

Abstract
The early intervention with endothelinA (ETA) receptor antagonists following coronary artery ligation has been shown to reduce the development of pulmonary hypertension, despite a lack of improvement in left ventricular function.
The present study examined the contribution of pulmonary vascular remodelling and the progression of lung fibrosis in the development of pulmonary hypertension and the subsequent role of endothelin-1 in these processes in a rat model of myocardial infarction (MI).
The administration of 60 mg kg−1 per day of the specific ETA receptor antagonist LU135253 ((+)-(S)-2-(4,6-dimethoxy-pyrimidin-2-yloxy)-3-methoxy-3,3-diphenyl-propionic acid) 24 h following coronary artery ligation, failed to improve left ventricular contractile indices, but reduced the extent of pulmonary hypertension, as reflected by the significant decrease in right ventricular systolic pressure.
The medial wall thickness of small pulmonary arteries (50–200 μm) was significantly increased 4 weeks following MI, albeit LU135253 treatment did not ameliorate this pattern of vascular remodelling.
The steady-state mRNA levels of collagen, fibronectin, transforming growth factor-β1, and -β3 were significantly increased in the lungs of MI rats. The treatment with LU135252 did not alter this pattern of gene expression.
Thus, these data demonstrate pulmonary vascular remodelling and the increased expression of extracellular matrix proteins represent underlying mechanisms implicated in the development of pulmonary hypertension in the MI rat.
Despite the amelioration of the pulmonary hypertensive state, ETA receptor blockade was insufficient to reverse pulmonary vascular remodelling, or the development of lung fibrosis in the MI rat.
Keywords: Coronary ligation; endothelin-1; ETA receptor; pulmonary hypertension; pulmonary vascular remodelling, extracellular matrix proteins; transforming growth factor-β
Introduction
A common pathophysiological feature of pulmonary hypertension (PH), regardless the etiology, is an increase in the circulating levels of endothelin-1 (ET-1) (Hay, 1999; MacLean, 1999; Stewart, 1993). In left ventricular failure, the most prevalent cause of secondary PH, elevated plasma ET-1 levels were found to relate directly with the severity of PH (Cody et al., 1992). Recent studies have confirmed the contribution of ET-1 in the development of left ventricular dysfunction and PH in the myocardial infarct (MI) rat model. The treatment with an ET receptor antagonist 1 week post-MI reduced mortality, improved cardiac function, and PH (Sakai et al., 1996a). By contrast, the early administration (24–48 h post-MI), did not have any beneficial effect or exacerbated the contractile dysfunction of the left ventricle, albeit an amelioration of PH was still evident (Sakai et al., 1996b; Nguyen et al., 1998). The apparent paradox with regard to the therapeutic efficacy of ET antagonists on left ventricular contractile function has been attributed to the suggested role of ET-1 in early scar formation and stabilization (Nguyen et al., 1998). Nonetheless, regardless of the time frame of ET-1 receptor antagonist administration, the beneficial effect on PH persists in the MI rat model.
The underlying mechanism(s) attributed to the beneficial effect of ET-1 receptor antagonist treatment on the development of PH in the MI rat remains unresolved. Pulmonary vascular remodelling and the progression of fibrosis represent pathophysiological events implicated in the genesis and/or maintenance of PH (Prié et al., 1997; Zhang & Phan, 1996). Indeed, several in vivo studies have shown the treatment with ET-1 receptor antagonists inhibited vascular smooth muscle hypertrophy in the DOCA-salt rat model of hypertension, and following angiotensin II infusion (Schiffrin, 1998; Moreau et al., 1997). Likewise, the administration of ET-1 receptor antagonists reduced the extent of fibrosis in the lungs of bleomycin-treated rats (Park et al., 1997). These latter data are consistent with the in vitro findings demonstrating the exogenous administration of ET-1 increased collagen levels in vascular smooth muscle cells, and fibroblasts (Rizvi and Myers, 1997; Guarda et al., 1993). Collectively, these observations suggest ET-1 receptor antagonists may improve PH via the modulation of vascular remodelling and the expression of extracellular matrix proteins. In this regard, the present study examined whether the development of PH in the MI rat was associated with pulmonary vascular remodelling, and a molecular phenotype of fibrosis. Secondly, the potential role of ET-1 in these pathophysiological events was examined via the administration of the ETA receptor antagonist LU135252 24 h post-MI for a period of 4 weeks.
