

Abstract
The products released by Helicobacter pylori (H. pylori) in the gastric antral and duodenal mucosa may be involved in mucosal ulceration by stimulating the local formation of cytotoxic factors such as nitric oxide (NO), superoxide or peroxynitrite.
The present study investigates the ability of purified H. pylori lipopolysaccharide (LPS) to induce nitric oxide synthase (iNOS) in rat duodenal epithelial cells following in vivo challenge and its interaction with superoxide in promoting cellular damage and apoptosis.
H. pylori LPS (0.75–3 mg kg−1 i.v. or 3–12 mg kg−1 p.o.) induced a dose–dependent expression of iNOS activity after 5 h in the duodenal epithelial cells, determined by [14C] arginine conversion to citrulline.
The epithelial cell viability, as assessed by Trypan Blue exclusion and MTT conversion, was reduced 5 h after challenge with H. pylori LPS, while the incidence of apoptosis was increased.
The iNOS activity and reduction in cell viability following H. pylori LPS challenge i.v. was inhibited by the selective iNOS inhibitor, 1400 W (0.2–5 mg kg−1 i.v.).
Concurrent administration of superoxide dismutase conjugated with polyethylene glycol (250–500 i.u. kg−1, i.v.), which did not modify the cellular iNOS activity, reduced the epithelial cell damage provoked by i.v. H. pylori LPS, and abolished the increased incidence of apoptosis.
These results suggest that expression of iNOS following challenge with H. pylori LPS provokes duodenal epithelial cell injury and apoptosis by a process involving superoxide, implicating peroxynitrite involvement. These events may contribute to the pathogenic mechanisms of H. pylori in promoting peptic ulcer disease.
Keywords: Helicobacter pylori, nitric oxide, inducible nitric oxide synthase, duodenal epithelial cells, duodenum, lipopolysaccharide
Introduction
Infection with Helicobacter pylori (H. pylori) is a dominant pathogenic factor in peptic ulcer disease (Blaser, 1990). This bacterium colonises the gastric antrum and sites of gastric metaplasia in the duodenum, and induces local inflammation (Carrick et al., 1989). H. pylori infection may provoke damage in the stomach and duodenum by releasing soluble factors that activate inflammatory cells such as neutrophils, to produce cytotoxic mediators such as superoxide (Mooney et al., 1991) and nitric oxide (NO) (McCall et al., 1989). High concentration of NO are known to be cytotoxic, and in combination with the superoxide radical, leads to the subsequent formation of the moieties, peroxynitrite and hydroxyl radicals, which are highly injurious to cells (Ischiropoulos et al., 1995; Beckman et al., 1990).
The inducible isoform of NO synthase (iNOS) is capable of the sustained production of high levels of NO (Knowles et al., 1990; Salter et al., 1991). This isoform can be expressed following challenge with endotoxin lipopolysaccharide (LPS), not only in inflammatory cells, but also in gastro-intestinal epithelial cells and its expression is associated with cytotoxicity (Brown et al., 1994; Tepperman et al., 1993; 1994). Since H. pylori can synthesize an endotoxin (Moran, 1996), expression of iNOS in gastro-duodenal epithelial cells could play a role in the pathogenesis of mucosal lesions related to infection by this organism. Studies on gastric mucosal biopsies from patients with gastritis associated with H. pylori infection exhibited increased antral mRNA for iNOS, as well as iNOS protein in epithelium, endothelium and inflammatory cells, compared with tissue from H. pylori–negative gastritis or controls (Fu et al., 1999). In another report, a correlation was found between the iNOS immunostaining, the degree of inflammation, the apoptotic index and the density of H. pylori infection, all of which decreased on eradication of the bacterium (Hahm et al., 1997).
Previous studies have shown that intravenous challenge with a water extract of H. pylori can express iNOS and lead to epithelial injury in the rat duodenum (Lamarque et al., 1998). These effects were inhibited by polymixin B, which binds LPS (Morrison & Jacobs, 1976), suggesting an important role for an LPS in this process. However, although in vitro studies have shown that H. pylori LPS can lead to the expression of iNOS in murine and human macrophage cell lines in culture, this LPS was only weakly active under those conditions (Perez-Perez et al., 1995). Such findings cast some doubt on the possibility that the expression of iNOS through the actions of the LPS is involved in the pathogenic processes associated with H. pylori infection, although in vitro studies of that nature do have limitations, including lack of cross-talk between different cell types and mediators. The aim of the present study was, therefore, to investigate the ability of a purified preparation of LPS from H. pylori to induce iNOS in duodenal epithelial cells and determine its association with cell damage and apoptosis following its administration in vivo to the rat. As the main objective was to evaluate the potential of the LPS to induce iNOS activity in an experimental setting in vivo, rather than provide a model of clinical infection, the intravenous route was utilized in the majority of the experiments. However, the ability of this LPS to induce iNOS activity and produce cellular injury after its intragastric instillation was also studied.
To evaluate the role of iNOS in the cytotoxic process, the effects of a highly selective inhibitor of iNOS, 1400 W (N-(3-(aminomethyl)benzyl)acetamidine; Garvey et al., 1997; Laszlo & Whittle, 1997) on epithelial cell injury were evaluated. In addition, to explore further the mechanisms underlying such cellular injury, the involvement of the superoxide, and hence peroxynitrite, on epithelial cell injury provoked by the H. pylori LPS was investigated. The effects of a conjugate of superoxide dismutase (SOD–PEG), which has previously been shown to reduce the mucosal injury provoked by local infusion of NO donors in the rat gastric mucosa (Lamarque & Whittle, 1995) was therefore evaluated on the cellular damage and increased apoptosis provoked by the LPS from H. pylori.
Methods
Preparation of LPS
Biomass of H. pylori (NCTC 11637 strain) was grown in brain–heart infusion containing 2% fœtal calf serum to ensure expression of high molecular weight LPS (Walsh & Moran, 1997) Extraction of LPS was performed using a phenol-water procedure (Westphal et al., 1952). Subsequently, extracted LPS was purified by treatment with RNase A, DNase II and proteinase K, and by ultracentrifugation at 100,000×g at 4°C for 18 h (Moran et al., 1992). For suspension, purified LPS was dispersed in endotoxin-free water by sonication.
Animal preparation
Male Wistar rats, weighing 200–250 g, were fasted overnight but allowed free access to water. In the majority of the experiments, purified LPS from H. pylori (0.75–3 mg kg−1) was administered via a tail vein under transient anaesthesia induced by ether. In control experiments, rats were pretreated with saline (0.5 ml kg−1, i.v.).
