (Noveral & Grunstein, 1992), phenylephrine (Noveral & Grunstein, 1994), endothelin-1 (Noveral et al., 1992; Glassberg et al., 1994; Panettieri et al., 1996; Whelchel et al., 1997) and 5-HT (Herschenson et al., 1995). The receptors for these ligands in ASM and other cell types are G protein-coupled to PLC and the activation of PKC (Yang et al., 1994; Panettieri et al., 1995; Noveral & Grunstein, 1994). This has led investigators to hypothesize that, in addition to a possible role in the regulation of contraction, activation of PKC may also be an important signalling event for the mitogenic response of ASM. In many other cell types, activation of PKC, either directly by phorbol diesters or indirectly by mitogens, has been examined in greater detail and found to be linked to several early events associated with the onset of mitogenesis. These include activation of the Na+/H+ exchanger, which promotes intracellular alkalinization, increased cellular protein synthesis and the induction of nuclear proto-oncogenes such as c-fos and c-myc.
Studies with PKC activators
Panettieri et al. (1991a) first demonstrated a possible role for PKC in human ASM mitogenesis by showing that PMA induced DNA synthesis and the expression of c-fos. Few subsequent studies have characterized the effect of direct stimulation of PKC using phorbol diesters or synthetic DAG analogues. Instead, many investigators have examined more physiological, receptor-mediated stimuli in ASM cells in which endogenous DAG levels have been manipulated or PKC depletion strategies based on prolonged pretreatment with phorbol esters have been used (Lew et al., 1997; Hirst et al., 1995a; Abe et al., 1994; 1998). In vascular smooth muscle, PKC depletion markedly attenuates phorbol diester-induced phosphorylation of a 76 kDa (p76) protein (presumably autophosphorylated PKC) and abolishes both the proliferative response and expression of c-fos (Kariya et al., 1987; 1989; Taubman et al., 1989; Rao & Berk, 1992). In bovine tracheal smooth muscle, stimulation of proliferation by β-hexosaminidases (mannosyl-rich lysomal hydrolases) acting at mannose-recognizing receptors, was enhanced by increasing endogenous DAG levels using the DAG kinase inhibitor R59022; while depletion of PKC by prolonged treatment with phorbol esters reduced proliferation (Lew et al., 1997). In our own studies, we have shown that prolonged pre-treatment of ASM cells with 4β-PMA, but not the inactive 4α-isomer, markedly attenuated the subsequent proliferative response induced by foetal bovine serum (Hirst et al., 1995a) or platelet-derived growth factor (PDGF)-BB (Hirst et al., 1994).
Studies with PKC inhibitors
In search of evidence that activation of PKC is a necessary event for proliferation of ASM, investigators have also examined the effect of inhibitors of PKC. Lew et al. (1997) found that pre-treatment of bovine tracheal smooth muscle cells with calphostin C prevented proliferation following activation of mannose receptors by β-hexosaminidase. Proliferation of rabbit tracheal smooth muscle cells induced by foetal calf serum was also inhibited by the bisindolylmaleimide, Ro31-8220 and the less potent analogue, Ro31-7549 (Hirst et al., 1995a). In the same study, these inhibitors also abolished Ca2+-dependent and -independent PKC activity purified from ASM cells. We have found that PKC inhibitors are effective in preventing proliferation of human (Hirst et al., 1995b) and rabbit (Hirst et al., 1994) ASM induced by PDGF isoforms acting through distinct PDGF receptors. Our findings contrast with those of others, using bovine tracheal smooth muscle cells, where calphostin C (Lew et al., 1997) and other selective PKC inhibitors such as GF109203X, Ro31-8220 and chelerythrine (Walker et al., 1998) failed to inhibit PDGF-stimulated DNA synthesis, but are in agreement with other findings reported in human ASM in which proliferation, induced by PDGF-BB, was dependent not only on tyrosine kinase-dependent mechanisms, but also on PLC-dependent processes in which PKC activation is believed to be a downstream event (De et al., 1996).
Functional role of PKC in proliferation
Little information is currently available on the PKC isoforms that control cell growth or the state of differentiation of ASM. Assender et al. (1994) reported that non-proliferating human and rat vascular smooth muscle cells showed a similar qualitative pattern of PKC isoform expression (α, δ and ζPKC) to proliferating smooth muscle cells in culture. Analysis of the mRNA, however, revealed that unlike their proliferating counterparts, contractile smooth muscle cells did not express mRNA for PKCε. Thus, while PKCα was found in smooth muscle cells in all states of differentiation, expression of PKCε was increased as smooth muscle cells became more proliferative. Expression of PKCβ was not detected in either freshly isolated or proliferating smooth muscle cells. In another study, PKCα was down-regulated when rat vascular smooth muscle cells assumed the proliferative-secretory phenotype (Haller et al., 1995). When the cells reassumed their contractile state, PKCα expression reverted to normal levels.
Similar evidence for phenotype-dependent expression of PKC isoforms is also emerging in ASM. For example, in canine tracheal smooth muscle, Halayko et al. (1996), using an antibody which recognizes the α and βPKC isoforms (note that PKCα was reported not to be expressed in canine trachealis (Donnelly et al., 1995)), reported that expression of PKCβ was increased 8–10 fold in proliferating compared to contractile cells. This is consistent with our own unpublished observations in human ASM in which we have found increased expression of PKCβ1, as well as PKCε in proliferating cells, compared to their serum-deprived, non-proliferating counterparts (Hirst, Webb and Giembycz, unpublished observations).
The functions of the various PKC isoforms expressed in ASM have not been investigated in any detail and we must look to other tissues for possible clues. In non-muscle cells, the PKCβII and ζ isoenzymes are reported to be essential for differentiation and proliferation (Murray et al., 1993; Berra et al., 1993). In human arterial smooth muscle cells, PKCα activation is necessary for progression from G1 to the S phase of the cell cycle and subsequent proliferation (Marrilley et al., 1996) and selective depletion of PKCα by antisense strategies prevents this response (Leszczynski et al., 1996). In contrast, PKCδ, also expressed in ASM, has previously been shown to retard cell cycle progression in G1 through an effect on cell cyclins (Fukumoto et al., 1997), suggesting that activation of different PKC isoforms in smooth muscle may lead either to growth acceleration or inhibition. Assender et al. (1994) reported that PKCζ was present in both proliferating and contractile vascular smooth muscle cells of the rat. A study examining PKCζ activity in chicken gizzard smooth muscle suggests that this isoform differs very markedly from other PKC isoforms in its susceptibility to down-regulation by phorbol diesters or inhibition by PKC inhibitors such as sphingosine, chelethyrine, staurosporine or calphostin C (Clemont-Chomienne & Walsh 1995). Given that ASM proliferation in response to a range of stimuli can be prevented by prolonged exposure to phorbol diesters or the presence of inhibitors of PKC, it might seem appropriate to conclude that at least in ASM, PKCζ is not involved directly in the proliferative process. However, a recent preliminary report documented that transfection of human ASM cells with a dominant negative PKCζ mutant inhibited PDGF-stimulated DNA synthesis by up to 80% (Black et al., 1998). In a follow-up study, the same investigators demonstrated that proliferation was associated with up-regulation of PKCζ without any change in the expression of other PKC isoforms (Carlin et al., 1999; Table 2).
While there seems little agreement over the patterns of expression of particular PKC isoforms in either contractile or proliferative vascular ASM, the ability of Ro31-8220 and Ro31-7549 to abolish Ca2+-dependent and -independent PKC activity purified from ASM, and to inhibit proliferation (Hirst et al., 1995a), is consistent with a role of at least one or more of these isoenzymes in the proliferative process. Furthermore, both Ro31-8220 and Ro31-7549 potently and selectively inhibit α, βI, βII and εPKC isoenzymes from other cell systems (Wilkinson et al., 1993), which are also expressed in ASM obtained from several species including humans (Table 2).
Interaction of PKC with other signalling molecules
Although most investigators now agree that activation of PKC is important in the cascade of biochemical events that lead to proliferation, not all agonists that activate PKC cause proliferation in smooth muscle cells. In rat cultured aortic smooth muscle cells, rendered quiescent in a defined serum-free medium, both angiotensin II (Geisterfer et al., 1989) and arginine-vasopressin (Geisterfer & Owens 1989) fail to induce proliferation, but cause marked increases in cellular protein synthesis with subsequent hypertrophy. Despite failure to induce proliferation, both angiotensin II and arginine-vasopressin stimulate early signal transduction events in cultured vascular smooth muscle cells such as activation of PLC, mobilization of intracellular Ca2+, activation of PKC and induction of proto-oncogenes (Berk et al., 1988; Griendling et al., 1987; Berk et al., 1989; Taubmann et al., 1989). Likewise, in human ASM cells, histamine and bradykinin promote Ca2+ mobilization (Yang et al., 1994; Twort & van Breemen, 1988; 1989), phosphatidylinositol-4,5-bisphosphate hydrolysis (Yang et al., 1994; Daykin et al., 1993) and c-fos expression, but only histamine increases DNA synthesis (Panettieri et al., 1991b). This suggests that while these events appear critical for force development in ASM, additional or more sustained regulatory signals are required to promote cell proliferation (Figure 3).
Figure 3.
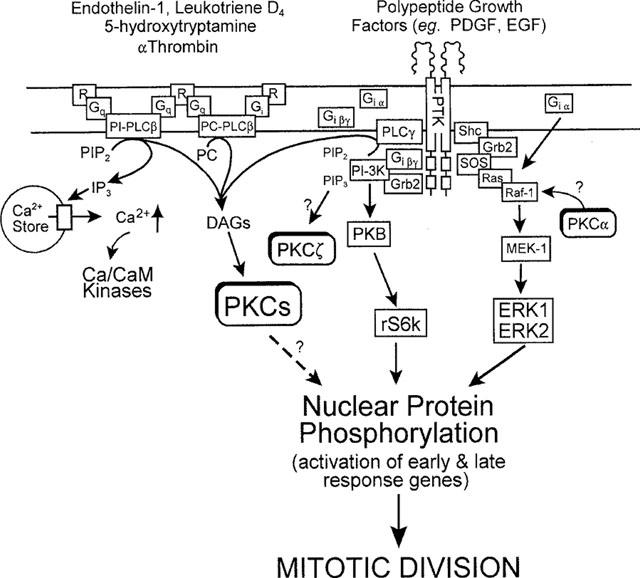
Possible mechanisms that regulate ASM mitogenesis induced by agonists that activate G-protein (G)-coupled receptors (R) and polypeptide growth factor receptor tyrosine kinases (PTK). G protein agonist-induced extracellular signal-regulated kinase (ERK) activation by Gi-coupled, but not PTK-linked receptors, occurs by a pertussis toxin-sensitive, Raf-1-dependent pathway, in which G1α subunits associate with Ras. Alternatively, interaction of polypeptide growth factors with their receptors results in recruitment of the guanine nucleotide releasing binding protein, Grb2 at the membrane. Grb2 localizes the guanine nucleotide exchange factor, Sos, to Ras. Activation of Ras results in recruitment to the membrane of Raf-1 and phosphorylation and activation of MEK-1 (MAP kinase/ERK-kinase activating kinase). MEK-1 directly phosphorylates and activates ERK1/ERK2 which translocate to the cell nucleus. Activation of PKC is implicated in ASM mitogenesis, though the importance of phosphatidylinositol (PI)- or phosphatidylcholine (PC)-dependent-PLC-derived diacylglycerols (DAGs) is disputed. Currently, no clear consensus for the site of integration of PKC activation in activation of ERK in ASM has been established, although in other systems PKCα may phosphorylate and activate Raf-1. The dotted arrow illustrates possible direct effects of PKC on nuclear transcription elements. Recent evidence suggests proliferation induced by certain Gi-coupled receptor agonists (α-thrombin) may be dependent on activation of PI3kinase by Giβγ subunits. Localized generation of PI 3,4,5-trisphosphate (PIP3) by PI-3kinase recruits protein kinase B (PKB) to the plasma membrane where it can be fully activated prior to activation of the S6 ribosomal kinase (p70s6k). PIP3 may also activate PKCζ and this may offer another integration site for PKC in ASM mitogenesis.