Methods
Experimental protocol
Myocardial infarction (MI) was induced in male Wistar rats (7 weeks old; 200–250 grams; Charles Rivers, St-Constant, Quebec, Canada) by ligating the left anterior coronary artery as previously described (Nguyen et al., 1998). The animals were randomized into four groups: (1) Sham-operated+0.9% saline, (2) Sham-operated+LU135252 (Knoll AG), (3) MI rats+0.9% saline, and (4) MI rats+LU135252. A dose of 60 mg kg−1 per day of LU135252 was administered twice daily by gavage 24 h following coronary-ligation or sham-operation for a period of 4 weeks. LU135252 is a novel nonpeptidic selective ETA receptor antagonist with high oral bioavailability and a long half-life (Münter et al., 1996). This dose and regimen has been previously shown to inhibit the increased expression of lung ET-1 protein levels and myocardial ET-1 mRNA levels, and decrease right ventricular systolic pressure in the MI rat (Nguyen et al., 1998; Picard et al., 1998). Likewise, in the monocrotaline-induced model of pulmonary hypertension, the administration of 50 mg g−1 day−1 of LU135252 by gavage ameliorated right ventricular systolic pressure and hypertrophy (Prié et al., 1997).
In the present study, the criteria used to categorize a large MI is a scar to body weight ratio >0.2 mg g−1, as previously described, and validated (Nguyen et al., 1998). The use and care of laboratory animals was according to the Canadian Council for Animal Care and approved by the Animal Care Committee of the Montreal Heart Institute.
Haemodynamic measurements
The haemodynamic profile of each rat was examined at 4 weeks post-infarction, as previously described (Nguyen et al., 1998). Briefly, rats were anaesthetized with a gas mixture containing 100% oxygen and halothane reduced from 2% to 0.5–0.8% 15 min before left and right heart haemodynamics were measured by a microtip pressure transducer catheter (model SPR-407, 2F, Millar instrument, Houston, Texas, U.S.A.).
Northern hybridization
Following haemodynamic measurements, total lung RNA was isolated by a modification of guanidine thiocyanate-phenol-chloroform extraction method, as previously described (Calderone et al., 1998). Total RNA (20 μg) was denatured with formaldehyde and formamide, and separated by size electrophoresis on a 1.3% agarose/4% formaldehyde gel, and subsequently transferred to nylon membranes (Genescreen Plus; Dupont-NEN) by vacuum blotting (model 785; Bio-Rad Laboratories).
A 0.985 kb fragment of rat TGF-β1 (American Type Culture Collection (ATCC); Rockville, MD, U.S.A.), a 1.2 kb fragment of mouse TGF-β3 (ATCC), a 0.6 kb fragment of rat fibronectin (courtesy of Dr R.O. Hynes), and a 0.3 kb fragment of prepro-collagen-α1 (type 1; ATCC) were labelled with 32P-dCTP (NEN) to a specific activity of 1–2×106 c.p.m./ng cDNA by the random hexamer (Pharmacia) priming method and hybridized to nylon membranes (Dupont-NEN) for 18–24 h at 42°C, as previously described (Calderone et al., 1998). The filters exposed to the cDNA probes were washed twice (15 min, room temperature) with 300 mmol/L NaCl/30 mmol/L trisodium citrate and 0.1% SDS and twice (15 min, 45°C) with 30 mmol/L NaCl/3 mmol/L trisodium citrate and 0.1% SDS. Nylon membranes were subsequently exposed to Kodak XAR film with an intensifying screen at −70°C, and films were scanned with a laser densitometer (Chemilmager 4000 I v4.04 software; Alphan Innotech Corporation). All levels of mRNA reported in this paper are normalized to the level of 28S rRNA.
Lung vascular morphometry
The pattern of pulmonary arterial bed remodelling was examined in a separate series of experiments. The scar/bw ratio of the MI rats+0.9% saline was 0.3±0.03 (n=7), and the MI rats+LU135252 was 0.32±0.03 (n=5). The pulmonary artery was injected at 50 cm H2O pressure with a hot barium-gelatin mixture (60°C) for 3 min, and the airways were distended with 10% formaldehyde solution at a pressure of 36 cm H2O.
The lungs were immersed in an inflated state in 10% formaldehyde solution for at least 2 days. For morphometric analysis, three sections were obtained from each rat lung (from the upper, median and lower parts), stained with haematoxylin and eosin, and microscopically assessed for medial wall thickness of pulmonary arteries. The measurements of luminal diameter and medial thickness on either side (m1 and m2) were made along the shortest diameter. Measurements were made at random on 10 muscular arteries (size ranges 50–100, 101–150 and >150 μm in external diameter) per lung section. For each artery, the medial wall thickness was related to the external diameter (d, luminal diameter+media on either side) and expressed as per cent wall thickness (%WT)=[(m1+m2)/2d]×100 (Prié et al., 1997).