In a further series of experiments to evaluate the ability of the LPS to induce iNOS after oral challenge, H. pylori LPS (3–12 mg kg−1) dissolved in saline (1.0 ml), was administered intragastrically through a smooth rubber feeding tube.
Duodenal epithelial cell isolation
Duodenal epithelial cells were isolated as described previously (Lamarque et al., 1998; Lentze et al., 1985). A 5 cm segment of duodenum was slowly flushed with 50 ml of a solution containing 0.15 M NaCl and 0.1 mM dithiothreitol (DTT). The segment was then filled with 5 ml of a solution containing (in mM): KCl 1.5, NaCl 96, sodium citrate 27, KH2PO4 8 and Na2HPO4 5.6 (pH 7.3), and the proximal and the distal ends were ligated. The segment was then immersed in phosphate-buffered saline (PBS) kept at 37°C, which was bubbled with 95% O2–5% CO2. After 15 min, the instilled solution was removed and another solution containing 1.5 mM EDTA and 0.5 mM DTT was instilled over 5 min, as described previously (Tepperman et al., 1993). The epithelial cells were collected in suspension in this solution. The cells were washed twice with PBS (pH 7.4) and centrifuged for 5 min at 800×g. The cells were suspended in a buffer containing N-2-hydroxyethylpiperazipine-N′-2-ethanesulphonic acid (HEPES) (10 mM), sucrose (320 mM), DTT (1 mM), soybean trypsin inhibitor (10 μg ml−1), leupeptin (10 μg ml−1), aprotonin (2 μg ml−1).
To assess the purity of epithelial cells in the aliquots isolated from the duodenum, in some experiments the cells were fixed with formaldehyde, stained by hematoxylin-eosin-safran and counted under light microscopy and expressed as the percentage of epithelial cells by fields.
NO synthase activity
NO synthase activity in duodenal epithelial cells was measured as the conversion of L-[14C]-arginine monohydrochloride to [14C]-citrulline, based on the method described previously (Knowles et al., 1990; Lamarque et al., 1998). Cells were homogenized (30 s, Ultra-Turrax; 5 mm blade) in buffer (pH 7.4) containing HEPES (10 mM), sucrose (32 mM), DTT (1 mM), leupeptin (10 μg ml−1), soybean trypsin inhibitor (10 μg ml−1) and aprotonin (2 μg ml−1).
Following centrifugation (10,000×g 4°C), an aliquot of the supernatant (40 μl) was used for the determination of the enzymatic activity and the remaining kept for protein content measurement by a modification of Bradford' method (Lamarque et al., 1998). The aliquot was placed in 100 μl of the pre-warmed incubation buffer containing (final concentration) potassium phosphate (50 mM; pH 7.4), L-valine (50 mM), MgCl2 (1 mM), CaCl2 (200 μM), DL-dithiothreitol (1 mM), L-citrulline (1 mM), NADPH (0.3 mM), FAD (3 μM), FMN (3 μM), BH4 (3 μM) L-[14C]arginine monohydrochloride (15.5 nM) and incubated for 10 min at 37°C. The incubation was terminated by binding arginine following the addition of 500 μl of 1:1 suspension of Dowex (AG 50W-8) in water. The resin was allowed to settle (30 min) and 975 μl of supernatant taken for scintillation counting in 3 ml of scintillation liquid.
Product formation that was inhibited by in vitro incubation with the NO synthase inhibitor NG-monomethyl-L-arginine (L-NMMA; 300 μM), but not by ethylene glycol-bis-(β-amino-ethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA; 1 mM), was taken as an index of iNOS activity (Salter et al., 1991). The constitutive NOS activity (designated cNOS as the nature of the isoform was not established) was taken as that activity inhibited by both L-NMMA and EGTA.
Duodenal epithelial cells viability
The viability of duodenal epithelial cells was determined in cells collected from rats that had been challenged, 5 h previously, with purified H. pylori LPS or saline. The viability of cells was determined by Trypan blue dye exclusion (0.5%, Trypan blue in PBS) as described previously (Tepperman et al., 1991). The number of viable cells was determined by light microscopy (×40 magnification) by counting those cells that excluded the dye. Cells were counted in a randomized manner using a haemocytometer.
Viability of duodenal epithelial cells was determined also by the conversion of the tetrazolium salt, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) to the formazan salt by mitochondrial dehydrogenases (Mosmann, 1983). Briefly, 300 μl of MTT was added to 50 μl of suspension of the homogenized cells prepared as described above and suspended in a buffer containing HEPES (10 mM), sucrose (320 mM), DTT (1 mM), soybean trypsin inhibitor (10 μg ml−1), leupeptin (10 μg ml−1), aprotonin (2 μg ml−1). After 4 h of incubation at 37°C, the suspension was centrifuged (10,000×g, 2 min), the pellet was solubilized in DMSO (1 ml) and centrifuged again. The spectrophotometric absorbance of the formazan salt was measured in the supernatant at 540 nm. Protein content in the initial suspension of homogenized cells was determined as above. Results were expressed as OD mg−1 of protein.
Effects of H. pylori LPS on NO synthase activity and viability in duodenal epithelial cells
At 5 h after administration of H. pylori LPS (0.75–3 mg kg−1 i.v or 3–12 mg kg−1 p.o.), the animals were killed by cervical dislocation. The duodenum was removed, and duodenal epithelial cells isolated for the determination of iNOS activity and cell viability. At this time after LPS (3 mg kg−1, i.v.) challenge, preliminary histological evaluation of the duodenal tissue indicated some areas of epithelial injury.
In further experiments, rats were treated with the selective iNOS inhibitor, 1400 W (0.2–5 mg kg−1 i.v.) or saline, concurrently administered with H. pylori LPS (3 mg kg−1, i.v.). The dose of 1400 W was taken from previous in vivo studies on rat gastrointestinal tissue (Laszlo & Whittle, 1997).
In a separate series of studies, the activity of H. pylori LPS (3 mg kg−1) on iNOS induction and cell viability was compared with that of E. coli LPS (3 mg kg−1), 5 h after intravenous administration.