In support of a requirement for additional mechanisms for full mitogenesis, Whelchel et al. (1997) showed that activation of ERK-2 was necessary for endothelin-1-stimulated rat tracheal smooth muscle proliferation. In the same study, these authors also demonstrated that activation of ERK-2 was prevented following PKC depletion (by phorbol diester pre-treatment), emphasizing a requirement for one or more of these enzymes. Similar results have also been reported using the PKC inhibitor, Ro31-8220, which partially reduced endothelin-1-stimulated activation of MAP kinase in bovine tracheal smooth muscle (Malarkey et al., 1995). Abe et al. (1994) had previously demonstrated a similar requirement for PKC in ERK-2 activation in ASM following stimulation with H2O2 but this is not a universal finding as other agonists of G protein-coupled receptors, such as thrombin, are able to activate ERK-2 in PKC-depleted cells. However, in other smooth muscles, thrombin is known to up-regulate the expression of both PDGF-A chain ligands and their corresponding α-receptors (Stouffer & Runge, 1998). These receptors have intrinsic tyrosine kinase activity and retain the ability to activate ERK-2 following PKC depletion (Whelchel et al., 1997). Concomitant up-regulation of PDGF ligands and receptors by thrombin may also help explain the potent and efficacious mitogenic activity of this proteinase on ASM.
Further evidence that PKC- and ERK-dependent processes are implicated in agonist-induced ASM mitogenesis is the observation that the MEK-1 inhibitor, PD 098059, prevents proliferation of rat tracheal myocytes elicited by endothelin-1 and PDGF (Whelchel et al., 1997). Thus, it would seem that for a full mitogenic stimulus to be propagated in ASM cells, PKC and ERK signalling cascades act co-operatively and converge at a locus upstream of MEK-1. Tolan et al. (1999) have also reported that PKC plays an intermediary step in the activation of the MEK-1/ERK cascade by PDGF in ASM. One possible mechanism that could explain the integration of these two signalling pathways is the activation of Raf-1 (a serine/threonine kinase that lies upstream of MEK-1) by PKC (Figure 3). Indeed, Kolch et al. (1993) have reported that Raf-1 is a substrate for PKCα and that phosphorylation results in an increase in catalytic activity. Evidence that such a mechanism operates in ASM derives from studies where phorbol diesters (Vichi et al., 1999) as well as agonists that act through G-protein-coupled receptors that utilize PKC such as endothelin-1 (Shapiro et al., 1996) and 5-HT (Hershenson et al., 1995), activate Raf-1. Furthermore, H2O2 is reported to promote ERK activation in ASM by successive activation of PKC, Raf-1 and MEK-1 (Abe et al., 1998). Finally, Lew et al. (1997) have demonstrated that induction of proliferation in bovine tracheal smooth muscle by β-hexosaminidases and activation of the ERK pathway was prevented by the PKC inhibitor calphostin C, an effect which appeared to be mediated at the Ras/Raf-1 locus.
In summary, the available data suggest that activation of PKC is necessary, but not sufficient, for the induction of G protein-coupled receptor-dependent ASM cell proliferation. Additional pathways clearly contribute to the proliferative response. In cultured ASM cells, the critical signalling event appears to be prolonged activation of ERKs. A component of this process is PKC-dependent, as indicated by PKC inhibitor and depletion studies. It is proposed that activation of ERKs by PKC could be mediated by direct phosphorylation of Raf-1, possibly by PKCα. As we learn more, other mechanisms will almost certainly be implicated in the proliferative response of ASM. In bovine trachealis, activation of phosphatidylinositol-3-kinase (PI3kinase) has also been proposed as a key event in mediating PDGF- and α-thrombin-stimulated proliferation (Scott et al., 1996; Walker et al., 1998). Novel (ε and ζ) and atypical (ζ and λ) isoforms of PKC have been shown to be regulated by PI3kinase. Thus, activation of PI3kinase may explain the marked reduction in PDGF-stimulated DNA synthesis reported in human ASM cells transfected with a dominant negative PKCζ (Black et al., 1998; Carlin et al., 1999). Initial data, however, show little involvement of PKC in the response, but a requirement for activation of MAP kinase-dependent mechanisms for a full mitogenesis was identified (Walker et al., 1998). The apparent PKC-independent mitogenic response may reflect the choice of agonist used to induce proliferation, possible secondary mediator interactions (Stouffer & Runge, 1998), or the species investigated. With the recognition of the diversity of agonists which induce proliferation of ASM in vitro and the involvement of multiple intracellular signalling pathways in this process, future studies are needed to characterize the relative importance of each of these intracellular mechanisms in mediating the mitogenic signal. Identification of the critical pathways that modulate ASM growth and division are essential before we can evaluate the role of increased smooth muscle content in the pathogenesis of diseases such as chronic severe asthma or develop novel therapeutic approaches to control these events.
Discussion
Since the discovery of PKC in 1977, much has been learned of the biochemistry and molecular biology of this expanding family of closely related enzymes. Fundamental advances have been made in understanding the regulation of these proteins and how they are positioned in various signalling pathways in a variety of organisms as diverse as budding yeast and mammals. However, the functional role of individual PKC isoenzymes, many of which are co-expressed in the same cell, are poorly studied and present a particular challenge for future research. The availability of small molecule inhibitors that have a degree of selectivity for the three main PKC sub-families have provided clues to their role in processes including smooth muscle contraction and mitogenesis (reviewed here). The use of antisense technology also has shed some light of the functions these enzymes subserve in various cells and tissues. Nevertheless, newer agents with high degrees of selectivity for individual PKC isoforms are urgently required. Until then, we can only speculate on the functions of the most extensive serine/threonine protein kinase family thus far identified.
Abbreviations
- ACh
acetylcholine
- ASM
airways smooth muscle
- [Ca2+]i
intracellular calcium concentration
- DAG
diacylglycerol
- DPB
12-deoxyphorbol 13-isobutyrate
- ERK
extracellular signal-regulated kinase
- H2O2
hydrogen peroxide
- KATP
ATP-sensitive K+ channels
- LC
light chain
- MAP
mitogen-activated protein
- MEK
mitogen-activated protein kinase kinase
- MLCK
myosin light chain kinase
- MLCP
myosin light chain phosphatase
- OAG
1-oleoyl-2-acetyl sn-glycerol
- PDA
phorbol-12, 13-diacetate
- PDBu
phorbol 12,13-dibutyrate
- PDGF
platelet-derived growth factor
- PDK
phosphatidylinositol-trisphosphate-dependent kinase
- PI
phosphatidylinositol
- PKC
protein kinase C
- PKD
protein kinase D
- PKM
protein kinase M
- PLC
phospholipase C
- PLD
phospholipase D
- PMA
phorbol 12-myristate 13-acetate
- PRK
protein kinase C-related kinase
- PS
phosphatidylserine
- RACK
receptor for activated C-kinase
- VDCC
voltage-dependent calcium channel
References
- ABE M.K., CHAO T.S., SOLWAY J., ROSNER M.R., HERSHENSON M.B. Hydrogen peroxide stimulates mitogen-activated protein kinase in bovine tracheal myocytes: Implications for human airway disease. Am. J. Resp. Cell Mol. Biol. 1994;11:577–585. doi: 10.1165/ajrcmb.11.5.7946386. [DOI] [PubMed] [Google Scholar]
- ABE M.K., KARTHA S., KARPOVA A.Y., LI J., LIU P.T., KUO W.L., HERSHENSON M.B. Hydrogen peroxide activates extracellular signal-regulated kinase via protein kinase C, Raf-1, and MEK1. Am. J. Resp. Cell Mol. Biol. 1998;18:562–569. doi: 10.1165/ajrcmb.18.4.2958. [DOI] [PubMed] [Google Scholar]
- ADAM L.P., HATHAWAY D.R. Identification of mitogen-activated protein kinase phosphorylation sequences in mammalian h-Caldesmon. FEBS Lett. 1993;322:56–60. doi: 10.1016/0014-5793(93)81110-l. [DOI] [PubMed] [Google Scholar]
- ADAM L.P., MILIO L., BRENGLE B., HATHAWAY D.R. Myosin light chain and caldesmon phosphorylation in arterial muscle stimulated with endothelin-1. J. Mol. Cell. Cardiol. 1990;22:1017–1023. doi: 10.1016/0022-2828(90)91041-5. [DOI] [PubMed] [Google Scholar]
- AHMED F., FOSTER R.W., SMALL R.C. Some effects of nifedipine in guinea-pig isolated trachealis. Br. J. Pharmacol. 1985;84:861–869. doi: 10.1111/j.1476-5381.1985.tb17380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AHMED S., LEE J., KOZMA R., BEST A., MONFRIES C., LIM L. A novel functional target for tumor-promoting phorbol esters and lysophosphatidic acid. The p21rac-GTPase activating protein n-chimaerin. J. Biol. Chem. 1993;268:10709–10712. [PubMed] [Google Scholar]
- AKIMOTO K., MIZUNO K., OSADA S.-I., HIRAI S.-I., TANUMA S.-I., SUZUKI K., OHNO S. A new member of the third class in the protein kinase C family, PKCλ, expressed predominantly in an undifferentiated mouse embryonal carcinoma cell line and also in many tissues and cells. J. Biol. Chem. 1994;269:12677–12683. [PubMed] [Google Scholar]
- AKOPOV S.E., ZHANG L., PEARCE W.J. Regulation of Ca2+ sensitization by PKC and rho proteins in ovine cerebral arteries: effects of artery size and age. Am. J. Physiol. 1998;275:H930–H939. doi: 10.1152/ajpheart.1998.275.3.H930. [DOI] [PubMed] [Google Scholar]
- AKSOY M.O., MURPHY R.A., KAMM K.E. Role of Ca2+ and myosin light chain phosphorylation in regulation of smooth muscle. Am. J. Physiol. 1982;242:C109–C116. doi: 10.1152/ajpcell.1982.242.1.C109. [DOI] [PubMed] [Google Scholar]