Data analysis
Data are represented as the mean±s.e.m. Statistical analysis was performed by Student' unpaired t-test (2-tailed), and by analysis of variance followed by Dunnett' test when appropriate. A value of P<0.05 was considered statistically significant.
Results
Morphologic and haemodynamic alterations in the myocardium of MI rats: The effect of the ETA receptor antagonist LU135252
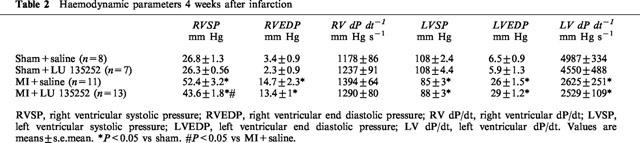
The ligation of the anterior descending coronary artery resulted in myocyte cell loss, as reflected by the significant decrease in left ventricular weight and scar formation in the MI group, as compared to sham (Table 1). A concomitant hypertrophic response was observed in the right ventricle, and lung wet weight was significantly increased in the MI rat group (Table 1). Left ventricular dysfunction was observed in the MI rat group, as reflected by the decrease in left ventricular systolic pressure, dP/dt index, and an increase in left ventricular end-diastolic pressure (Table 2). Moreover, PH was evident in the MI rat group, as reflected by the significant increase of right ventricular systolic pressure (Table 2). Long-term therapy of the MI rat group with the ETA receptor antagonist LU135252 resulted in a significant reduction of right ventricular systolic pressure in the MI rats, without a concomitant change in ventricular hypertrophy, lung wet weight, scar size or left ventricular contractile function (Table 2).
Table 1.
Morphology parameters of myocardial infarction rats
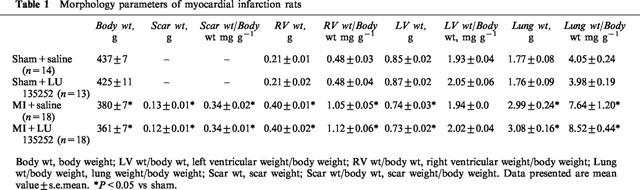
Table 2.
Haemodynamic parameters 4 weeks after infarction
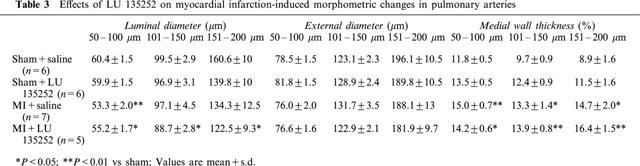
Pulmonary artery remodelling in the MI rats
Morphometric changes of the pulmonary arteries are shown in Table 3. The external diameter of the caliber of arteries examined was similar in all sham and MI rats with or without LU135252 treatment. The luminal diameter of 50–100 μm pulmonary arteries was significantly decreased in the MI rats, as compared to sham. By contrast, in the 101–150 and 151–200 μm caliber arteries, a non-significant decrease in luminal diameter was observed in the MI rats, as compared to sham. The treatment with LU135252 did not reverse the decrease in luminal diameter observed in the MI rats. Medial wall thickness was significantly increased in all the caliber of arteries examined in the MI rats, as compared to sham, regardless the arterial caliber. LU135252 treatment did not reverse this pattern of remodelling.
Table 3.
Effects of LU 135252 on myocardial infarction-induced morphometric changes in pulmonary arteries
A molecular phenotype of pulmonary fibrosis
To assess whether the MI rats were associated with the development of lung fibrosis, the steady-state mRNA levels of the extracellular matrix proteins collagen and fibronectin were assessed. In the lungs of the MI rats, the steady-state mRNA levels of prepro-collagen-α1 and fibronectin were increased 3.7±1.0 fold (P<0.05 versus sham; n=6) and 2.9±0.2 fold (P<0.05 versus sham; n=6) respectively, in the lungs of MI rats, as compared to sham rats (Figure 1). The transforming growth factor-β (TGF-β) family has been implicated as a local autocrine/paracrine factor involved in the progression and/or maintenance of fibrosis. Consistent with this premise, the increased mRNA expression of the extracellular matrix proteins was associated with a concomitant significant increase in the steady-state mRNA levels of TGF-β1 (3.7±0.8 fold increase; P<0.05 versus sham; n=6) and TGF-β3 (2.7±0.7 fold increase; P<0.05 versus sham; n=6) in the lungs of the MI rats, as compared to the sham rats (Figure 1). Long-term therapy of sham rats with the ETA receptor antagonist LU135252 did not alter the expression of these transcripts, as compared to sham (Figure 1). In the MI rat group, LU135252 treatment did not prevent the increased expression of either collagen (3.0±0.8 fold increase; P<0.05 versus sham+LU135252; n=6), fibronectin (2.4±0.6 fold increase; P<0.05 versus sham+LU135252; n=6), TGF-β1 (4±0.8 fold increase; P<0.05 versus sham+LU135252; n=6), or TGF-β3 (3.2±0.8 fold increase; P<0.05 versus sham+LU135252; n=6) mRNAs, as compared to untreated MI rats (Figure 1).