In a further group of rats, a systemically acting conjugate of polyethylene glycol and superoxide dismutase (SOD–PEG; 250–500 i.u. kg−1) or isotonic saline was administered by an intravenous bolus injection, 15 min prior to H. pylori LPS administration (3 mg kg−1, i.v.). The doses of SOD–PEG were taken from previous studies on its inhibitory action on the inflammatory response in the rat skin following systemic administration (Boughton-Smith et al., 1993) and its action in preventing gastric mucosal injury induced by local intra-arterial infusion of NO donors (Lamarque & Whittle, 1995). The viability, and iNOS activity was determined in duodenal cells from rats treated by SOD–PEG or saline, 5 h after H. pylori LPS administration.
Determination of apoptosis
The degree of apoptosis was determined in duodenal epithelial cells collected from rats that had been challenged, 5 h previously, with purified H. pylori LPS (3 mg kg−1, i.v.) or saline (n=5 for each). The cells were fixed with buffered formaldehyde for 10 min and then incubated with 1 mg ml−1 4,6-diamidino-2-phenylindole-dihydrochloride (DAPI) for 15 min at 37°C. Cells were evaluated by fluorescent microscopy, and nuclei with highly condensed and fragmented chromatin were considered apoptotic. The percentage of apoptotic cells was determined in 15 different fields per preparation of duodenal cells, with 40 cells per field being evaluated.
In a further group (n=5), the effect of pretreatment with SOD–PEG (500 i.u. kg−1 i.v.) on the degree of apoptosis was also evaluated.
Materials
All chemical compounds were obtained from Sigma Chemical Co (Sigma France, St Quentin Fallavier) excepted L-[14C]arginine monohydrochloride obtained from Amersham France (Les Ulis), the scintillation liquid Ready safe from Beckman (Division Biorecherche, Paris Nord II, Villepinte, France) and 4,6-diamidino-2-phenylindole-dihydrochloride (DAPI) from Boehringer, Mannheim, Germany. The SOD–PEG was obtained from Oxis international Inc (NewYork, U.S.A.). 1400 W (N-(3-(aminomethyl)benzyl)acetamidine) was provided as a gift from GlaxoWellcome (Stevenage, U.K).
Statistical analysis
The data are expressed as the mean±s.e.m. of (n) rats per group. Statistical comparisons were made by analysis of variance with the Bonferroni test where P<0.05 was taken as significant.
Results
Induction of iNOS in duodenal epithelial cells after H. pylori LPS
No significant increase in the low basal iNOS activity, determined as that NOS activity which was inhibited by L-NMMA but not by in vitro incubation with EGTA (1 mM), could be detected in the supernatants of the lysed duodenal epithelial cells obtained from animals challenged with saline alone. Following i.v. administration of LPS (0.75–3 mg kg−1), a dose-dependent increase in iNOS activity was detected, determined after 5 h, as shown in Figure 1. This iNOS activity remained at a similar level when determined 7 h after challenge. The basal cNOS activity (705±152 pmol min−1 mg protein−1; n=10) did not change 5 h after challenge with 3 mg kg−1 of LPS (728±172 pmol min−1 mg protein−1; n=10).
Figure 1.
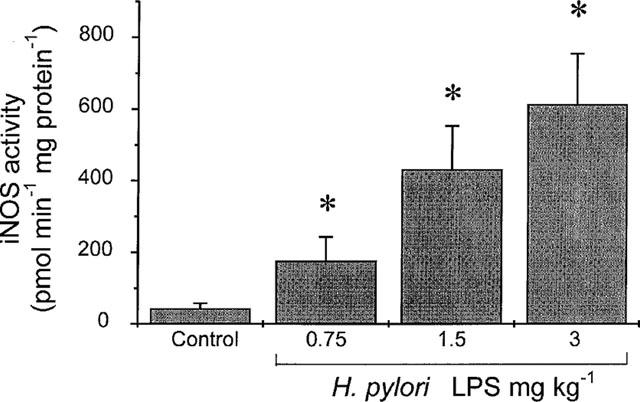
Dose-dependent increase in inducible nitric oxide (iNOS) activity in isolated duodenal epithelial cells, harvested 5 h following challenge with H. pylori lipopolysaccharide (LPS; 0.75–3 mg kg−1, i.v.) in rat. Data, shown as the iNOS activity (pmol min−1 mg protein−1), are mean±s.e.mean of 5–10 experiments, where * denotes a significant difference from the control group (P<0.05).
A significant dose-dependent increase in iNOS activity was also detected 5 h after intragastric administration of LPS, reaching 78±56, 136±82 and 244±82 pmol min−1 mg protein−1 (P<0.05 for each; n=8) following the dose of 3, 6 and 12 mg kg−1 respectively.
Effect of H. pylori LPS on duodenal epithelial cell viability
The proportion of epithelial cells in the cell suspension isolated from the duodenum by dispersion was 98±2% (n=4), as determined by microscopy, the other cells identified by morphological analysis being mastocytes.
The proportion of non-viable intestinal cells isolated from control rats, assessed by Trypan Blue staining, was 13±1%; n=12. The intravenous administration of H. pylori LPS (0.75–3 mg kg−1) provoked a significant dose-dependent increase of the number of non-viable cells when assessed by dye exclusion 5 h later (Figure 2). Likewise, following H. pylori LPS (0.75–3 mg kg−1, i.v.) administration, the percentage of non-viable cells, estimated by MTT conversion, was dose-dependently increased, as shown in Figure 2.
Figure 2.
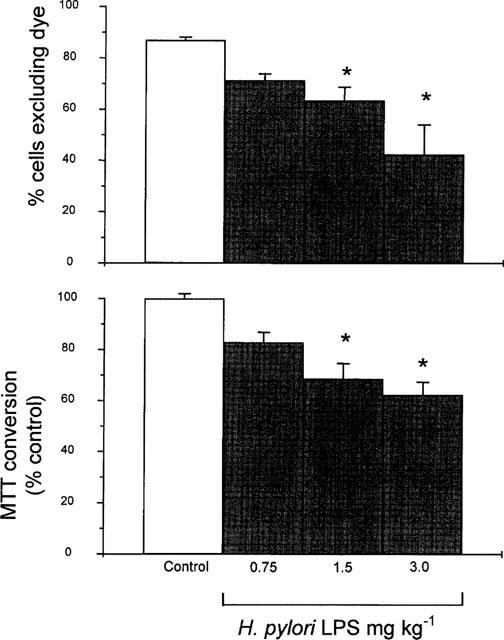
Percentage of viable isolated duodenal epithelial cells, as assessed by Trypan blue exclusion (per cent cells excluding dye; upper graph) or MTT conversion (per cent control; lower graph), 5 h following challenge with H. pylori lipopolysaccharide (LPS; 0.75–3 mg kg−1, i.v.). Data are mean±s.e.mean of 5–12 experiments where * denotes a significant difference from the saline control group (P<0.01).