- AKSOY M.O., WILLIAMS D., SHARKEY E.M., HARTSHORNE D.J. A relationship between Ca2+ sensitivity and phosphorylation of gizzard actomyosin. Biochem. Biophys. Res. Comm. 1976;69:35–41. doi: 10.1016/s0006-291x(76)80268-9. [DOI] [PubMed] [Google Scholar]
- ALKON D.L. Memory storage and neural systems. Scientific Am. 1989;261:42–50. doi: 10.1038/scientificamerican0789-42. [DOI] [PubMed] [Google Scholar]
- ALLEN B.G., ANDREA J.E., WALSH M.P. Identification and characterisation of protein kinase Cζ-immunoreactive proteins. J. Biol. Chem. 1994;269:29288–29298. [PubMed] [Google Scholar]
- AMANO M., ITO M., KIMURA K., FUKATA Y., CHIHARA K., NAKANO T., MATSUURA Y., KAIBUCHI K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J. Biol. Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- ASHENDEL C.L. The phorbol ester receptor: A phospholipid-regulated kinase. Biochim. Biophys. Acta. 1985;822:219–242. doi: 10.1016/0304-4157(85)90009-7. [DOI] [PubMed] [Google Scholar]
- ASSENDER J.W., KONTNY E., FREDHOLM B.B. Expression of protein kinase C isoforms in smooth muscle cells in various states of differentiation. FEBS Lett. 1994;342:76–80. doi: 10.1016/0014-5793(94)80588-1. [DOI] [PubMed] [Google Scholar]
- BACHER N., ZISMAN Y., BERENT E, LIVNEH E. Isolation and characterisation of PKC-L, a new member of the protein kinase C-related gene family specifically expressed in lung, skin and heart. Mol. Biol. Cell. 1991;12:1404–1404. doi: 10.1128/mcb.12.3.1404-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAIER G., TELFORD D., GIAMPA L., COGGESHALL K.M., BAIER-BITTERLICH G., ISAKOV N., ALTMAN A. Molecular cloning and characterization of PKCθ, a novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J. Biol. Chem. 1993;268:4997–5004. [PubMed] [Google Scholar]
- BAZZI M.D., NELSESTUAN G.L. Association of protein kinase C with phospholipid vesicles. Biochemistry. 1987;26:115–122. doi: 10.1021/bi00375a017. [DOI] [PubMed] [Google Scholar]
- BAZZI M.D., NELSESTUAN G.L. Autophosphorylation of protein kinase C may require a high order of protein-phospholipid aggregates. J. Biol. Chem. 1992;267:22891–22896. [PubMed] [Google Scholar]
- BEECH D.J. Actions of neurotransmitters and other messengers on Ca2+ channels and K+ channels in smooth muscle cells. Pharmacol. Ther. 1997;73:91–119. doi: 10.1016/s0163-7258(97)87271-3. [DOI] [PubMed] [Google Scholar]
- BELL R.M., BURNS D.J. Lipid activation of protein kinase C. J. Biol. Chem. 1991;266:4461–4464. [PubMed] [Google Scholar]
- BENGUR A.R., ROBINSON E.A., APPELLA E., SELLERS J.R. Sequence of the sites phosphorylated by protein kinase C in the smooth muscle myosin light chain. J. Biol. Chem. 1987;5:7613–7617. [PubMed] [Google Scholar]
- BERK B.C., ARONOV M.S., BROCK T.A., CRAGOE E., GIMBRONE M.A., ALEXANDER R.W. Angiotensin II-stimulated Na+/H+ exchange in cultured vascular smooth muscle cells: Evidence for protein kinase C-dependent and -independent pathways. J. Biol. Chem. 1988;262:5057–5064. [PubMed] [Google Scholar]
- AZZI A., BOSCOBOINIK D., HENSEY C. The protein kinase C family. Eur. J. Biochem. 1992;208:547–557. doi: 10.1111/j.1432-1033.1992.tb17219.x. [DOI] [PubMed] [Google Scholar]
- BERK B.C., VEKSHTEIN V., GORDON H.M., TSUDA T. Angiotensin II-stimulated protein synthesis in cultured vascular smooth muscle cells. Hypertension. 1989;13:305–314. doi: 10.1161/01.hyp.13.4.305. [DOI] [PubMed] [Google Scholar]
- BERRA E., DIAZ-MECO M.T., DOMINGUEZ I., MUNICIO M.M., SANZ L., LOZANO J., CHAPKIN R.S., MOSCAT J. Protein kinase Cζ isoform is critical for mitogenic signal transduction. Cell. 1993;74:555–563. doi: 10.1016/0092-8674(93)80056-k. [DOI] [PubMed] [Google Scholar]
- BLACK J., CARLIN S., JOHNSON P., COOK D., CARPENTER L., BIDEN T., PORONNIK P. A dominant negative mutant PKCζ inhibits growth of human airway smooth muscle (HASM) cells in culture. Am. J. Resp. Crit. Care. 1998;157:A425. [Google Scholar]
- BORNANCIN F., PARKER P.J. Phosphorylation of threonine 638 critically controls the dephosphorylation and inactivation of protein kinase Cα. Current Biol. 1996;6:1114–1123. doi: 10.1016/s0960-9822(02)70678-7. [DOI] [PubMed] [Google Scholar]
- BORNANCIN F., PARKER P.J. Phosphorylation of protein kinase Cα on Serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J. Biol. Chem. 1997;272:3544–3549. doi: 10.1074/jbc.272.6.3544. [DOI] [PubMed] [Google Scholar]
- BREMERICH D.H., KAI T., WARNER D.O., JONES K.A. Effect of phorbol esters on Ca2+ sensitivity and myosin light-chain phosphorylation in airway smooth muscle. Am. J. Physiol. 1998;274:C1253–C1260. doi: 10.1152/ajpcell.1998.274.5.C1253. [DOI] [PubMed] [Google Scholar]
- BREMERICH D.H., WARNER D.O., LORENZ R.R., SHUMWAY R., JONES K.A. Role of protein kinase C in calcium sensitization during muscarinic stimulation in airway smooth muscle. Am. J. Physiol. 1997;273:L775–L781. doi: 10.1152/ajplung.1997.273.4.L775. [DOI] [PubMed] [Google Scholar]
- BRUMFELD V., LESTER D.S. Protein kinase C penetration into lipid bilayers. Archiv. Biochem. Biophys. 1990;277:318–323. doi: 10.1016/0003-9861(90)90586-n. [DOI] [PubMed] [Google Scholar]
- CARLIN S., YANG K.X.F., DONNELLY R., BLACK J.L. Protein kinase C isoforms in human airway smooth muscle cells: activation of PKCζ during proliferation. Am. J. Physiol. 1999;276:L506–L512. doi: 10.1152/ajplung.1999.276.3.L506. [DOI] [PubMed] [Google Scholar]
- CHATTERJEE M., MURPHY R.A. Calcium-dependent maintenance without myosin phosphorylation in skinned smooth muscle. Science. 1983;221:464–466. doi: 10.1126/science.6867722. [DOI] [PubMed] [Google Scholar]
- CHAUHAN V.P.S., CHAUHAN A., DESHMUKH D.S., BROCKERHOFF H. Lipid activators of protein kinase C. Life Sci. 1990;47:981–986. doi: 10.1016/0024-3205(90)90469-8. [DOI] [PubMed] [Google Scholar]
- CHEN S.G., KULJU D., HALT S., MURAKAMI K. Phosphatidylcholine-dependent protein kinase C activation. Biochem. J. 1992;284:221–226. doi: 10.1042/bj2840221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG J.B., TOWNLEY R.G. Pharmacological characterization of effects of nifedipine on isolated guinea-pig and rat tracheal smooth muscle. Arch. Int. Pharmacodyn. Ther. 1983;263:228–244. [PubMed] [Google Scholar]
- CHOPRA L.C., TWORT C.H.C., WARD J.P.T. Differences in sensitivity to the specific protein kinase C inhibitor Ro31-8220 between small and large bronchioles of the rat. Br. J. Pharmacol. 1994;113:1237–1242. doi: 10.1111/j.1476-5381.1994.tb17130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK T., NGAI P.K., SUTHERLAND C., GROSCHEL-STEWART U., WALSH M.P. Vascular smooth muscle caldesmon. J. Biol. Chem. 1986;261:8028–8035. [PubMed] [Google Scholar]
- CLEMONT-CHOMIENNE O., WALSH M.P. Identification of protein kinase C isoenzymes in smooth muscle: partial purification and characterization of chicken gizzard PKC. Biochem. Cell Biol. 1995;74:51–65. doi: 10.1139/o96-006. [DOI] [PubMed] [Google Scholar]
- COBURN R.F. Electromechanical coupling in canine trachealis muscle: acetylcholine contractions. Am. J. Physiol. 1979;236:C177–C184. doi: 10.1152/ajpcell.1979.236.3.C177. [DOI] [PubMed] [Google Scholar]
- COFFER P.J., WOODGETT J.R. Molecular cloning and characterization of a novel putative protein serine kinase related to the cAMP-dependent and protein kinase C families. Eur. J. Biochem. 1991;201:475–481. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- COHEN P., NOVERAL J.P., BHALA A., NUNN S.E., HERRICK D.J., GRUNSTEIN M.M. Leukotriene D4 facilitates airway smooth muscle cell proliferation via modulation of the IGF axis. Am. J. Physiol. 1995;269:L151–L157. doi: 10.1152/ajplung.1995.269.2.L151. [DOI] [PubMed] [Google Scholar]
- COLLINS E.M., WALSH M.P., MORGAN K.G. Contraction of single vascular smooth muscle cells by phenylephrine at constant [Ca2+]i. Am. J. Physiol. 1992;262:H754–H762. doi: 10.1152/ajpheart.1992.262.3.H754. [DOI] [PubMed] [Google Scholar]
- COUSSENS L., PARKER P.J., RHEE L., YANG-FENG T.L., CHEN E., WATERFIELD M.D., FRANCKE U., ULLRICH A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signalling pathways. Science. 1986;233:859–866. doi: 10.1126/science.3755548. [DOI] [PubMed] [Google Scholar]
- COUSSENS L., RHEE L., PARKER P.J., ULLRICH A. Alternative splicing increases the diversity of the human protein kinase C family. DNA. 1987;6:389–394. doi: 10.1089/dna.1987.6.389. [DOI] [PubMed] [Google Scholar]
- CUTLER R.E., JR, MAIZELS E.T., BROOKS E.J., MIZUNO K., OHNO S., HUNZICKER-DUNN M. Regulation of δ protein kinase C during rat ovarian differentiation. Biochim. Biophys. Acta. 1993;1179:260–270. doi: 10.1016/0167-4889(93)90081-y. [DOI] [PubMed] [Google Scholar]
- DANIEL E.E., BOURREAU J.P., ABELA A., JURY J. The internal calcium store in airway muscle: emptying, refilling and chloride. Possible new directions for drug development. Biochem. Pharmacol. 1992;43:29–37. doi: 10.1016/0006-2952(92)90657-5. [DOI] [PubMed] [Google Scholar]
- DAYKIN K., WIDDOP S., HALL I.P. Control of histamine-induced inositol phospholipid hydrolysis in human tracheal smooth muscle cells. Eur. J. Pharmacol. 1993;246:135–140. doi: 10.1016/0922-4106(93)90090-v. [DOI] [PubMed] [Google Scholar]
- DE S., ZELAZNY E.T., SOUHRADA J.F., SOUHRADA M. Role of phospholipase C and tyrosine kinase systems in growth response of human airway smooth muscle. Am. J. Physiol. 1996;270:L795–L802. doi: 10.1152/ajplung.1996.270.5.L795. [DOI] [PubMed] [Google Scholar]
- DEKKER L.V., PARKER P.J. Protein kinase C: A question of specificity. Trends Biochem. Sci. 1994;19:73–77. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- DESSY C., KIM I., SOUGNEZ C.I., LAPORTE R., MORGAN K.G. A role for MAP kinase in differentiated smooth muscle contraction evoked by alpha-adrenoceptor stimulation. Am. J. Physiol. 1998;275:C1081–C1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- DILLON P.F., AKSOY M.O., DRISKA S.P., MURPHY R.A. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science. 1981;211:495–497. doi: 10.1126/science.6893872. [DOI] [PubMed] [Google Scholar]