Figure 1.
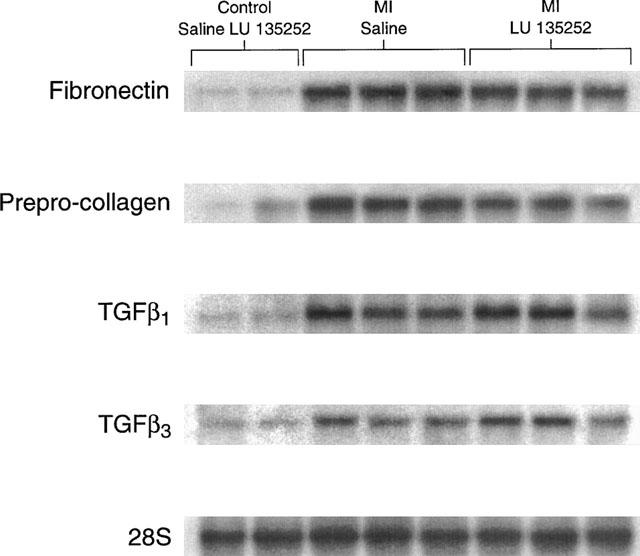
A Northern hybridization depicting changes in the steady-state mRNA levels of prepro-collagen α1, fibronectin, transforming growth factor-β1 (TGF-β1), and TGF-β3 in the total RNA isolated from rat lungs. The pattern of gene expression in the sham-operated rats treated with the ETA receptor antagonist LU135252 was similar to the sham-operated rats administered saline. The steady-state mRNA levels of prepro-collagen α1, fibronectin, TGF-β1, and TGF-β3 were increased in the lungs of rats with myocardial infarction (MI). The long-term therapy with LU135252 did not reverse the pattern of gene expression in the MI rats. mRNA levels were normalized to the level of 28S ribosomal RNA.
Discussion
The development of PH, secondary to MI in the rat, was associated with pulmonary artery remodelling, and a pattern of gene expression consistent with the presence of fibrosis. The long-term administration of the ETA receptor antagonist LU135252 did not reverse these phenotypic events in the lungs, despite a significant reduction of right ventricular systolic pressure. Collectively, these data suggest ET-1, acting at least partially via the ETA receptor, may in part contribute to the development of PH. However, the absence of a beneficial effect of LU135252 on either pulmonary vascular remodelling or the fibrotic pattern of gene expression in the lungs of the MI rat, suggests ET-1 activation of the ETA receptor was not implicated in the progression of these pathophysiological events.
Data from several studies support the concept that ET-1, acting via the ETA receptor, is implicated in the development of PH. Evidence to support this premise includes increased expression of ET-1 in the lungs of various models of PH, and the identification of ETA receptors on the pulmonary arterial vasculature (Soma et al., 1999; Nguyen et al., 1998; Park et al., 1997; Sakai et al., 1996b; Nakamichi et al., 1992). Consistent with this concept, the administration of structurally distinct ETA receptor antagonists were shown to reverse the increase in pulmonary artery pressure, and the associated hypertrophic response of the right ventricle in hypoxia- and monocrotaline-induced models of PH (Prié et al., 1997; DiCarlo et al., 1995). In the lungs of the MI rat, ET-1 peptide levels were found elevated, and the long-term treatment with LU135252 completely suppressed the increased expression (Sakai et al., 1996b; Nguyen et al., 1998). These latter findings are consistent with a role of the ETA receptor in the synthesis of endothelin-1 (Barton et al., 1998). Moreover, coincident with LU135252 treatment, a significant reduction in right ventricular systolic pressure was observed in the MI rats. Likewise, the treatment of MI rats with the structurally distinct ETA receptor antagonist BQ-123 was found to ameliorate right ventricular systolic pressure when administered 24 h post-infarction (Sakai, 1996b). By contrast, neither LU135252 nor BQ-123 treatment ameliorated left ventricular contractile indices, or attenuated scar size in the MI rats (Sakai, 1996b). Collectively, these data suggest that ET-1 may have a pathophysiological role in the development of PH in the MI rat.