Following intragastric administration of H. pylori LPS (3, 6 and 12 mg kg−1), the proportion of non-viable intestinal cells as assessed by Trypan blue staining, was 12±1, 15±1 and 20±1% (n=12) respectively, being significantly greater (P<0.05) than the control at doses of 6 and 12 mg kg−1.
Effects of 1400 W
The increase in iNOS activity, determined in the epithelial cells 5 h after intravenous challenge with H. pylori LPS, was inhibited dose-dependently by concomitant administration of 1400 W (0.2–5 mg kg−1, i.v.) as shown in Figure 3. The cNOS activity measured 5 h after LPS challenge was not significantly modified by the treatment with 1400 W (5 mg kg−1, i.v.), being 643±234 and 684±176 pmol min−1 mg protein−1 (n=8) respectively.
Figure 3.
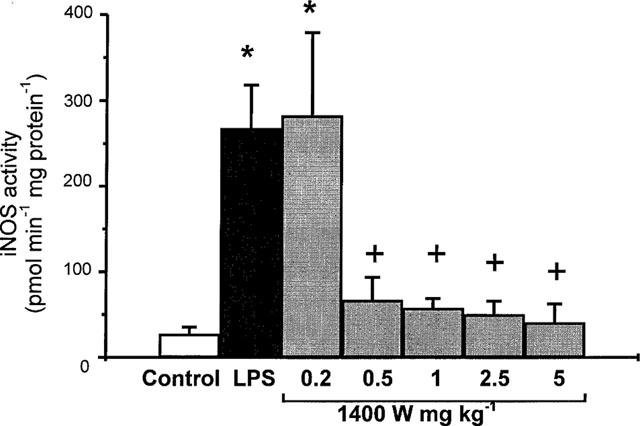
Inducible nitric oxide synthase (iNOS) activity in duodenal epithelial cells 5 h following challenge with H. pylori lipopolysaccharide (LPS; 3 ml kg−1, i.v.) in rat treated concurrently with saline (control) or with N-(3-(aminomethyl)benzyl)acetamidine (1400 W; 0.2–5 mg kg−1, i.v.). Data, shown as iNOS activity (pmol min−1 mg protein−1), are mean±s.e.mean of 5–9 experiments, where * denotes a significant difference from the control (P<0.01) and + a significant difference from the LPS alone group (P<0.01).
The increase in non-viable cells, estimated by Trypan blue dye exclusion or MTT conversion 5 h after the LPS injection, was dose-dependently reduced by concomitant treatment of the rats with 1400 W (0.2–5 mg kg−1, i.v.), as shown in Figure 4.
Figure 4.
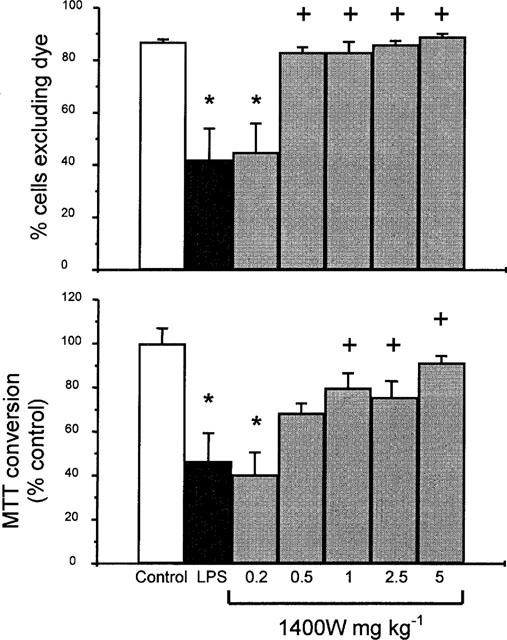
Percentage of viable isolated duodenal epithelial cells, as assessed by Trypan blue exclusion (per cent cells excluding dye; upper graph), or MTT conversion (per cent control; lower graph), 5 h following challenge with H. pylori lipopolysaccharide (LPS; 3 ml kg−1, i.v.) in rat treated concurrently with saline (control) or with N-(3-(aminomethyl)benzyl)acetamidine (1400 W; 0.2–5 mg kg−1, i.v.). Data are mean±s.e.mean of 5–9 experiments, where * denotes a significant difference from the control (P<0.01) and + a significant difference from the LPS alone group (P<0.05).
Comparison of the activity of H. pylori LPS and E. coli LPS on cellular iNOS and damage
In a single-dose comparative study, the increase in iNOS activity in duodenal epithelial cells was Δ 224±34 pmol min−1 mg protein−1 (n=8) 5 h following H. pylori LPS administration (3 mg kg−1 i.v.), whereas that observed after E. coli LPS administration (3 mg kg−1 i.v.) was Δ 463± 27 pmol min−1 mg protein−1 (n=8).
The proportion of non-viable cells, estimated by Trypan blue staining, that had been isolated from rats challenged with H. pylori LPS (27±5%, n=6) was not significantly different than that of E. coli LPS-challenged rats (25±4%, n=6). Likewise, a comparable injurious effect was also found in both groups when the percentage of non-viable cells was assessed by MTT conversion (38±3%, n=6, and 38±4%, n=6, respectively).
Effects of SOD–PEG
Administration of SOD–PEG (500 i.u. kg−1, i.v.), 15 min prior to challenge with H. pylori LPS, did not significantly affect the increase in iNOS activity in duodenal epithelial cells , determined 5 h after challenge (Figure 5). The cNOS activity measured 5 h after H. pylori LPS challenge was likewise not affected following SOD–PEG administration (652±228 and 796±175 pmol min−1 mg protein−1, n=10, respectively).
Figure 5.