- DONNELLY R., YANG K., OMARY M.B., BLACK J.L. Expression of multiple isoenzymes of protein kinase C in airway smooth muscle. Am. J. Resp. Cell Mol. Biol. 1995;13:253–256. doi: 10.1165/ajrcmb.13.3.7654381. [DOI] [PubMed] [Google Scholar]
- DUNCAN P.G., DOUGLAS J.S. Sensitivity and responsiveness of tracheal and bronchial tissues from young and old guinea pigs: effect of calcium antagonists. J. Pharmacol. Exp. Ther. 1984;228:612–619. [PubMed] [Google Scholar]
- ETO A., AKITA Y., SAIDO T.C., SUZUKI K., KAWASHIMA S. The role of the calpain-calpastatin system in thyrotropin-releasing hormone-induced selective down-regulation of a protein kinase C isozyme, nPKCε, in rat pituitary GH4C1 cells. J. Biol. Chem. 1995a;270:25115–25120. doi: 10.1074/jbc.270.42.25115. [DOI] [PubMed] [Google Scholar]
- ETO M., OHMORI T., SUZUKI M., FURUYA K., MORITA F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J. Biochem. (Tokyo) 1995b;118:1104–1107. doi: 10.1093/oxfordjournals.jbchem.a124993. [DOI] [PubMed] [Google Scholar]
- ETO M., SENBA S., MORITA F., YAZAWA M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI17) in smooth muscle: its specific localization in smooth muscle. FEBS Lett. 1997;410:356–360. doi: 10.1016/s0014-5793(97)00657-1. [DOI] [PubMed] [Google Scholar]
- FARLEY J.M., MILES P.R. The sources of calcium for acetylcholine-induced contractions of dog tracheal smooth muscle. J. Pharmacol. Exp. Ther. 1978;207:340–346. [PubMed] [Google Scholar]
- FARMER S.G.Bradykinin Airways Smooth Muscle: Peptide Receptors, Ion Channels and Signal Transduction 1995Basel, Switzerland: Birkhauser Verlag; 51–65.eds, Raeburn, D. & Giembycz, M.A. pp [Google Scholar]
- FLINT A.J., PALADINI R.D., KOSHLAND D., JR Autophosphorylation of protein kinase C at three separated regions of its primary sequence. Science. 1990;249:408–411. doi: 10.1126/science.2377895. [DOI] [PubMed] [Google Scholar]
- FREISEWINKEL I., RICHTMACHER D., STABEL S. Downregulation of protein kinase Cγ is independent of a functional kinase domain. FEBS Lett. 1991;280:262–266. doi: 10.1016/0014-5793(91)80307-o. [DOI] [PubMed] [Google Scholar]
- FUJIHARA H., WALKER L.A., GONG M.C., LEMICHEZ E., BOQUET P., SOMLYO A.V., SOMLYO A.P. Inhibition of RhoA translocation and calcium sensitization by in vivo ADP-ribosylation with the chimeric toxin DC3B. Mol. Biol. Cell. 1997;8:2437–2447. doi: 10.1091/mbc.8.12.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUJITA A., TAKEUCHI T., NAKAJIMA H., NISHIO H., HATA F. Involvement of heterotrimeric GTP-binding protein and rho protein, but not protein kinase C, in agonist-induced Ca2+ sensitization of skinned muscle of guinea pig vas deferens. J. Pharmacol. Exp. Ther. 1995;274:555–561. [PubMed] [Google Scholar]
- FUKUMOTO S., NISHIKAWA Y., HOSOI M., KOYAMA H., YAMAKAWA K., OHNO S., MORII H. Protein kinase C-δ inhibits the proliferation of vascular smooth muscle cells by suppressing G1 cyclin expression. J. Biol. Chem. 1997;272:13816–13822. doi: 10.1074/jbc.272.21.13816. [DOI] [PubMed] [Google Scholar]
- GEISTERFER A., OWENS G.K. Arginine vasopressin induced hypertrophy of cultured rat aortic smooth muscle cells. Hypertension. 1989;14:413–420. doi: 10.1161/01.hyp.14.4.413. [DOI] [PubMed] [Google Scholar]
- GEISTERFER A., PEACH M.J., OWENS G.K. Angiotensin II induces hypertrophy, not hyperplasia of cultured rat aortic smooth muscle cells. Circ. Res. 1989;62:749–756. doi: 10.1161/01.res.62.4.749. [DOI] [PubMed] [Google Scholar]
- GERTHOFFER W.T. Calcium dependence of myosin phosphorylation and airway smooth muscle contraction and relaxation. Am. J. Physiol. 1986;250:C597–C604. doi: 10.1152/ajpcell.1986.250.4.C597. [DOI] [PubMed] [Google Scholar]
- GERTHOFFER W.T. Regulation of the contractile element of airway smooth muscle. Am. J. Physiol. 1991;261:L15–L28. doi: 10.1152/ajplung.1991.261.2.L15. [DOI] [PubMed] [Google Scholar]
- GERTHOFFER W.T. Agonist synergism in airway smooth muscle contraction. J. Pharmacol. Exp. Ther. 1996;278:800–807. [PubMed] [Google Scholar]
- GERTHOFFER W.T., MURPHEY K.A., GUNST S.J. Aequorin luminescence, myosin phosphorylation, and active stress in tracheal smooth muscle. Am. J. Physiol. 1989;257:C1062–C1068. doi: 10.1152/ajpcell.1989.257.6.C1062. [DOI] [PubMed] [Google Scholar]
- GERTHOFFER W.T., POHL J. Caldesmon and calponin phosphorylation in regulation of smooth muscle contraction. Can. J. Physiol. Pharmacol. 1994;72:1410–1414. doi: 10.1139/y94-203. [DOI] [PubMed] [Google Scholar]
- GERTHOFFER W.T., YAMBOLIEV I.A., POHL J., HAYNES R., DANG S., MCHUGH J. Activation of MAP kinases in airway smooth muscle. Am. J. Physiol. 1997;272:L244–L252. doi: 10.1152/ajplung.1997.272.2.L244. [DOI] [PubMed] [Google Scholar]
- GIEMBYCZ M.A., RAEBURN D. Current concepts on mechanisms of force generation and maintenance in airways smooth muscle. Pulmon. Pharmacol. 1992;5:101–119. doi: 10.1016/0952-0600(92)90071-n. [DOI] [PubMed] [Google Scholar]
- GLASSBERG M.K., ERGUL A., WANNER A., PUETT D. Endothelin-1 promotes mitogenesis in airway smooth muscle cells. Am. J. Resp. Cell Mol. Biol. 1994;10:316–321. doi: 10.1165/ajrcmb.10.3.7509612. [DOI] [PubMed] [Google Scholar]
- GONG M.C., FUJIHARA H., SOMLYO A.V., SOMLYO A.P. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J. Biol. Chem. 1997;272:10704–10709. doi: 10.1074/jbc.272.16.10704. [DOI] [PubMed] [Google Scholar]
- GONG M.C., IIZUKA K., NIXON G., BROWNE J.P., HALL A., ECCLESTON J.F., SUGAI M., KOBAYASHI S., SOMLYO A.V., SOMLYO A.P. Role of guanine nucleotide-binding proteins – ras-family or trimeric proteins or both – in Ca2+ sensitization of smooth muscle. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1340–1345. doi: 10.1073/pnas.93.3.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOODE N.T., NASSER HAJIBAGHERI M.A., PARKER P.J. Protein kinase C (PKC)-induced PKC down-regulation. Association with up-regulation of vesicle traffic. J. Biol. Chem. 1995;270:2669–2673. doi: 10.1074/jbc.270.6.2669. [DOI] [PubMed] [Google Scholar]
- GOODE N.T., NASSER HAJIBAGHERI M.A., WARREN G., PARKER P.J. Expression of mammalian protein kinase C in Schizosaccharomyces pombe: isotype-specific induction of growth arrest, vesicle formation, and endocytosis. Mol. Biol. Cell. 1994;5:907–920. doi: 10.1091/mbc.5.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPALAKRISHNA R., ANDERSON W.B. Susceptibility of protein kinase C to oxidative inactivation: loss of both phosphotransferase activity and phorbol diester binding. FEBS Lett. 1987;225:233–237. doi: 10.1016/0014-5793(87)81164-x. [DOI] [PubMed] [Google Scholar]
- GOPALAKRISHNA R., ANDERSON W.B. Ca2+- and phospholipid independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc. Natl. Acad. Sci. U.S.A. 1989;86:6758–6762. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPALAKRISHNA R., ANDERSON W.B. Reversible oxidative activation and inactivation of protein kinase C by the mitogen/tumour promoter periodate. Archiv. Biochem. Biophys. 1991;285:382–387. doi: 10.1016/0003-9861(91)90377-u. [DOI] [PubMed] [Google Scholar]
- GOPALAKRISHNA R., CHEN Z.-H., GUNDIMEDA U. Nitric oxide and nitric oxide-generating agents induce a reversible inactivation of protein kinase C activity and phorbol ester binding. J. Biol. Chem. 1993;268:27180–27185. [PubMed] [Google Scholar]
- GOPALAKRISHNA R., CHEN Z.H., GUNDIMEDU U. Modifications of cysteine-rich regions in PKC induced by oxidant tumour promoters and enzyme-specific inhibitors. Methods Enzymol. 1995;252:132–146. doi: 10.1016/0076-6879(95)52016-3. [DOI] [PubMed] [Google Scholar]
- GRANGER B.L., LAZARIDES E. Synemin: a new high molecular weight protein associated with desmin and vimentin filaments in muscle. Cell. 1980;22:727–738. doi: 10.1016/0092-8674(80)90549-8. [DOI] [PubMed] [Google Scholar]
- GRIENDLING K.K., BERK B.C., GANZ P., GIMBRONE M.A., ALEXANDER R.W. Angiotensin II stimulation of vascular smooth muscle phosphoinositide metabolism. Hypertension. 1987;9:181–185. doi: 10.1161/01.hyp.9.6_pt_2.iii181. [DOI] [PubMed] [Google Scholar]
- GRUNSTEIN M.M., ROSENBERG S.M., SCHRAMM C.M., PAWLOWSKI N.A. Mechanisms of action of endothelin 1 in maturing rabbit airway smooth muscle. Am. J. Physiol. 1991;260:L434–L443. doi: 10.1152/ajplung.1991.260.6.L434. [DOI] [PubMed] [Google Scholar]
- GUNST S.J., AL-HASSANI M.H., ADAM L.P. Regulation of isotonic shortening velocity by second messengers in tracheal smooth muscle. Am. J. Physiol. 1994;266:C684–C691. doi: 10.1152/ajpcell.1994.266.3.C684. [DOI] [PubMed] [Google Scholar]
- GUNST S.J., PISONI J.M. Effects of extracellular calcium on canine tracheal smooth muscle. J. Appl. Physiol. 1986;61:706–711. doi: 10.1152/jappl.1986.61.2.706. [DOI] [PubMed] [Google Scholar]
- HALAYKO A.J., SALARI H., MA X., STEPHENS N.L. Markers of airway smooth muscle cell phenotype. Am. J. Physiol. 1996;270:L1040–L1051. doi: 10.1152/ajplung.1996.270.6.L1040. [DOI] [PubMed] [Google Scholar]
- HALLER H., LINDSCHAU C., QUASS P., DISTLER A., LUFT F.C. Differentiation of vascular smooth muscle cells and regulation of protein kinase C-α. Circ. Res. 1995;76:21–29. doi: 10.1161/01.res.76.1.21. [DOI] [PubMed] [Google Scholar]
- HALLER H., SMALLWOOD J.I., RASMUSSEN H. Protein kinase C translocation in intact vascular smooth muscle strips. Biochem. J. 1990;270:375–381. doi: 10.1042/bj2700375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANKS S.K., QUINN A.M., HUNTER T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science. 1998;241:45–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- HANSSON A., SERHAN C.N., HAEGGSTROM J., INGELMAN-SUNDBERG M., SAMUELSSON B. Activation of protein kinase C by lipoxin A and other eicosanoids. Intracellular action of oxygenation products of arachidonic acid. Biochim. Biophys. Acta. 1986;134:1215–1222. doi: 10.1016/0006-291x(86)90380-3. [DOI] [PubMed] [Google Scholar]