Although both LU135252 and BQ-123 ameliorated right ventricular systolic pressure in the MI, BQ-123 rather than LU135252 caused a significant decrease in right ventricular hypertrophy in the MI rats (Sakai et al., 1996b). The reason for this dichotomy between two ETA antagonists on right ventricular hypertrophy in the MI rat remains presently unknown. An important caveat which may in part explain these latter findings is that the reduction of right ventricular pressures with BQ-123 was greater than LU135252, and in this regard, the decrease by LU135252 may not have been sufficient to have any measurable effect on the hypertrophic response. This difference may be in part explained by the pharmacological profiles of these two antagonists. It has been demonstrated BQ-123 has a greater selectivity (500–2000 fold) for the ETA receptor versus the ETB receptor (Battistini & Dussault, 1998). By contrast, LU135252 has a 140 fold greater selectivity for ETA than ETB (Battistini & Dussault, 1998). Consequently, LU135252 treatment may have elicited some antagonism of the ETB receptor sufficient to counteract the beneficial effect of exclusively blocking ETA receptors. The ETB receptor is found predominantly on endothelial cells, and is coupled to the synthesis of nitric oxide and prostacyclin, two important vasodilatory substances (Love & McMurray, 1996). Thus, any antagonism of the ETB receptor by LU135252 would diminish the beneficial vasodilatory effect, thereby partially offsetting the decrease in right ventricular pressures mediated by ETA antagonism leading to less effective diminution of right ventricular hypertrophy. Moreover, nitric oxide has been shown to exert an antihypertrophic action on cardiac myocyte growth (Calderone et al., 1998). Thus, the concomitant blockade of endothelial ETB receptors in the myocardium could diminish the local growth-inhibiting action of nitric oxide, an effect less feasible with the highly selective ETA antagonist (Battistini & Dussault, 1998). Nonetheless, despite this difference, the ability of two structurally distinct ETA receptor antagonists (LU135252 and BQ-123) to ameliorate right ventricular systolic pressure supports the involvement of ET-1 in the development of PH in the MI rat.
It has been suggested that endothelin-1 may be involved in the development of pulmonary and systemic hypertension, and subsequent vascular bed remodelling. In the DOCA-salt rat model of hypertension, ETA receptor blockade markedly reduced elevated blood pressure, and improved vascular remodelling (Schiffrin, 1998). Likewise, in an angiotensin II-infused experimental rat model of hypertension, the treatment with the ETA antagonist LU135252 reduced blood pressure and attenuated vascular hypertrophy (Moreau et al., 1997). In the hypoxia-induced model of PH, BQ-123 treatment reversed the increase in pulmonary artery pressure and medial wall thickness of 50–100 μm caliber pulmonary arteries (DiCarlo et al., 1995). Consistent with these in vivo findings, the exogenous administration of ET-1 to cultured vascular smooth muscles cells promoted proliferation and hypertrophy via the recruitment of the small GTP-binding protein ras (Hafizi et al., 1999; Schieffer et al., 1997). Interestingly, in human vascular smooth muscle cells, ET-1 had no direct effect on growth, but potentiated the growth response to platelet-derived growth factor (Yang et al., 1999). This latter effect was blocked by the ETA receptor antagonist LU135252. In the present study, the development of PH in the MI rat was associated with the concomitant remodelling of the pulmonary arterial bed. A significant decrease in luminal diameter in the 50–100 caliber arteries was observed, whereas a similar tendency, but non-significant effect was observed in the 101–150 and 151–200 μm caliber arteries. In all groups of caliber arteries examined, medial wall thickness was significantly increased in the MI rat, as compared to the sham-operated rat. However, despite the beneficial effect on right ventricular systolic pressure, pulmonary vascular remodelling was not attenuated in the MI rat following LU135252 therapy. Likewise, in the monocrotaline-induced model of PH, LU135252 treatment improved right ventricular systolic pressure, but did not reverse pulmonary artery remodelling (Prié et al., 1997). Thus, it is possible that alternative vascular smooth muscle mitogens, such as angiotensin II, and platelet-derived growth factor may be instrumental in pulmonary artery remodelling in the MI rat model, as well as the monocrotaline rat model of PH (Yang et al., 1999; Moreau et al., 1997).
Lung fibrosis represent a common pathophysiological feature of various pulmonary diseases (Kovacs & DiPietro, 1994), and both in vivo and in vitro studies have characterized ET-1 as a profibrotic factor. In the bleomycin-induced rat model of pulmonary fibrosis, immunoreactive ET-1 levels were increased, and the treatment with the non-selective ET receptor antagonist bosentan, reduced the extent of fibrosis (Park et al., 1997). In vascular smooth muscle cells, and fibroblasts, ET-1 administration led to the synthesis of collagen, and further decreased collagenase activity in fibroblasts (Rizvi & Myers, 1997; Guarda et al., 1993). Lastly, in pulmonary bronchial epithelial cells, and cultured rabbit synoviocytes, ET-1 increased fibronectin gene expression via activation of the ETA receptor (Gutierrez et al., 1996; Marini et al., 1996). In the present study, the presence of PH in the MI rat was associated with an increase in the steady-state lung mRNA levels of collagen and fibronectin. The long-term therapy with LU135252 did not reverse the increased expression of these extracellular matrix proteins. Consequently, these data are, to the best of our knowledge, the first to highlight a molecular phenotype of fibrosis in the lungs of the MI rat, which may in part contribute to the development of PH. The inability of LU135252 to block this molecular fibrotic phenotype suggests the ETA receptor is not involved in this process, at least in the lungs of the MI rat. By contrast, it is possible that there may exist a certain degree of redundancy in this process, such that despite the blockade of the ETA receptor, alternative local and/or hormonal factors may initiate the development of extracellular matrix remodelling.