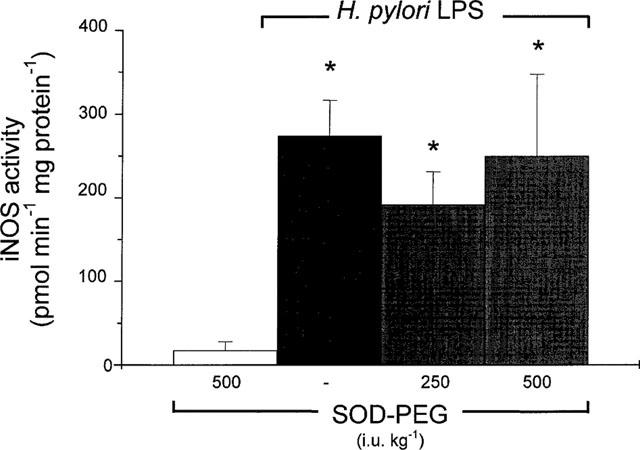
Inducible nitric oxide (iNOS) activity in duodenal epithelial cells, 5 h following challenge with H. pylori lipopolysaccharide (LPS; 3 mg kg−1, i.v.) in rat treated concurrently with saline or with superoxide dismutase conjugated with polyethylene glycol (SOD–PEG; 250–500 i.u. kg−1, i.v.). Data, expressed as iNOS activity (pmol min−1 mg protein−1) are mean±s.e.mean of 5–10 experiments, where * denotes a significant difference from the control (P<0.01). In control experiments, a group of rats received SOD–PEG (500 i.u. kg−1, i.v.). There was no significant difference (P>0.05) between values for LPS alone and LPS with either dose of SOD–PEG.
The proportion of non-viable intestinal cells, assessed by Trypan blue staining, that was isolated from rats treated 5 h previously with SOD–PEG (500 i.u. kg−1, i.v.) alone, was not different from those of cells taken from control rats (12±1%; n=6 compared with 12±2%, n=5). However, the increase in non-viable cells 5 h after H. pylori LPS administration was prevented by pretreatment of the rats with SOD–PEG (250–500 i.u. kg−1, i.v.), 15 min prior to challenge (Figure 6). Likewise, the increase in non-viable cells assessed by MTT conversion after H. pylori LPS challenge was prevented by pretreatment with these doses of SOD–PEG (Figure 6).
Figure 6.
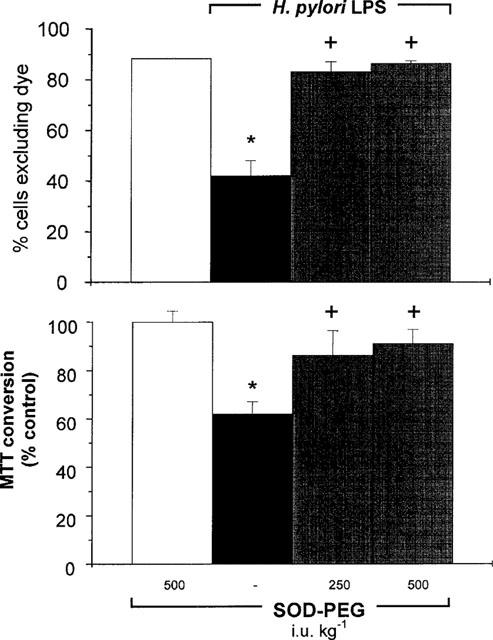
Percentage of viable isolated duodenal epithelial cells, as assessed by Trypan blue exclusion (per cent of cells excluding dye; upper graph), or MTT conversion (per cent control; lower graph), 5 h following challenge with H. pylori lipopolysaccharide (LPS; 3 mg kg−1, i.v.) in rat treated concurrently with saline or with superoxide dismutase conjugated with polyethylene glycol (SOD–PEG; 250–500 i.u. kg−1, i.v.). In control experiments, a group of rats received SOD–PEG (500 i.u. kg−1, i.v.). Data are mean±s.e.mean of 5–10 experiments, where * denotes a significant difference from the control (P<0.05) and + denotes a significant difference from the H. pylori LPS group alone (P<0.05).
Effects of H. pylori LPS and SOD–PEG on apoptosis
The percentage of apoptotic epithelial cells, assessed by the condensed chromatin fragments in the nuclei and by the segmentation of the nuclei after DNA staining, 5 h following H. pylori LPS injection, was significantly increased to 11.0±0.6% (n=5; P<0.05) as compared to the control group (5.3±0.8%; n=5). This increase in the incidence of apoptosis in the cells following LPS challenge was prevented by the pretreatment with SOD–PEG (500 i.u. kg−1, i.v.), 15 min prior to challenge (5.6±0.4%; n=5; P<0.05 compared with LPS alone).
Discussion
In a previous study, iNOS activity was detected in rat duodenal tissue, as well as in isolated duodenal epithelial cells after the intravenous administration of an H. pylori water extract (Lamarque et al., 1998). In the present investigation, a dose-dependent elevation of iNOS activity was observed in rat duodenal epithelial cells 5 h after intravenous challenge with a purified LPS from H. pylori. In addition, dose-dependent iNOS induction following a single intragastric administration of H. pylori LPS was likewise observed in the current study. The higher doses required by this latter route may reflect the requirement of penetration of the LPS through the mucus barrier overlying the epithelium.
Expression of iNOS in colonic and small intestinal epithelial cells following E. coli endotoxin challenge is associated with a reduction in epithelial cell viability (Tepperman et al., 1993; 1994). Likewise, in the present study, a reduction in duodenal epithelial cell viability, determined ex vivo using both Trypan blue dye exclusion and the MTT mitochondrial assay on harvested cells, was observed 5 h after intravenous administration of the H. pylori LPS. An increase in the incidence of apoptosis was also observed in these duodenal cells following challenge with this LPS. Duodenal epithelial cell injury was likewise observed after the single intragastric administration of the LPS. Recent studies have also shown that the repeated intragastric administration of an H. pylori LPS preparation, that induced iNOS in the rat gastric mucosa, could also provoke apoptosis in the gastric epithelial cells (Slomiany et al., 1998a,1998b).
Other studies have demonstrated that duodenal perfusion of an extract of H. pylori reduced alkaline secretion in the rat, considered to reflect inhibition of NO formation by agents formed from local peptidase activity on proteins contained in the crude extract (Fandriks et al., 1997). Comparable effects on alkaline secretion were not seen with extracts of E. coli. However, any such inhibitors would be unlikely to influence the present results obtained following parenteral administration of purified LPS.
Our results suggest that the activity of H. pylori LPS on iNOS expression, as well as cytotoxicity, in duodenal epithelial cells were of a similar order of magnitude to that observed with E. coli, at the dose investigated, although dose-response studies were not conducted. Such a finding in vivo is in contrast to previous studies with LPS in vitro (Perez-Perez et al., 1995; Shapiro & Hotchkiss, 1996). Thus, although H. pylori LPS induced the production of NO from macrophages in culture, it was 2×104 fold less potent than the LPS from E. coli, which may reflect both the nature of the in vitro study and the bone-marrow derived cell type utilized for iNOS expression in those studies.