- HEMRIC M.E., CHALOVICH J.M. Effect of caldesmon on the ATPase activity and the binding of smooth and skeletal myosin subfragments to actin. J. Biol. Chem. 1988;263:1878–1885. [PubMed] [Google Scholar]
- HERSHENSON M.B., CHAO T.S., ABE M.K., GOMES I., KELLEHER M.D., SOLWAY J., ROSNER M.R. Histamine antagonises serotonin and growth factor-induced mitogen-activated protein kinase activation in bovine tracheal smooth muscle cells. J. Biol. Chem. 1995;270:19908–19913. doi: 10.1074/jbc.270.34.19908. [DOI] [PubMed] [Google Scholar]
- HIGASHIDA H. ADP-ribosyl-cyclase coupled with receptors via G proteins. FEBS Lett. 1997;418:355–356. doi: 10.1016/s0014-5793(97)01410-5. [DOI] [PubMed] [Google Scholar]
- HIRATA K., KIKUCHI A., SASAKI T., KURODA S., KAIBUCHI K., MATSUURA Y., SEKI H., SAIDA K., TAKAI Y. Involvement of rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. J. Biol. Chem. 1992;267:8719–8722. [PubMed] [Google Scholar]
- HIRST S.J., BARNES P.J., TWORT C.H.C. Differential proliferation of rabbit tracheal smooth muscle by PDGF isoforms: Inhibition by selective inhibitors of protein kinase and protein tyrosine kinase. Br. J. Pharmacol. 1994;112:81P. [Google Scholar]
- HIRST S.J., BARNES P.J., TWORT C.H.C. Protein kinase inhibitors (PKC and PTK) inhibit PDGF isoform-stimulated proliferation of human and rabbit airway smooth muscle. Am. J. Resp. Crit. Care Med. 1995b;151:A46. [Google Scholar]
- HIRST S.J., WEBB B.L.J., GIEMBYCZ M.A., BARNES P.J., TWORT C.H.C. Inhibition of fetal calf serum-stimulated proliferation of rabbit cultured tracheal smooth muscle cells by selective inhibitors of protein kinase C and protein tyrosine kinase. Am. J. Resp. Cell. Mol. Biol. 1995a;12:149–161. doi: 10.1165/ajrcmb.12.2.7865214. [DOI] [PubMed] [Google Scholar]
- HOROWITZ A., CLEMENT-CHOMIENNE O., WALSH M.P., MORGAN K.G. Epsilon-isoenzyme of protein kinase C induces a Ca2+-independent contraction in vascular smooth muscle. Am. J. Physiol. 1996a;271:C589–C594. doi: 10.1152/ajpcell.1996.271.2.C589. [DOI] [PubMed] [Google Scholar]
- HOROWITZ A., CLEMENT-CHOMIENNE O., WALSH M.P., TAO T., KATSUYAMA H., MORGAN K.G. Effects of calponin on force generation by single smooth muscle cells. Am. J. Physiol. 1996b;270:H1858–H1863. doi: 10.1152/ajpheart.1996.270.5.H1858. [DOI] [PubMed] [Google Scholar]
- HUANG K.-P. The mechanism of protein kinase C activation. Trends Neurosci. 1989;12:425–432. doi: 10.1016/0166-2236(89)90091-x. [DOI] [PubMed] [Google Scholar]
- HUG H., SARRE T.H. Protein kinase C isoenzymes: divergence in signal transduction. Biochem. J. 1993;291:329–343. doi: 10.1042/bj2910329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUWILER A., FABBRO D., PFEILSCHIFTER J. Comparison of different tumour promoters and bryostatin 1 on protein kinase C activation and down-regulation in rat renal mesangial cells. Biochem. Pharmacol. 1994;48:689–700. doi: 10.1016/0006-2952(94)90046-9. [DOI] [PubMed] [Google Scholar]
- HUXLEY A.F., NIEDERGERKE R. Structural changes in muscle during shortening. Nature. 1954;173:971–973. doi: 10.1038/173971a0. [DOI] [PubMed] [Google Scholar]
- HUXLEY H.E., HANSON J. Changes in the cross striations of muscle during contraction and stretch and their structural interpretation. Nature. 1954;173:973–976. doi: 10.1038/173973a0. [DOI] [PubMed] [Google Scholar]
- IIZUKA K., DOBASHI K., YOSHII A., HORIE T., SUZUKI H., NAKAZAWA T., MORI M. Receptor-dependent G protein-mediated Ca2+ sensitization in canine airway smooth muscle. Cell Calcium. 1997;22:21–30. doi: 10.1016/s0143-4160(97)90086-5. [DOI] [PubMed] [Google Scholar]
- IIZUKA K., YOSHII A., SAMIZO K., TSUKAGOSHI H., ISHIZUKA T., DOBASHI K., NAKAZAWA T., MORI M. A major role for the Rho-associated coiled coil forming protein kinase in G-protein-mediated Ca2+ sensitization through inhibition of myosin phosphatase in rabbit trachea. Br. J. Pharmacol. 1999;128:925–933. doi: 10.1038/sj.bjp.0702864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IKEBE M., HARTSHORNE D.J., ELZINGA M. Phosphorylation of the 20,000-dalton light chain of smooth muscle myosin by the calcium-activated, phospholipid-dependent protein kinase. Phosphorylation sites and effects of phosphorylation. J. Biol. Chem. 1987;15:9569–9573. [PubMed] [Google Scholar]
- IKEBE M., REARDON S. Binding of caldesmon to smooth muscle myosin. J. Biol. Chem. 1988;263:3055–3058. [PubMed] [Google Scholar]
- INOUE M., KISHIMOTO A., TAKAI Y., NISHIZUKA Y. Studies on cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. I. Proenzyme and its activation by calcium-dependent protease from rat brain. J. Biol. Chem. 1977;252:7610–7616. [PubMed] [Google Scholar]
- ITOH T., SUZUKI A., WATANABE Y. Effect of a peptide inhibitor of protein kinase C on G-protein-mediated increase in myofilament Ca2+-sensitivity in rabbit arterial skinned muscle. Br. J. Pharmacol. 1994;111:311–317. doi: 10.1111/j.1476-5381.1994.tb14061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHANNES F.J., PRESTLE J., EIS S., OBERHAGEMANN P., PFIZENMAIER K. PKCμ is a novel, atypical member of the protein kinase C family. J. Biol. Chem. 1994;269:6140–6148. [PubMed] [Google Scholar]
- JONES P.F., JAKUBOWICZ T., PITOSSI F.J., MAURER F., HEMMINGS B.A. Molecular cloning and identification of a serine threonine protein kinase of the second messenger subfamily. Proc. Natl. Acad. Sci. U.S.A. 1991;88:4171–4175. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUNCO M., WEBSTER C., CRAWFORD C., BOSCA L., PARKER P.J. Protein kinase C V3 domain mutants with differential sensitivities to m-calpain are not resistant to phorbol-ester-induced down-regulation. Eur. J. Biochem. 1994;223:259–263. doi: 10.1111/j.1432-1033.1994.tb18990.x. [DOI] [PubMed] [Google Scholar]
- KAMISHIMA T., NELSON M.T., PATLAK J.B. Carbachol modulates voltage sensitivity of calcium channels in bronchial smooth muscle of rats. Am. J. Physiol. 1992;263:C69–C77. doi: 10.1152/ajpcell.1992.263.1.C69. [DOI] [PubMed] [Google Scholar]
- KAMM K.E., STULL J.T. Regulation of smooth muscle contractile elements by second messengers. Ann. Rev. Physiol. 1989;51:299–313. doi: 10.1146/annurev.ph.51.030189.001503. [DOI] [PubMed] [Google Scholar]
- KARIYA K., FUKUMOTO Y., TSUDA T., YAMAMOTO T., KAWAHARA Y., FUKUZAKI H., TAKAI Y. Antiproliferative action of protein kinase C in cultured rabbit aortic smooth muscle cells. Exp. Cell Res. 1987;173:504–514. doi: 10.1016/0014-4827(87)90290-4. [DOI] [PubMed] [Google Scholar]
- KARIYA K.-I., KAWAHARA Y., FUKUZAKI H., HAGIWARA M., HIDAKA H., FUKUMOTO Y., TAKAI Y. Two types of protein kinase C with different functions in rabbit aortic smooth muscle cells. Biochem. Biophys. Res. Comm. 1989;161:1020–1027. doi: 10.1016/0006-291x(89)91345-4. [DOI] [PubMed] [Google Scholar]
- KATOCH S.S. Endothelin-1 induced sustained contractions of tracheal smooth muscle involves an activation of protein kinase C. J. Biosci. 1997;22:299–314. [Google Scholar]
- KAZANIETZ M.G., LEWIN N.E., BRUNS J.D., BLUMBERG P.M. Characterization of the cysteine-rich region of the Caenorhabditis elegans protein Unc-13 as a high affinity phorbol ester receptor. Analysis of ligand-binding interactions, lipid cofactor requirements, and inhibitor sensitivity. J. Biol. Chem. 1995;270:10777–10783. doi: 10.1074/jbc.270.18.10777. [DOI] [PubMed] [Google Scholar]
- KHALIL R.A., LAJOIE C., MORGAN K.G. In situ determination of [Ca2+]i threshold for translocation of the alpha-protein kinase C isoform. Am. J. Physiol. 1994;266:C1544–C1551. doi: 10.1152/ajpcell.1994.266.6.C1544. [DOI] [PubMed] [Google Scholar]
- KHALIL R.A., LAJOIE C., RESNICK M.S., MORGAN K.G. Ca2+ independent isoforms of protein kinase C differentially translocate in smooth muscle. Am. J. Physiol. 1992;263:C714–C719. doi: 10.1152/ajpcell.1992.263.3.C714. [DOI] [PubMed] [Google Scholar]
- KHALIL R.A., MORGAN K.G. Phenylephrine-induced translocation of protein kinase C and shortening of two types of vascular cells of the ferret. J. Physiol. 1992;455:585–599. doi: 10.1113/jphysiol.1992.sp019317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KISHIMOTO A., KAJIKAWA N., SHIOTA M., NISHIZUKA Y. Proteolytic activation of calcium-activated, phospholipid-dependent protein kinase by calcium-dependent neutral protease. J. Biol. Chem. 1983;258:1156–1164. [PubMed] [Google Scholar]
- KISHIMOTO A., MIKAWA K., HASHIMOTO K., YASUDA I., TANAKA S., TOMINAGA M., KURODA T., NISHIZUKA Y. Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain) J. Biol. Chem. 1989;264:4088–4092. [PubMed] [Google Scholar]
- KITAGAWA M., MUKAI H., SHIBATA H., ONO Y. Purification and characterization of a fatty-acid-activated protein kinase (PKN) from rat testis. Biochem. J. 1995;310:657–664. doi: 10.1042/bj3100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITAMURA S., ANDO S., SHIBATA M., TANABE K., SATO C., INAGAKI M. Protein kinase C phosphorylation of desmin at four serine residues within the non-alpha-helical head domain. J. Biol. Chem. 1989;264:5674–5678. [PubMed] [Google Scholar]
- KITAZAWA T., GAYLINN B.D., DENNEY G.H., SOMLYO A.P. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. J. Biol. Chem. 1991a;266:1708–1715. [PubMed] [Google Scholar]
- KITAZAWA T., KOBAYASHI S., HORIUTI K., SOMLYO A.V., SOMLYO A.P. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ J. Biol. Chem. 1989;264:5339–5342. [PubMed] [Google Scholar]
- KITAZAWA T., MASUO M., SOMLYO A.P. G protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proc. Natl. Acad. Sci. U.S.A. 1991b;88:9307–9310. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITAZAWA T., TAKIZAWA N., IKEBE M. Reconstitution of protein kinase C-induced contractile Ca2+ sensitization in Triton X-100-demembranated rabbit arterial smooth muscle. J. Physiol. (Lond). 1999;520:139–152. doi: 10.1111/j.1469-7793.1999.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOX A.J., BALDWIN D.R., CRAGOE E.J., JR, AJAO P. The effect of sodium transport and calcium channel inhibitors on phorbol ester-induced contraction of bovine airway smooth muscle. Pulmon. Pharmacol. 1993;6:241–246. doi: 10.1006/pulp.1993.1032. [DOI] [PubMed] [Google Scholar]