In vitro studies have demonstrated that the exogenous administration of the transforming growth factor-β (TGF-β) isoforms to lung fibroblasts increased collagen expression, with TGF-β3 the most potent profibrotic agent (Eickelberg et al., 1999; Coker et al., 1997). Consistent with the in vitro effects of TGF-β, an increased expression of TGF-β isoforms, as well as a direct role of TGF-β1 in the development of lung fibrosis has been documented (Nakao et al., 1999; Coker et al., 1997; Sime et al., 1997). In the lungs of the MI rats, the steady-state mRNA levels of TGF-β1 and TGF-β3 were increased, as compared to sham rats. The long-term therapy of MI rats with LU135252 did not prevent the increased expression of either TGF-β1 or TGF-β3 mRNA, thereby suggesting ET-1 activation of the ETa receptor was not involved in their regulation. Nonetheless, the concomitant increased mRNA expression of extracellular matrix proteins and TGF-β isoforms is consistent with a role of these peptide growth factors in the progression and/or maintenance of lung fibrosis in the MI rat. The cellular origin of the increased expression of the TGF-β isoforms, and the hormonal and/or local factors implicated in their expression remain to be determined.
In conclusion, the long-term therapy of MI rats with the ETA selective antagonist LU135252 caused a significant reduction in right ventricular systolic pressure, suggesting ET-1, acting at least partially via the ETA receptor, contributes in part to the development of PH. However, neither pulmonary artery remodelling nor the increased expression of lung collagen and fibronectin mRNAs were prevented by long-term LU135252 treatment in the MI rat. Lastly, the profibrotic agents, TGF-β1 and TGF-β3 mRNAs were increased in the MI rats, and these peptide growth factors may play an instrumental role in the progression and/or maintenance of fibrosis in the lung of MI rats.
Acknowledgments
This work was supported by the Medical Research Council of Canada, Heart & Stroke Foundations of Canada and Quebec, and les Fonds de Recherche en Santé du Québec.
Abbreviations
- ET-1
endothelin-1
- ETA
endothelin-A receptor
- LU135253
(+)-(S)-2-(4,6-dimethoxy-pyrimidin-2-yloxy)-3-methoxy-3,3-diphenyl-propionic acid
- MI
myocardial infarction
- PH
pulmonary hypertension
- TGF-β
transforming growth factor-β
References
- BARTON M., D'USCIO L.V., SHAW S., MEYER P., MOREAU P., LUSCHER T.F. ET(A) receptor blockade prevents increased tissue endothelin-1, vascular hypertrophy, and endothelial dysfunction in salt-sensitive hypertension. Hypertension. 1998;31(1 pt 2):499–504. doi: 10.1161/01.hyp.31.1.499. [DOI] [PubMed] [Google Scholar]
- BATTISTINI B., DUSSAULT P. Blocking of the endothelin system: The development of receptor antagonists. Pulmon. Pharmacol. Therapeut. 1998;11:97–112. doi: 10.1006/pupt.1998.0154. [DOI] [PubMed] [Google Scholar]
- CALDERONE A., TAKAHASHI N., THAIK C., COLUCCI W.S. Atrial natriuretic peptide, nitric oxide and cGMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J. Clin. Invest. 1998;101:812–818. doi: 10.1172/JCI119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CODY R.J., HAAS G.J., BINKLEY P.F., CAPERS Q., KELLEY R. Plasma endothelin correlates with the extent of pulmonary hypertension in patients with chronic congestive heart failure. Circulation. 1992;85:504–509. doi: 10.1161/01.cir.85.2.504. [DOI] [PubMed] [Google Scholar]
- COKER R.K., LAURENT G.J., SHAHZEIDI S., LYMPANY P.A., DU BOIS R.M., JEFFERY P.K., MCANULTY R.J. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am. J. Pathol. 1997;150:981–991. [PMC free article] [PubMed] [Google Scholar]