It has been established that administration in vivo of preparations of H. pylori LPS can provoke the release of pro-inflammatory cytokines, which may also be involved in the pathological processes (Slomiany et al., 1998b). These cytokines released by H. pylori may also act through NO-dependent pathways (Crabtree, 1998) since they are potent inducers of the iNOS enzyme. It is feasible that the initiation of a cascade of pro-inflammatory mediators in vivo that can induce iNOS, may in part, explain the activity seen under these present conditions compared with the low potency of the H. pylori LPS for iNOS expression in vitro, confirming the importance of in vivo models in the understanding of these pathological processes.
In the present study, the reduction in cell viability and the iNOS activity that followed challenge with the H. pylori LPS was inhibited by concurrent administration of 1400 W, known to be a highly selective inhibitor of iNOS (Garvey et al, 1997; Laszlo & Whittle, 1997). Indeed, at the doses of 1400 W that abolished iNOS activity, no greater cellular injury than under control conditions was detected by dye-exclusion or the MTT assay. These findings strongly implicate the involvement of the iNOS activity in the process that lead to the epithelial cell injury following challenge with the LPS. The data also suggest that the levels of iNOS activity, rather than the total NO synthase activity, is of importance in the process of cell injury. This may reflect the prevailing conditions of the microenvironment which lead to iNOS expression, especially the presence of other cytotoxic moieties and the subsequent interactions of the NO so generated.
Intravenous administration of the systemically active conjugate, SOD–PEG, that scavenges superoxide, did not prevent the expression of iNOS activity, but did inhibit the cell damage induced by H. pylori LPS. Such findings, therefore, support the involvement of the superoxide anion, along with NO, in the cell damage induced by H. pylori LPS. These findings also imply that a close correlation between only one potential cytotoxic process, such as iNOS expression alone, would not be anticipated, especially if these processes interact synergistically. In recent studies, the damage induced by intravenous challenge with E. coli LPS in rat small intestinal epithelial cells, using similar techniques as described previously (Lamarque et al., 1998), has also been shown to be attenuated by administration of a SOD-mimetic (Salvamini et al., 1999).
Sonicates of H. pylori have been shown to induce an oxidative burst in human polymorphonuclear and monocytes (Nielsen & Andersen, 1992), and purified LPS has been shown to prime neutrophils for increased activity on subsequent stimulation (Nielsen et al., 1994). In addition, an increased luminol chemiluminesence, which reflects the generation of reactive oxygen species, has been found in the gastric antrum of patients infected by H. pylori (Davies et al., 1994). The local release of these cytotoxic oxygen radicals has hence been suggested to play a role in the mucosal lesions observed in peptic ulcer disease associated with H. pylori (Nielsen & Andersen, 1992; Davies et al., 1994). Increased iNOS expression has also be observed in gastric biopsies from patients with H. pylori-associated gastritis (Hahm et al., 1997; Fu et al., 1999). By interacting with NO formed by iNOS, these oxygen species may form further damaging products such as peroxynitrite that can induce lipid peroxidation (Beckman et al., 1990). Such reactive species may also provoke epithelial cell injury by activating poly (ADP-ribose) synthase that depletes the intracellular energy store (Kennedy et al., 1998). Although superoxide production has not determined in the present study using freshly isolated duodenal epithelial cells, it has been demonstrated in previous studies in rat gastric epithelial cells in culture following ethanol challenge (Hirashi et al., 1999). Moreover, the damage induced by intravenous challenge with E. coli LPS in rat small intestinal epithelial cells has been shown to be reduced by agents that act as peroxynitrite decomposition catalysts (Salvemini et al., 1999), providing support to the involvement of peroxynitrite in such cytotoxicity.
The increase in DNA fragmentation, as an index of apoptosis, observed in duodenal epithelial cells from rats challenged with H. pylori LPS in the present study, was suppressed in cells from rats pretreated with SOD–PEG. These results suggest the involvement of peroxynitrite in the NO-dependent apoptopic process. The mechanism of the apoptosis induced by peroxynitrite in neuronal cells is considered to involve the Bcl-2 pathway and the impairment of the mitochondrial function (Almeida et al., 1998; Keller et al., 1998). It is therefore relevant that mitochondrial function assessed by MTT conversion was diminished after H. pylori LPS challenge which was prevented SOD–PEG pretreatment, suggesting that this cytotoxic process also operates in the duodenal epithelial cells.
The expression of iNOS in duodenal epithelial cells could reflect a host–defence mechanism against colonization by H. pylori, since NO can exert bactericidal actions (Evans et al., 1996; Granger et al., 1988). Furthermore, the present findings indicate that induction of iNOS can provoke local epithelial cytotoxicity as well as stimulating apoptosis, which would thus lead to the clearance of the epithelial cells on which H. pylori was adhering (Kim et al., 1998).
The current findings thus give support to the concept that release of H. pylori LPS in vivo may lead to the local production of elevated concentrations of NO from duodenal epithelium and possibly other mucosal cells, through the expression of iNOS. The findings that the cytotoxic actions and apoptosis in these cells following challenge with H. pylori LPS can be attenuated both by the selective iNOS inhibitor, 1400 W, and by SOD–PEG supports an interactive role of NO and superoxide. Either radical may act independently to cause injury, interacting in a synergistic manner, or may combine to form the reactive species, peroxynitrite, which may underlie them in the cellular injury. If such mechanisms play a role in the pathogenesis of peptic ulceration associated with H. pylori infection, or the subsequent development of mucosal atrophy and increased cancer risk (Hahm et al., 1997; Fu et al., 1999; Tatemichi et al., 1998), pharmacological intervention to attenuate the production of these reactive moieties may be of therapeutic benefit.
Acknowledgments
D. Lamarque was a recipient of a grant from Institut De Recherche Des Maladies de l'Appareil Digestif and A.P. Moran received a grant from the Irish Health Research Board.