- KOLCH W., HEIDECKER G., KOCHS G., HUMMEL R., VAHIDI H., MISCHAK H., FINKENZELLER G., MARME D., RAPP U. Protein kinase C activates Raf-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- KOTLIKOFF M.I. Calcium currents in isolated canine airway smooth muscle cells. Am. J. Physiol. 1988;254:C793–C801. doi: 10.1152/ajpcell.1988.254.6.C793. [DOI] [PubMed] [Google Scholar]
- LANGLANDS J.M., DIAMOND J. Translocation of protein kinase C in bovine tracheal smooth muscle strips: the effects of methacholine and isoprenaline. Eur. J. Pharmacol. 1992;227:131–138. doi: 10.1016/0922-4106(92)90120-k. [DOI] [PubMed] [Google Scholar]
- LANGLANDS J.M., DIAMOND J. The effect of Ca2+ on the translocation of protein kinase C in bovine tracheal smooth muscle. Eur. J. Pharmacol. 1994;15:229–236. doi: 10.1016/0922-4106(94)90131-7. [DOI] [PubMed] [Google Scholar]
- LAPETINA E.G., REEP B., GANONG B.R., BELL R.M. Exogenous sn-1,2-diacylglycerols containing saturated fatty acids function as bioregulators of protein kinase C in human platelets. J. Biol. Chem. 1985;260:1358–1361. [PubMed] [Google Scholar]
- LEE H.W., SMITH L., PETTIT G.R., VINITSKY A., SMITH J.B. Ubiquitination of protein kinase C-a and degradation by the proteasome. J. Biol. Chem. 1996;271:20973–20976. [PubMed] [Google Scholar]
- LEE H.-W., SMITH L., PETTIT G.R., SMITH J.B. Bryostatin 1 and phorbol ester down-modulate protein kinase C-a and -e via the ubiquitin/proteasome pathway in human fibroblasts. Mol. Pharmacol. 1997;51:439–447. [PubMed] [Google Scholar]
- LEE Y.H., KIM I., LAPORTE R., WALSH M.P., MORGAN K.G. Isozyme-specific inhibitors of protein kinase C translocation: effects on contractility of single permeabilized vascular muscle cells of the ferret. J. Physiol. (Lond). 1999;517:709–720. doi: 10.1111/j.1469-7793.1999.0709s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LESTER D.S., DOLL L., BRUMFELD V., MILLER I.R. Lipid dependence of surface conformations of protein kinase C. Biochem. Biophys. Acta. 1990;1039:33–41. doi: 10.1016/0167-4838(90)90223-3. [DOI] [PubMed] [Google Scholar]
- LESZCZYNSKI D., JOENVAARA S., FOEGH M.L. Protein kinase Cα regulates proliferation but not apoptosis in rat coronary vascular smooth muscle cells. Life Sci. 1996;58:599–606. doi: 10.1016/0024-3205(95)02329-1. [DOI] [PubMed] [Google Scholar]
- LEW D.B., BROWN E.R., DEMPSEY K., WRIGHT H.M., MALIK K.U. Contribution of PKC to β-hexosaminidase-induced airway smooth muscle proliferation. Am. J. Physiol. 1997;272:L639–L643. doi: 10.1152/ajplung.1997.272.4.L639. [DOI] [PubMed] [Google Scholar]
- LEW B.D., DEMPSEY B.K., ZHAO Y., MUTHALIF M., FATIMA S., MALIK K. β-Hexosaminidase-induced activation of p44/p42 mitogen-activated protein kinase is dependent on p21Ras and protein kinase C and mediates bovine airway smooth muscle proliferation. Am. J. Respir. Cell Mol. Biol. 1999;21:111–118. doi: 10.1165/ajrcmb.21.1.3542. [DOI] [PubMed] [Google Scholar]
- LI L., ETO M., LEE M.R., MORITA F., YAZAWA M., KITAZAWA T. Possible involvement of the novel CPI-17 protein in protein kinase C signal transduction of rabbit arterial smooth muscle. J. Physiol. (Lond). 1998;508:871–881. doi: 10.1111/j.1469-7793.1998.871bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI W., ZHANG J., BOTTARO D.P., LI W., PIERCE J.H. Identification of serine 643 as an important autophosphorylation site for its enzymatic activity. J. Biol. Chem. 1997;272:24550–24555. doi: 10.1074/jbc.272.39.24550. [DOI] [PubMed] [Google Scholar]
- MALARKEY K., CHILVERS E., LAWSON M.F., PLEVIN R. Stimulation by endothelin-1 of mitogen-activated protein kinases and DNA synthesis in bovine tracheal smooth muscle cells. Br. J. Pharmacol. 1995;116:2267–2273. doi: 10.1111/j.1476-5381.1995.tb15063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAMOON A.M., SMITH J., BAKER R.C., FARLEY J.M. Activation of muscarinic receptors in porcine airway smooth muscle elicits a transient increase in phospholipase D activity. J. Biomed. Sci. 1999;6:97–105. doi: 10.1007/BF02256440. [DOI] [PubMed] [Google Scholar]
- MARRILLEY D., MOSIENIAK G., BOSCOBINIK D., AZZI A. Correlation between human vascular smooth muscle cell proliferation and protein kinase Cα expression: effect of d-α-tocopherol. Biochem. Mol. Biol. Int. 1996;40:699–707. doi: 10.1080/15216549600201303. [DOI] [PubMed] [Google Scholar]
- MASUO M., REARDON S., IKEBE M., KITAZAWA T. A novel mechanism for the Ca2+-sensitizing effect of protein kinase C on vascular smooth muscle: inhibition of myosin light chain phosphatase. J. Gen. Physiol. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELLONI E., PONTREMOLI S., MICHETTI M., SACCO O., SPARATORE B., HORECKER B.L. The involvement of calpain in the activation of protein kinase C in neutrophils stimulated by phorbol myristic acid. J. Biol. Chem. 1986;261:4101–4105. [PubMed] [Google Scholar]
- MELLOR H., PARKER P.J. The extended protein kinase C superfamily. Biochem. J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENICE C.B., HULVERSHORN J., ADAM L.P., WANG C.A., MORGAN K.G. Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. J. Biol. Chem. 1997;272:25157–25161. doi: 10.1074/jbc.272.40.25157. [DOI] [PubMed] [Google Scholar]
- MENKES H., BARABAN J.M., SNYDER S.H. Protein kinase C regulates smooth muscle tension in guinea-pig trachea and ileum. Eur. J. Pharmacol. 1986;122:19–27. doi: 10.1016/0014-2999(86)90153-6. [DOI] [PubMed] [Google Scholar]
- MEYER ZU HERINGDORF D., LASS H., ALEMANY R., LASER K.T., NEUMANN E., ZHANG C., SCHMIDT M., RAUEN U., JAKOBS K.H., VAN KOPPEN C.J. Sphingosine kinase-mediated Ca2+ signalling by G-protein-coupled receptors. EMBO J. 1998;17:2830–2837. doi: 10.1093/emboj/17.10.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MICHETTI M., SALAMINO F., MELLONI E., PONTREMOLI S. Reversible inactivation of calpain isoforms by nitric oxide. Biochem. Biophys. Res. Comm. 1995;207:1009–1014. doi: 10.1006/bbrc.1995.1285. [DOI] [PubMed] [Google Scholar]
- MIETUSSNYDER M., FRIERA A., GLASS C.K., PITAS R.E. Regulation of scavenger receptor expression in smooth muscle cells by protein kinase C: a role for oxidative stress. Arterioscler. Thromb. Vasc. Biol. 1997;17:969–978. doi: 10.1161/01.atv.17.5.969. [DOI] [PubMed] [Google Scholar]
- MINO T., YUASA U., NAKA M., TANAKA T. Phosphorylation of calponin mediated by protein kinase C in association with contraction in porcine coronary artery. Biochem. Biophys. Res. Commun. 1995;208:397–404. doi: 10.1006/bbrc.1995.1351. [DOI] [PubMed] [Google Scholar]
- MOCHLY-ROSEN D., HENRICH C.J., CHEEVER L., KHANER H., SIMPSON P.C. A protein kinase C isozyme is translocated to cytoskeletal elements on activation. Cell Regulation. 1990;1:693–706. doi: 10.1091/mbc.1.9.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOCHLY-ROSEN D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- MORRISON K.J., VANHOUTTE P.M. Inhibition of airway smooth muscle tone by a phorbol ester in the guinea pig trachea: role of epithelium and receptor reserve of the contractile agent. J. Pharmacol. Exp. Ther. 1991;259:198–204. [PubMed] [Google Scholar]
- MOSIER M., MCLAUGHLIN S. Peptides that mimic the pseudosubstrate binding site of protein kinase C bind to acidic lipids in membranes. Biophys. J. 1991;60:149–159. doi: 10.1016/S0006-3495(91)82038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY R.A., REMBOLD C.M., HAI C.M. Contraction in smooth muscle – what is latch. New York: Wiley Liss Inc; Frontiers in Smooth Muscle Research. 1990;327:39–50. [PubMed] [Google Scholar]
- MURRAY N.R., BAUMGARDNER G.P., BURNS D.J., FIELDS A.P. Protein kinase C isotypes in human erythroleukaemia (K562) cell proliferation and differentiation. J. Biol. Chem. 1993;268:15847–15853. [PubMed] [Google Scholar]
- NEWTON A.C. Protein kinase C: structure, function, and regulation. J. Biol. Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- NEWTON A.C., KOSHLAND D.E. Phosphatidylserine affects specificity of protein kinase C substrate phosphorylation and autophosphorylation. Biochemistry. 1990;29:6656–6661. doi: 10.1021/bi00480a015. [DOI] [PubMed] [Google Scholar]
- NGAI P.K., WALSH M.P. Inhibition of smooth muscle actin-activated myosin Mg2+ ATPase activity by caldesmon. J. Biol. Chem. 1984;259:13656–13659. [PubMed] [Google Scholar]
- NISHIZUKA Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- NISHIZUKA Y. Intracellular signalling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- NISHIZUKA Y. Protein kinase C and lipid signalling for sustained cellular responses. FASEB. 1995;9:484–495. [PubMed] [Google Scholar]
- NOVERAL J.P., GRUNSTEIN M.M. Role and mechanism of thromboxane-induced proliferation of cultured airway smooth muscle cells. Am. J. Physiol. 1992;263:L555–L561. doi: 10.1152/ajplung.1992.263.5.L555. [DOI] [PubMed] [Google Scholar]
- NOVERAL J.P., GRUNSTEIN M.M. Adrenergic receptor-mediated regulation of cultured airway smooth muscle cell proliferation. Am. J. Physiol. 1994;267:L291–L299. doi: 10.1152/ajplung.1994.267.3.L291. [DOI] [PubMed] [Google Scholar]
- NOVERAL J.P., ROSENBERG S.M., ANBAR R.A., PAWLOWSKI N.A., GRUNSTEIN M.M. Role of endothelin-1 in regulating proliferation of cultured rabbit airway smooth muscle cells. Am. J. Physiol. 1992;263:L317–L324. doi: 10.1152/ajplung.1992.263.3.L317. [DOI] [PubMed] [Google Scholar]
- NUTTLE L.C., FARLEY J.M. Muscarinic receptors inhibit ATP-sensitive K+ channels in swine tracheal smooth muscle. Am. J. Physiol. 1997;273:L478–L484. doi: 10.1152/ajplung.1997.273.2.L478. [DOI] [PubMed] [Google Scholar]