- DICARLO V.S., CHEN S.J., MENG Q.C., DURAND J., YANO M., CHEN Y.F., OPARIL S. ETA-receptor antagonist prevents and reverses chronic hypoxia-induced pulmonary hypertension in rat. Am. J. Physiol. 1995;269:L690–L697. doi: 10.1152/ajplung.1995.269.5.L690. [DOI] [PubMed] [Google Scholar]
- EICKELBERG O., KOHLER E., REICHENBERGER F., BERTSCHIN S., WOODTLI T., ERNE P., PERRUCHOUD A.P., ROTH M. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-beta3. Am. J. Physiol. 1999;276:L814–824. doi: 10.1152/ajplung.1999.276.5.L814. [DOI] [PubMed] [Google Scholar]
- GUARDA E., KATWA L.C., MYERS P.R., TYAGI S.C., WEBER K.T. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc. Res. 1993;27:2130–2134. doi: 10.1093/cvr/27.12.2130. [DOI] [PubMed] [Google Scholar]
- GUTIERREZ S., PALACIOS I., EGIDO J., GOMEZ-GARRE D., HERNANDEZ P., GONZALEZ E., HERRERO-BEAUMONT G. Endothelin-1 induces loss of proteoglycans and enhances fibronectin and collagen production in cultured rabbit synovial cells. Eur. J. Pharmacol. 1996;302:191–197. doi: 10.1016/0014-2999(96)00068-4. [DOI] [PubMed] [Google Scholar]
- HAFIZI S., ALLEN S.P., GOODWIN A.T., CHESTER A.H., YACOUB M.H. Endothelin-1 stimulates proliferation of human coronary smooth muscle cells via the ET(A) receptor and is co-mitogenic with growth factors. Atherosclerosis. 1999;146:351–359. doi: 10.1016/s0021-9150(99)00178-1. [DOI] [PubMed] [Google Scholar]
- HAY D.W. Putative mediator role of endothelin-1 in asthma and other lung diseases. Clin. Exp. Pharmacol. Physiol. 1999;26:168–171. doi: 10.1046/j.1440-1681.1999.03009.x. [DOI] [PubMed] [Google Scholar]
- KOVACS E.J., DIPIETRO L.A. Fibrogenic cytokines and connective tissue production. FASEB J. 1994;8:854–861. doi: 10.1096/fasebj.8.11.7520879. [DOI] [PubMed] [Google Scholar]
- LOVE M.P., MCMURRAY J.J.V. Endothelin in chronic heart failure: current position and future prospects. Cardiovas. Res. 1996;31:665–674. doi: 10.1016/0008-6363(96)00055-7. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R. Endothelin-1 and serotonin: mediators of primary and secondary pulmonary hypertension. J. Lab. Clin. Med. 1999;134:105–114. doi: 10.1016/s0022-2143(99)90114-2. [DOI] [PubMed] [Google Scholar]
- MARINI M., CARPI S., BELLINI A., PATALANO F., MATTOLI S. Endothelin-1 induces increased fibronectin expression in human bronchial epithelial cells. Biochem. Biophys. Res. Commun. 1996;220:896–899. doi: 10.1006/bbrc.1996.0502. [DOI] [PubMed] [Google Scholar]
- MOREAU P., D'USCIO L.V., SHAW S., TAKASE H., BARTON M., LUSCHER T.F. Angiotensin II increases tissue endothelin and induces vascular hypertrophy: reversal by ET(A)-receptor antagonist. Circulation. 1997;96:1593–1597. doi: 10.1161/01.cir.96.5.1593. [DOI] [PubMed] [Google Scholar]
- MÜNTER K., HERGENRÖDER S., UNGER L., KIRCHENGAST M. Oral treatment with an ETA-receptor antagonist inhibits neointima formation induced by endothelial injury. Pharm. Pharmacol. Lett. 1996;2:90–92. [Google Scholar]
- NAKAMICHI K., IHARA M., KOBAYASHI M., SACKI T., ISHIKAWA K., YANO M. Different distribution of endothelin receptor subtypes in pulmonary tissues revealed by the novel selective ligand BQ-123 and (ALA1,3,11,15)ET-1. Biochim. Biophys. Res. Commun. 1992;182:144–150. doi: 10.1016/s0006-291x(05)80123-8. [DOI] [PubMed] [Google Scholar]
- NAKAO A., FUJII M., MATSUMURA R., KUMANO K, SAITO Y., MIYAZONO K., IWAMOTO I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J. Clin. Invest. 1999;104:5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NGUYEN Q.T., CERNACEK P., CALDERONE A., STEWART D.J., PICARD P., SIROIS P., WHITE M., ROULEAU J.L. Endothelin A receptor blockade causes adverse left ventricular remodeling but improves pulmonary artery pressure after infarction in the rat. Circulation. 1998;98:2323–2330. doi: 10.1161/01.cir.98.21.2323. [DOI] [PubMed] [Google Scholar]