Abbreviations
- 1400 W
N-(3-(aminomethyl)benzyl)acetamidine
- cNOS
constitutive isoforms of NO synthase
- DTT
dithiothreitol
- HEPES
N-2-hydroxyethylpiperazipine-N′-2-ethanesulfonic acid
- HES
hematoxylin-eosin-safran
- iNOS
inducible isoform of NO synthase
- LPS
lipopolysaccharide
- PBS
phosphate-buffered saline
- SOD–PEG
conjugate of polyethylene glycol and superoxide dismutase
References
- ALMEIDA A., HEALES S.J.R., BOLANOS J.P., MEDINA J.M. Glutamate neurotoxicity is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione depletion. Brain Res. 1998;790:209–216. doi: 10.1016/s0006-8993(98)00064-x. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., BECKMAN T.W., CHEN J., MARSHALL P.A., FREEMAN B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLASER M.J. Helicobacter pylori and the pathogenesis of gastroduodenal inflammation. J. Infect. Dis. 1990;161:626–633. doi: 10.1093/infdis/161.4.626. [DOI] [PubMed] [Google Scholar]
- BOUGHTON-SMITH N.K., DEAKIN A.M., FOLLENFANT R.L., WHITTLE B.J., GARLAND L.G. Role of oxygen radicals and arachidonic acid metabolites in the reverse passive Arthus reaction and carrageenin paw oedema in the rat. Br. J. Pharmacol. 1993;110:896–902. doi: 10.1111/j.1476-5381.1993.tb13897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN J.F., TEPPERMAN B.L., HANSON P.J., WHITTLE B.J.R. Lipopolysaccharide induces Ca2+-independent nitric oxide synthase activity in rat gastric mucosal cells. Eur. J. Pharmacol. 1994;292:111–114. doi: 10.1016/0926-6917(94)90033-7. [DOI] [PubMed] [Google Scholar]
- CARRICK W.J.A., LEE A., HAZELL S., RALSTON M., DASKALOPOULOS G. Campylobacter pylori, duodenal ulcer, and gastric metaplasia: possible role of functional heterotopic tissue in ulcerogenesis. Gut. 1989;30:790–797. doi: 10.1136/gut.30.6.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRABTREE J.E. Role of cytokines in pathogenesis of Helicobacter pylori-induced mucosal damage. Dig. Dis, Sci. 1998;43:46S–55S. [PubMed] [Google Scholar]
- DAVIES G.R., SIMMONDS N.J., STEVENS T.R.J., SHEAFF M.T., BANATVALA N., LAURENSON I.F., BLAKE D.R., RAMPTON D.S. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut. 1994;35:179–185. doi: 10.1136/gut.35.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS T.J., BUTTERY L.D., CARPENTER A., SPRINGALL D.R., POLAK J.M., COHEN J. Cytokine-treated human neutrophils contain inducible nitric oxide synthase that produces nitration of ingested bacteria. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9553–9558. doi: 10.1073/pnas.93.18.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FANDRIKS L., VON BOTHMER C., JOHANSSON B., HOLM M., BOLIN I., PETTERSSON A. Water extract of Helicobacter pylori inhibits duodenal mucosal alkaline secretion in anesthetized rats. Gastroenterology. 1997;113:1570–1575. doi: 10.1053/gast.1997.v113.pm9352859. [DOI] [PubMed] [Google Scholar]
- FU S., RAMANUJAM K.S., WONG A., FANTRY G.T., DRACHENBERG C.B., JAMES S.P., MELTZER S.J., WILSON K.T. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology. 1999;116:1319–1329. doi: 10.1016/s0016-5085(99)70496-8. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J., KNOWLES R.G. 1400 W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- GRANGER D., HIBBS J.B., PERFECT J.R., DURACK D.T. Specific amino acid (L-arginine) requirement for the microbiostatic activity of murine macrophage. J. Clin. Invest. 1988;81:1129–1136. doi: 10.1172/JCI113427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAHM K.B., LEE K.J., CHOI S.Y., KIM J.H., CHO S.W., YIM H., PARK S.J., CHUNG M.H. Possibility of chemoprevention by the eradication of Helicobacter pylori: oxidative DNA damage and apoptosis in H. pylori infection. Am. J. Gastroenterol. 1997;92:1853–1857. [PubMed] [Google Scholar]
- HIRAISHI H., SHIMADA T., IVEY K.J., TERANO A. Role of antioxidant defenses against ethanol-induced damage in cultured rat gastric epithelial cells. J. Pharmacol. Exptl. Ther. 1999;289:103–109. [PubMed] [Google Scholar]
- ISCHIROPOULOS H., AL-MEDHI A.B., FISHER A.B. Reactive species in ischemic rat lung injury: contribution of peroxynitrite. Am. J. Physiol. 1995;269:L158–L164. doi: 10.1152/ajplung.1995.269.2.L158. [DOI] [PubMed] [Google Scholar]
- KELLER J.N., KINDY M.S., HOLTSBERG F.W., ST CLAIR D.K., YEN H.C., GERMEYER A., STEINER S.M., BRUCE-KELLER A.J., HUTCHINS J.B., MATTSON M.P. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNEDY M., DENENBERG A.G., SZABO C., SALZMAN A.L. Poly(ADP-ribose) synthetase activation mediates increased permeability induced by peroxynitrite in Caco-2BBe cells. Gastroenterology. 1998;114:510–518. doi: 10.1016/s0016-5085(98)70534-7. [DOI] [PubMed] [Google Scholar]
- KIM J.M., ECKMANN L., SAVIDGE T.C., LOWE D.C., WITTHOFT T., KAGNOFF M.F. Apoptosis of human intestinal epithelial cells after bacterial invasion. J. Clin. Invest. 1998;102:1815–1823. doi: 10.1172/JCI2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNOWLES R.G., MERRET M., SALTER M., MONCADA S. Differential induction of brain, lung and liver nitric oxide synthase by endotoxin in the rat. Biochem. J. 1990;270:833–836. doi: 10.1042/bj2700833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMARQUE D., KISS J., TANKOVIC J., FLEJOU J.F., DELCHIER J.C., WHITTLE B.J. Induction of nitric oxide synthase in vivo and cell injury in rat duodenal epithelium by a water soluble extract of Helicobacter pylori. Br. J. Pharmacol. 1998;123:1073–1078. doi: 10.1038/sj.bjp.0701706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMARQUE D., WHITTLE B.J.R. Involvement of superoxide and xanthine oxidase in neutrophil-independent rat gastric damage induced by NO donors. Br. J. Pharmacol. 1995;116:1843–1848. doi: 10.1111/j.1476-5381.1995.tb16672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LASZLO F., WHITTLE B.J. Actions of isoform-selective and non-selective nitric oxide synthase inhibitors on endotoxin-induced vascular leakage in rat colon. Eur. J. Pharmacol. 1997;334:99–102. doi: 10.1016/s0014-2999(97)01163-1. [DOI] [PubMed] [Google Scholar]