- OISHI K., MITA M., ONO T., HASHIMOTO T., UCHIDA M.K. Protein kinase C-independent sensitization of contractile proteins to Ca2+ in α-toxin-permeabilized smooth muscle cells from the guinea-pig stomach. Br. J. Pharmacol. 1992;107:908–909. doi: 10.1111/j.1476-5381.1992.tb13383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ONO Y., FUJII T., OGITA K., KIKKAWA U., IGARASHI K., NISHIZUKA Y. The structure, expression and properties of additional members of the protein kinase C family. J. Biol. Chem. 1988;263:6927–6932. [PubMed] [Google Scholar]
- OTTE A.P., KRAMER I.M., DURSTON A.J. Protein kinase C and the regulation of the local competence of xenopus ectoderm. Science. 1991;251:570–573. doi: 10.1126/science.1990433. [DOI] [PubMed] [Google Scholar]
- OTTO B., STEUSLOFF A., JUST I., AKTORIES K., PFITZER G. Role of Rho proteins in carbachol-induced contractions in intact and permeabilized guinea-pig intestinal smooth muscle. J. Physiol. (Lond). 1996;496:317–329. doi: 10.1113/jphysiol.1996.sp021687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALMER R.H., RIDDEN J., PARKER P.J. Identification of multiple, novel, protein kinase C-related gene products. FEBS Lett. 1994;356:5–8. doi: 10.1016/0014-5793(94)01202-4. [DOI] [PubMed] [Google Scholar]
- PALUMBO E.J., SWEATT J.D., CHEN S.-J., KLANN E. Oxidation-induced persistent activation of protein kinase C in hippocampal homogenates. Biochem. Biophys. Res. Comm. 1992;187:1439–1445. doi: 10.1016/0006-291x(92)90463-u. [DOI] [PubMed] [Google Scholar]
- PANETTIERI R.A., GOLDIE R.G., RIGBY P.J., ESZTERHAS A.J., HAY D.W.P. Endothelin-1 induced potentiation of human airway smooth muscle proliferation: an ETA receptor-mediated phenomenon. Br. J. Pharmacol. 1996;118:191–197. doi: 10.1111/j.1476-5381.1996.tb15385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANETTIERI R.A., HALL I.P., MAKI C.S., MURRAY R.K. α-Thrombin increases cytosolic calcium and induces human airway smooth muscle cell proliferation. Am. J. Resp. Crit. Care Med. 1995;151:205–216. doi: 10.1165/ajrcmb.13.2.7626288. [DOI] [PubMed] [Google Scholar]
- PANETTIERI R.A., RUBINSTEIN N.A., FEUERSTEIN B., KOTLIKOFF M.I. Beta-adrenergic inhibition of airway smooth muscle proliferation. Am. Rev. Resp. Dis. 1991a;43:A608. [Google Scholar]
- PANETTIERI R.A., RUBINSTEIN N.A., KELLY A.M., KOTLIKOFF M.I. The specificity of c-fos expression in the induction of airway smooth muscle proliferation by contractile agonists. Am. Rev. Resp. Dis. 1991b;143:A934. [Google Scholar]
- PANETTIERI R.A., YADVISH P.A., KELLY A.M., RUBINSTEIN N.A., KOTLIKOFF M.I. Histamine stimulates proliferation of airway smooth muscle and induces c-fos expression. Am. J. Physiol. 1990;259:L365–L371. doi: 10.1152/ajplung.1990.259.6.L365. [DOI] [PubMed] [Google Scholar]
- PARK S., RASMUSSEN H. Activation of tracheal smooth muscle contraction: synergism between Ca2+ and activators of protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 1985;82:8835–8839. doi: 10.1073/pnas.82.24.8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARK S., RASMUSSEN H. Carbachol-induced protein phosphorylation changes in bovine tracheal smooth muscle. J. Biol. Chem. 1986;261:15734–15739. [PubMed] [Google Scholar]
- PARKER P.J.Protein kinase C: a structurally related family of enzymes Protein kinase C: current concepts and future perspectives 1992Ellis Horwood; 3–24.eds, Epand RM, Lester DS. pp [Google Scholar]
- PARKER C.A., TAKAHASHI K., TANG J.X., TAO T., MORGAN K.G. Cytoskeletal targeting of calponin in differentiated, contractile smooth muscle cells of the ferret. J. Physiol. (Lond). 1998;508:187–198. doi: 10.1111/j.1469-7793.1998.187br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKER C.A., TAKAHASHI K., TAO T., MORGAN K.G. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. Am. J. Physiol. 1994;267:C1262–C1270. doi: 10.1152/ajpcell.1994.267.5.C1262. [DOI] [PubMed] [Google Scholar]
- PEIPER U., KNIPP S.C., THIES B., HENKE R. Activation of protein kinase C accelerates contraction kinetics of airway smooth muscle. Pflugers. Arch. 1996;432:R47–R52. [PubMed] [Google Scholar]
- POHL J., WINDER S.J., ALLEN B.G., WALSH M.P., SELLERS J.R., GERTHOFFER W.T. Phosphorylation of calponin in airways smooth muscle. Am. J. Physiol. 1997;272:L115–L123. doi: 10.1152/ajplung.1997.272.1.L115. [DOI] [PubMed] [Google Scholar]
- PYNE S., PYNE N.J. Differential effects of β2 receptor antagonists upon bradykinin-stimulated phospholipase C and D in guinea-pig cultured tracheal smooth muscle. Br. J. Pharmacol. 1993a;110:477–481. doi: 10.1111/j.1476-5381.1993.tb13835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PYNE S., PYNE N.J. Bradykinin stimulates phospholipase D in primary cultures of guinea-pig tracheal smooth muscle. Biochem. Pharmacol. 1993b;45:593–603. doi: 10.1016/0006-2952(93)90132-g. [DOI] [PubMed] [Google Scholar]
- RAO G.N., BERK B.C. Active oxygen species stimulates vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res. 1992;70:593–599. doi: 10.1161/01.res.70.3.593. [DOI] [PubMed] [Google Scholar]
- RASMUSSEN H., TAKUWA Y., PARK S. Protein kinase C in the regulation of smooth muscle contraction. FASEB. 1987;1:177–185. [PubMed] [Google Scholar]
- RECHSTEINER M., ROGERS S.W. PEST sequences and regulation by proteolysis. Trends. Biochem. Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- REMBOLD C.M., MURPHY R.A. [Ca2+]-dependent myosin phosphorylation in phorbol diester stimulated smooth muscle contraction. Am. J. Physiol. 1988;255:C719–C723. doi: 10.1152/ajpcell.1988.255.6.C719. [DOI] [PubMed] [Google Scholar]
- RON D., KAZANIETZ M.D. New insight into the regulation of protein kinase C and novel phorbol ester receptors. FASEB J. 1999;13:1658–1676. [PubMed] [Google Scholar]
- ROSSETTI M., SAVINEAU J.P., CREVAL H., MARTHAN R. Role of protein kinase C in nonsensitized and passively sensitized human isolated bronchial smooth muscle. Am. J. Physiol. 1995;268:L966–L971. doi: 10.1152/ajplung.1995.268.6.L966. [DOI] [PubMed] [Google Scholar]
- RYVES W.J., EVANS A.T., OLIVIER A.R., PARKER P.J., EVANS F.J. Activation of the PKC-isotypes α, β1, γ, δ and ε by phorbol esters of different biological activities. FEBS Lett. 1991;288:5–9. doi: 10.1016/0014-5793(91)80989-g. [DOI] [PubMed] [Google Scholar]
- SASAGURI T., WATSON S.P. Protein kinase C regulates the tonic but not the phasic component of contraction in guinea pig ileum. Br. J. Pharmacol. 1989;98:791–798. doi: 10.1111/j.1476-5381.1989.tb14607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHRAMM C.M., GRUNSTEIN M.M. Mechanisms of protein kinase C regulation of airway contractility. J. App. Physiol. 1989;66:1935–1941. doi: 10.1152/jappl.1989.66.4.1935. [DOI] [PubMed] [Google Scholar]
- SCOTT P.H., BELHAM C.M., AL-HAFIDH J., CHILVERS E.R., PEACOCK A.J., GOULD G.W., PLEVIN R. A regulatory role for cAMP in phosphatidylinsositol 3-kinase/p70 ribosomal S6 kinase-mediated DNA synthesis in platelet-derived growth factor-stimulated bovine airway smooth muscle cells. Biochem. J. 1996;318:965–971. doi: 10.1042/bj3180965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAPIRO P.S., EVAN J.N., DAVIS R.J., POSADA J.A. The seven-transmembrane-spanning receptors for endothelin and thrombin cause proliferation of airway smooth muscle cells and activation of the extracellular signal-regulated kinase and c-Jun NH2-terminal kinase groups of mitogen-activated protein kinases. J. Biol. Chem. 1996;271:5750–5754. doi: 10.1074/jbc.271.10.5750. [DOI] [PubMed] [Google Scholar]
- SHEA T.B., BEERMANN M.L., GRIFFIN W.R., LELI U. Degradation of protein kinase Cα and its free catalytic subunit, protein kinase M, in intact human neuroblastoma cells and under cell-free conditions. FEBS Lett. 1994;350:223–229. doi: 10.1016/0014-5793(94)00769-1. [DOI] [PubMed] [Google Scholar]
- SHIRAZI A., IIZUKA K., FADDEN P., MOSSE C., SOMLYO A.P., SOMLYO A.V., HAYSTEAD T.A. Purification and characterization of the mammalian myosin light chain phosphatase holoenzyme. The differential effects of the holoenzyme and its subunits on smooth muscle. J. Biol. Chem. 1994;269:31598–31606. [PubMed] [Google Scholar]
- SHIRINSKY V.P., BIRYUKOV K.G., HETTASCH J.M., SELLERS J.R. Inhibition of the relative movement of actin and myosin by caldesmon and calponin. J. Biol. Chem. 1992;267:15886–15892. [PubMed] [Google Scholar]
- SHISTIK E., IVANINA T., BLUMENSTEIN Y., DASCAL N. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. J. Biol. Chem. 1998;273:17901–17909. doi: 10.1074/jbc.273.28.17901. [DOI] [PubMed] [Google Scholar]
- SINGER H.A., OREN J.W., BENSCOTER H.A. Myosin light chain phosphorylation in 32P-labeled rabbit aorta stimulated by phorbol 12,13-dibutyrate and phenylephrine. J. Biol. Chem. 1989;15:21215–21222. [PubMed] [Google Scholar]
- SMALL J.V., FURST D.O., DEMAY J. Localization of filamin in smooth muscle. J. Cell Biol. 1986;102:210–220. doi: 10.1083/jcb.102.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMALL J.V., SOBIESZEK A. Ca-regulation of mammalian smooth muscle actomyosin via a kinase-phosphatase-dependent phosphorylation and dephosphorylation of the 20 000-Mr light chain of myosin. Eur. J. Biochem. 1977;15:521–530. doi: 10.1111/j.1432-1033.1977.tb11622.x. [DOI] [PubMed] [Google Scholar]
- SMALL R.C., FOSTER R.W.The electrophysiology of calcium channels in airways smooth muscle Airways Smooth Muscle: Development and Regulation of Contractility 1994Basle: Birkhauser Verlag; 137–161.eds. Giembycz, M.A. & Raeburn, D. pp [Google Scholar]
- SMITH J.B., SMITH L., PETTIT G.R. Bryostatins: potent, new mitogens that mimic phorbol ester tumor promoters. Biochem. Biophys. Res. Commun. 1985;132:939–945. doi: 10.1016/0006-291x(85)91898-4. [DOI] [PubMed] [Google Scholar]
- SOBIESZEK A., SMALL J.V. Regulation of actin-myosin interaction in vertebrate smooth muscle: activation via a myosin light chain kinase and the effects of tropomyosin. J. Mol. Biol. 1977;112:559–576. doi: 10.1016/s0022-2836(77)80164-2. [DOI] [PubMed] [Google Scholar]