- PARK S.H., SALEH D., GIAID A., MICHEL R.P. Increased endothelin-1 in bleomycin-induced pulmonary fibrosis and the effect of an endothelin receptor antagonist. Am. J. Respir. Crit. Care. Med. 1997;156:600–608. doi: 10.1164/ajrccm.156.2.9607123. [DOI] [PubMed] [Google Scholar]
- PICARD P., SMITH P.J.W., MONGE J.C., ROULEAU J.L., NGUYEN Q.T., CALDERONE A., STEWART D.J. Coordinated upregulation of the cardiac endothelin system in a rat model of heart failure. J. Cardiovas. Res. 1998;31 Suppl. 1:S294–S297. doi: 10.1097/00005344-199800001-00082. [DOI] [PubMed] [Google Scholar]
- PRIÉ S., LEUNG T.K., CERNACEK P., RYAN J.W., DUPUIS J. The orally active ET(A) receptor antagonist (+)-(S)-2-(4,6-dimethoxy-pyrimidin-2-yloxy)-3-methoxy-3,3-diphe nyl-propionic acid ( LU135252) prevents the development of pulmonary hypertension and endothelial metabolic dysfunction in monocrotaline-treated rats. J. Pharmacol. Exp. Ther. 1997;282:1312–1318. [PubMed] [Google Scholar]
- RIZVI M.A., MYERS P.R. Nitric oxide modulates basal and endothelin-induced coronary artery vascular smooth muscle cell proliferation and collagen levels. J. Mol. Cell. Cardiol. 1997;29:1779–1789. doi: 10.1006/jmcc.1996.0480. [DOI] [PubMed] [Google Scholar]
- SAKAI S., MIYAUCHI T., KOBAYASHI M., YAMAGUCHI I., GOTO K., SUGISHITA Y. Remarkable improvement in long-term mortality and prevention of unfavourable ventricular remodelling by inhibition of the upregulated myocardial endothelin pathway in rats. Nature. 1996a;384:353–355. doi: 10.1038/384353a0. [DOI] [PubMed] [Google Scholar]
- SAKAI S., MIYAUCHI T., SAKURAI T., YAMAGUCHI I., KOBAYASHI M., GOTO K., SUGISHITA Y. Pulmonary hypertension caused by congestive heart failure is ameliorated by long-term application of an endothelin receptor antagonist: Increased expression of endothelin-1 messenger ribonucleic acid and endothelin-1-like immunoreactivity in the lung in congestive heart failure in rats. J. Am. Coll. Cardiol. 1996b;28:1580–1588. doi: 10.1016/s0735-1097(96)00336-1. [DOI] [PubMed] [Google Scholar]
- SCHIEFFER B., DREXLER H., LING B.N., MARRERO M.B. G protein coupled receptors control vascular smooth muscle cell proliferation via pp60c-src and p21ras. Am. J. Physiol. 1997;272:C2019–C2030. doi: 10.1152/ajpcell.1997.272.6.C2019. [DOI] [PubMed] [Google Scholar]
- SCHIFFRIN E.L. Endothelin and endothelin antagonists in hypertension. J. Hypertens. 1998;16:1891–1895. doi: 10.1097/00004872-199816121-00007. [DOI] [PubMed] [Google Scholar]
- SIME P.J., XING Z., GRAHAM F.L., CSAKY K.G., GAULDIE J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOMA S., TAKAHASHI H., MURAMATSU M., OKA M., FUKUCHI Y. Localization and distribution of endothelin receptor subtypes in pulmonary vasculature of normal and hypoxia-exposed rats. Am. J. Respir. Cell. Mol. Biol. 1999;20:620–630. doi: 10.1165/ajrcmb.20.4.3356. [DOI] [PubMed] [Google Scholar]
- STEWART D.J. Endothelin in cardiopulmonary disease: factor paracrine vs neurohumoral. Eur. Heart. J. 1993;14 Suppl I:48–54. [PubMed] [Google Scholar]
- YANG Z., KRASNICI N., LUSCHER T.F. Endothelin-1 potentiates human smooth muscle cell growth to PDGF: effects of ETA and ETB receptor blockade. Circulation. 1999;100:5–8. doi: 10.1161/01.cir.100.1.5. [DOI] [PubMed] [Google Scholar]
- ZHANG K., PHAN S.H. Cytokines and pulmonary fibrosis. Biol. Signals. 1996;5:232–239. doi: 10.1159/000109195. [DOI] [PubMed] [Google Scholar]