- LENTZE M.J., COLONY P.C., TRIER J.S. Glucocorticoid receptors in isolated intestinal epithelial cells in rats. Am. J. Physiol. 1985;249:G58–G65. doi: 10.1152/ajpgi.1985.249.1.G58. [DOI] [PubMed] [Google Scholar]
- MCCALL T.B., BOUGHTON-SMITH N.G., PALMER R.M.J., WHITTLE B.J.R., MONCADA S. Synthesis of nitric oxide from L-arginine by neutrophils. Release and interaction with superoxide anion. Biochem. J. 1989;261:293–296. doi: 10.1042/bj2610293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOONEY C, KEENAN J., MUNSTER D., WILSON I., ALLARDYCE R., BAGSHAW P., CHAPMAN B., CHADWICK V. Neutrophil activation by Helicobacter pylori. Gut. 1991;32:853–857. doi: 10.1136/gut.32.8.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORAN A.P. The role of lipopolysaccharide in Helicobacter pylori pathogenesis. Aliment. Pharmacol. Ther. 1996;10 Suppl 1:39–500. doi: 10.1046/j.1365-2036.1996.22164004.x. [DOI] [PubMed] [Google Scholar]
- MORAN A.P., HELANDER I.M., KOSUNEN T.U. Compositional analysis of Helicobacter pylori rough-form lipopolysaccharides. J. Bacteriol. 1992;174:1370–1377. doi: 10.1128/jb.174.4.1370-1377.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORRISON D.C., JACOBS D.M. Binding of polymyxin B to the lipid portion of bacterial lipopolysaccharides. Immunochem. 1976;13:813–818. doi: 10.1016/0019-2791(76)90181-6. [DOI] [PubMed] [Google Scholar]
- MOSMANN T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- NIELSEN H., ANDERSEN L.P. Chemotactic activity of Helicobacter pylori sonicate for human polymorphonuclear leukocytes and monocytes. Gut. 1992;33:738–742. doi: 10.1136/gut.33.6.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIELSEN H., BIRKHOLZ S., ANDERSEN L.P., MORAN A.P. Neutrophil activation by Helicobacter pylori lipopolysaccharides. J. Infect. Dis. 1994;170:135–139. doi: 10.1093/infdis/170.1.135. [DOI] [PubMed] [Google Scholar]
- PEREZ-PEREZ G.I., SHERPHERD V.L., MORROW J.D., BLASER M.J. Activation of human THP-1 cells and rat bone marrow-derived macrophages by Helicobacter pylori lipopolysaccharide. Infect. Immun. 1995;63:1183–1187. doi: 10.1128/iai.63.4.1183-1187.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALTER M., KNOWLES R.G., MONCADA S. Widespread tissue distribution, species distribution and changes in activity of Ca2+ dependent and Ca2+ independent nitric oxide synthases. FEBS Lett. 1991;291:145–149. doi: 10.1016/0014-5793(91)81123-p. [DOI] [PubMed] [Google Scholar]
- SALVEMINI D., RILEY D.P., LENNON P.J., WANG Z-Q., CURRIE M.G., MACARTHUR H., MISKO T.P. Protective effects of a superoxide dismutase mimetic and peroxynitrite decomposition catalysts in endotoxin-induced intestinal damage. Br. J Pharmacol. 1999;127:685–692. doi: 10.1038/sj.bjp.0702604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAPIRO K.B., HOTCHKISS J.H. Induction of nitric oxide synthesis in murine macrophages by Helicobacter pylori. Cancer Lett. 1996;102:49–56. doi: 10.1016/0304-3835(96)04154-7. [DOI] [PubMed] [Google Scholar]
- SLOMIANY B.L., PIOTROWSKI J., SLOMIANY A. Effect of sucralfate on gastric mucosal inflammatory responses induced by Helicobacter pylori lipopolysaccharide. Scand. J. Gastroenterol. 1998a;33:916–922. doi: 10.1080/003655298750026912. [DOI] [PubMed] [Google Scholar]
- SLOMIANY B.L., PIOTROWSKI J., SLOMIANY A. Induction of caspase-3 and nitric oxide synthase-2 during gastric mucosal inflammatory reaction to Helicobacter pylori lipopolysaccharide. Biochem. Mol. Biol. Int. 1998b;46:1063–1070. doi: 10.1080/15216549800204612. [DOI] [PubMed] [Google Scholar]
- TATEMICHI M., OGURA T., NAGATA H., ESUMI H. Enhanced expression of inducible nitric oxide synthase in chronic gastritis with intestinal metaplasia. J. Clin. Gastroenterol. 1998;27:240–245. doi: 10.1097/00004836-199810000-00012. [DOI] [PubMed] [Google Scholar]
- TEPPERMAN B.L., BROWN J.F., KOROLKIEWICZ R., WHITTLE B.J.R. Nitric oxide synthase activity, viability and cyclic GMP levels in rat colonic epithelial cells. Effects of endotoxin challenge. J. Pharmacol. Exp. Ther. 1994;271:1477–1482. [PubMed] [Google Scholar]
- TEPPERMAN B.L., BROWN J.F., WHITTLE B.J.R. Nitric oxide synthase induction and intestinal epithelial cell viability in rats. Am. J. Physiol. 1993;265:G214–G218. doi: 10.1152/ajpgi.1993.265.2.G214. [DOI] [PubMed] [Google Scholar]
- TEPPERMAN B.L., TAN S.Y., WHITTLE B.J.R. Effects of calcium-modifying agents on integrity of rabbit isolated gastric mucosal cells. Am. J. Physiol. 1991;261:G119–G127. doi: 10.1152/ajpgi.1991.261.1.G119. [DOI] [PubMed] [Google Scholar]
- WALSH E.J., MORAN A.P. Influence of medium composition on the growth and antigen expression of Helicobacter pylori. J. Appl. Microbiol. 1997;83:67–75. doi: 10.1046/j.1365-2672.1997.00164.x. [DOI] [PubMed] [Google Scholar]
- WESTPHAL O., LÜDERITZ O., BISTER F. Uber die extraktion von bakterien mit phenol/wasser. Z. Naturforsch. Sect. B. Chem. Sci. 1952;7:148–155. [Google Scholar]