- SOBUE K., MURAMOTO Y., INUI M., KANDA K., KAKIUCHI S. Purification of a calmodulin-binding protein from chicken gizzard that interacts with F-actin. Proc. Natl. Acad. Sci. U.S.A. 1981;78:5652–5655. doi: 10.1073/pnas.78.9.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOBUE K., SELLERS J.R. Caldesmon, a novel regulatory protein in smooth muscle and nonmuscle actomyosin systems. J. Biol. Chem. 1991;266:12115–12118. [PubMed] [Google Scholar]
- SODERLING T.R. Protein kinases. Regulation by autoinhibitory domains. J. Biol. Chem. 1990;265:1823–1826. [PubMed] [Google Scholar]
- SOMLYO A.P., HIMPENS B. Cell calcium and its regulation in smooth muscle. FASEB J. 1989;3:2266–2276. doi: 10.1096/fasebj.3.11.2506092. [DOI] [PubMed] [Google Scholar]
- SOMLYO A.P., SOMLYO A.V. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- SOUHRADA M., SOUHRADA J.F. Sodium and calcium influx induced by phorbol esters in airway smooth muscle cells. Am. Rev. Resp. Dis. 1989a;139:927–932. doi: 10.1164/ajrccm/139.4.927. [DOI] [PubMed] [Google Scholar]
- SOUHRADA M., SOUHRADA J.F. The role of protein kinase-C in sensitization and antigen response of airway smooth muscle. Am. Rev. Respir. Dis. 1989b;140:1567–1572. doi: 10.1164/ajrccm/140.6.1567. [DOI] [PubMed] [Google Scholar]
- STABEL S., PARKER P.J. Protein kinase C. Pharmacol. Ther. 1991;51:71–95. doi: 10.1016/0163-7258(91)90042-k. [DOI] [PubMed] [Google Scholar]
- STEMPKA L., GIROD A., MULLER H.-J., RINCKE G., MARKS F., GSCHWENDT M., BOSSEMEYER D. Phosphorylation of protein kinase Cδ at threonine 505 is not a prerequisite for enzymatic activity. J. Biol. Chem. 1997;272:6805–6811. doi: 10.1074/jbc.272.10.6805. [DOI] [PubMed] [Google Scholar]
- STOUFFER G.A., RUNGE M.S. The role of secondary growth factor production in thrombin-induced proliferation of vascular smooth muscle cells. Sem. Throm. Hem. 1998;24:145–150. doi: 10.1055/s-2007-995833. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI K., HIWADA K., KOKUBU T. Isolation and characterisation of a 34,000 dalton calmodulin- and F-actin-binding protein from chicken gizzard smooth muscle. Biochem. Biophys. Res. Comm. 1986;141:20–26. doi: 10.1016/s0006-291x(86)80328-x. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI K., HIWADA K., KOKUBU T. Occurrence of anti-gizzard P34K antibody cross-reactive components in bovine smooth muscles and non-smooth muscle tissues. Life Sci. 1987;41:291–296. doi: 10.1016/0024-3205(87)90151-2. [DOI] [PubMed] [Google Scholar]
- TAKAI Y., KISHIMOTO A., INOUE M., NISHIZUKA Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. I. Purification and characterization of an active enzyme from bovine cerebellum. J. Biol. Chem. 1977;252:7603–7609. [PubMed] [Google Scholar]
- TAKUWA Y., TAKUWA N., RASMUSSEN H. Carbachol induces a rapid and sustained hydrolysis of phosphoinositide in bovine tracheal smooth muscle: measurements of the mass of polyphosphoinositides, 1,2-diacylglycerol, and phosphatidic acid. J. Biol. Chem. 1986;261:14670–14675. [PubMed] [Google Scholar]
- TAUBMAN M.B., BERK B.C., IZUMO S., TSUDA T., ALEXANDER R.W., NADAL-GINARD B. Angiotensin II induces c-fos mRNA in aortic smooth muscle. Role of Ca2+ mobilisation. J. Biol. Chem. 1989;264:526–530. [PubMed] [Google Scholar]
- TOGASHI H., HIRSHMAN C.A., EMALA C.W. Qualitative immunoblot analysis of PKC isoforms expressed in airway smooth muscle. Am. J. Physiol. 1997;272:L603–L607. doi: 10.1152/ajplung.1997.272.4.L603. [DOI] [PubMed] [Google Scholar]
- TOLAN D., CONWAY A.-M., RAKHIT S., PYNE N.J., PYNE S. Assessment of the extracellular and intracellular actions of sphingosine 1-phosphate by using the p42/p44 mitogen-activated protein kinase cascade as a model. Cell Signal. 1999;11:349–354. doi: 10.1016/s0898-6568(99)00005-4. [DOI] [PubMed] [Google Scholar]
- TOMASIC M., BOYLE J.P., WORLEY J.F., KOTLIKOFF M.I. Contractile agonists activate voltage-dependent calcium channels in airway smooth muscle cells. Am. J. Physiol. 1992;263:C106–C113. doi: 10.1152/ajpcell.1992.263.1.C106. [DOI] [PubMed] [Google Scholar]
- TWORT C.H.C., VAN BREEMEN C. Human airway smooth muscle in culture. Tissue Cell. 1988;20:339–344. doi: 10.1016/0040-8166(88)90069-9. [DOI] [PubMed] [Google Scholar]
- TWORT C.H.C., VAN BREEMEN C. Human airway smooth muscle in culture: Control of the intracellular calcium store. Pulm. Pharmacol. 1989;2:45–53. doi: 10.1016/s0952-0600(89)80009-2. [DOI] [PubMed] [Google Scholar]
- UMEMOTO S., BENGUR A.R., SELLERS J.R. Effect of multiple phosphorylations of smooth muscle and cytoplasmic myosins on movement in an in vitro motility assay. J. Biol. Chem. 1989;264:1431–1436. [PubMed] [Google Scholar]
- VALVERDE A.M., SINNETT-SMITH J., VAN LINT J., ROZENGURT E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. U.S.A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VICHI P., WHELCHEL A., KNOT H., NELSON M., KOLCH W., POSADA J. Endothelin-stimulated ERK-activation in airway smooth muscle cells requires calcium influx and Raf activation. Am. J. Respir. Cell Mol. Biol. 1999;20:99–105. doi: 10.1165/ajrcmb.20.1.3210. [DOI] [PubMed] [Google Scholar]
- WALKER T.R., MOORE S., LAWSON M.F., PANETTIERI R.A., CHILVERS E.R. Platelet-derived growth factor-BB and thrombin activate phosphoinositide 3-kinase and protein kinase B: Role in mediating airway smooth muscle proliferation. Mol. Pharmacol. 1998;54:1007–1015. doi: 10.1124/mol.54.6.1007. [DOI] [PubMed] [Google Scholar]
- WALSH M.P. Smooth muscle caldesmon. Prog. Clin. Biol. Res. 1990;327:127–140. [PubMed] [Google Scholar]
- WALSH M.P., HOROWITZ A., CLEMENT-CHOMIENNE O., ANDREA J.E., ALLEN B.G., MORGAN K.G. Protein kinase C mediation of Ca2+-independent contractions of vascular smooth muscle. Biochem. Cell. Biol. 1996;74:485–502. doi: 10.1139/o96-053. [DOI] [PubMed] [Google Scholar]
- WATTS S.W., COX D.A., JOHNSON B.G., SCHOEPP D.D., COHEN M.L. Contractile serotonin-2A receptor signal transduction in guinea pig trachea: importance of protein kinase C and extracellular and intracellular calcium but not phosphoinositide hydrolysis. J. Pharmacol. Exp. Ther. 1994;271:832–844. [PubMed] [Google Scholar]
- WEBB B.L.J., LINDSAY M.A., BARNES P.J., GIEMBYCZ M.A. Protein kinase C isoenzymes in airway smooth muscle. Biochem. J. 1997a;324:167–175. doi: 10.1042/bj3240167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEBB B.L.J., LINDSAY M.A., SEYBOLD J.S., BRAND N.J., YACOUB M.H., HADDAD E.-B., BARNES P.J., ADCOCK I.M., GIEMBYCZ M.A. Identification of the protein kinase C isoenzymes in human lung and airways smooth muscle at the protein and mRNA level. Biochem. Pharmacol. 1997b;54:199–205. doi: 10.1016/s0006-2952(97)00165-2. [DOI] [PubMed] [Google Scholar]
- WHELCHEL A., EVANS J., POSADA J. Inhibition of ERK activation attenuates endothelin-stimulated airway smooth muscle proliferation. Am. J. Resp. Cell Mol. Biol. 1997;16:589–596. doi: 10.1165/ajrcmb.16.5.9160841. [DOI] [PubMed] [Google Scholar]
- WILKINSON S.E., PARKER P.J., NIXON J.S. Isozyme-specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem. J. 1993;294:335–337. doi: 10.1042/bj2940335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WINDER S.J., PATO M.D., WALSH M.P. Purification and characterization of calponin phosphatase from smooth muscle. Effect of dephosphorylation on calponin function. Biochem. J. 1992;286:197–203. doi: 10.1042/bj2860197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WINDER S.J., WALSH M.P. Structural and functional characterization of calponin fragments. Biochem. Int. 1990;22:335–341. [PubMed] [Google Scholar]
- XU S., COLLINS M.A., CHANG K.-J. Phorbol esters induce oscillatory contractions of intestinal smooth muscles. Eur. J. Pharmacol. 1991;201:215–222. doi: 10.1016/0014-2999(91)90348-t. [DOI] [PubMed] [Google Scholar]
- YAMAKAGE M. Direct inhibitory mechanisms of halothane on canine tracheal smooth muscle contraction. Anaesthesiol. 1992;77:546–553. doi: 10.1097/00000542-199209000-00022. [DOI] [PubMed] [Google Scholar]
- YAMAKAGE M., HIRSHMAN C.A., CROXTON T.L. Cholinergic regulation of voltage-dependent Ca2+ channels in porcine tracheal smooth muscle cells. Am. J. Physiol. 1995;269:L776–L782. doi: 10.1152/ajplung.1995.269.6.L776. [DOI] [PubMed] [Google Scholar]
- YAMASHITA T., KOKUBUN S. Muscarinic inhibition of L-type Ca2+ channels in guinea-pig tracheal smooth muscle cells. J. Smooth Muscle Res. 1997;33:175–185. doi: 10.1540/jsmr.33.175. [DOI] [PubMed] [Google Scholar]
- YANG C.M., YO Y.-L., ONG R., HSAI H.-C. Endothelin- and sarafotoxin-induced phosphoinositide hydrolysis in cultured canine tracheal smooth muscle cells. J. Neurochem. 1994;62:1440–1448. doi: 10.1046/j.1471-4159.1994.62041440.x. [DOI] [PubMed] [Google Scholar]
- YANG K.X.F., BLACK J.D. The involvement of protein kinase C in the contraction of human airway smooth muscle. Eur. J. Pharmacol. 1995;275:283–289. doi: 10.1016/0014-2999(94)00785-6. [DOI] [PubMed] [Google Scholar]
- YANG K.X.F., BLACK J.L. Protein kinase C-induced changes in human airway smooth muscle tone the effects of Ca2+ and Na+ transport. Eur. J. Pharmacol. 1996;315:65–71. doi: 10.1016/s0014-2999(96)00595-x. [DOI] [PubMed] [Google Scholar]
- YOSHIDA M., SUZUKI A., ITOH T. Mechanisms of vasoconstriction induced by endothelin-1 in smooth muscle of rabbit mesenteric artery. J. Physiol. (Lond). 1994;477:253–265. doi: 10.1113/jphysiol.1994.sp020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHII A., IIZUKA K., DOBASHI K., HORIE T., HARADA T., NAKAZAWA T., MORI M. Relaxation of contracted rabbit tracheal and human bronchial smooth muscle by Y-27632 through inhibition of Ca2+ sensitization. Am. J. Respir. Cell Mol. Biol. 1999;20:1190–1200. doi: 10.1165/ajrcmb.20.6.3441. [DOI] [PubMed] [Google Scholar]
- ZIDOVETZKI R., LESTER D.S. The mechanism of activation of protein kinase C: a biophysical perspective. Biochem. Biophys. Acta. 1992;1134:261–272. doi: 10.1016/0167-4889(92)90185-e. [DOI] [PubMed] [Google Scholar]