

Abstract
We investigated the regulatory mechanisms responsible for release of eosinophil cationic protein (ECP) from eosinophils activated by platelet-activating factor (PAF) and monitored intra-cellular pH (pHi) changes using a pH-sensitive fluorescent probe. We also explored the mechanisms by which eosinophils suppress T-lymphocyte proliferation induced by phytohaemagglutinin (PHA). In these experiments, a separated culture to investigate the ECP-mediated pathway and a coculture to identify the adhesion molecules involved in eosinophil-lymphocyte interactions were employed.
Chymostatin (1×10−6 M) inhibited ECP release by about 50% via stimulation by PAF or recombinant interleukin 5(rIL-5) plus IgG.
PAF (1×10−7 M) raised eosinophil pHi from 6.9 to 7.3 within 20 s and pretreatment of these cells with chymostatin (1×10−6 M), but not with leupeptin or E64-d, completely prevented this increase.
Calcium ionophore A23187 (1×10−7 M) induced ECP release and raised pHi to within a range similar to that of PAF, however, chymostatin had no effect on either.
Chymostatin reversed ECP-mediated suppression of PHA-induced T-lymphocyte proliferation in separated cultures, but not in cocultures.
In coculture, eosinophils exhibited the same level of suppression of both CD4+ and CD8+ T-cell proliferation in response to PHA.
Monoclonal antibodies against CD11a, CD18 and CD54, but not CD11b, restored eosinophil suppression of T-lymphocyte proliferation which was chymostatin-resistant in coculture.
Eosinophils were unable to suppress the proliferative response to lymphocytes to anti-CD3 stimulation.
In conclusion, chymostatin specifically inhibited both the eosinophil pHi increase and ECP release induced by PAF. Eosinophils regulate PHA-induced T-lymphocyte proliferation via the ECP-mediation associated with chymotrypsin-like protease activity. These cells also control interactions with lymphocyte between adhesion molecules, CD11a, CD18 and CD54.
Keywords: Adhesion molecules, chymotrypsin-like protease activity, eosinophils, intra-cellular pH, lymphocyte proliferation
Introduction
Eosinophils are implicated in the pathology of allergic reactions in view of their release of various kinds of granule-derived proteins, including eosinophil peroxidase (EPO) (Carlson et al., 1985), eosinophil cationic protein (ECP) (Dahl et al., 1978), eosinophil-derived neurotoxin (EDN) (Durack et al., 1981), and major basic protein (MBP) (Gleich et al., 1979). These act as chemical mediators and their release is induced by certain classes of immunoglobulins, PAF or calcium ionophore (Smith & Cook, 1993). Secretion of these granular proteins is the result of a sequence of cellular events including induction of a Ca2+ influx, involvement of the cyclic AMP-dependent signal transduction pathway (Kita et al., 1991), translocation and fusion of the secretory granules with the plasma membrane, and the swelling of granules (Lawson et al., 1977). It has been reported that a high concentration of diisopropylfluorophosphate, a serine protease inhibitor, can suppress the degranulation process in mast cells (Ishizaka et al., 1985). We have previously reported that eosinophils contain a chymotrypsin-type serine protease with an apparent molecular mass of 28 kDa, and that this protease contributes to degranulation of the EPO from activated human eosinophils (Matsunaga et al., 1994). In the present study, we monitored PAF-induced intracellular pH(pHi) changes in eosinophils, and found that chymostatin exerts a suppressive effect on the pHi increase. Eosinophils may be active in the regulation of T-cell mediated-reactions by virtue of their granule proteins, and they may also control excessive activation of inflammatory cells. It has also been reported that ECP and EDN have an inhibitory effect on mitogen-induced T-lymphocyte proliferation as well as on the mixed leucocyte reaction which occurs concomitantly with eosinophil activation, however, the mechanism(s) involved are unknown (Peterson et al., 1986). We considered possible mechanisms whereby eosinophils inhibit mitogen-induced T-lymphocyte proliferation. For this purpose, we employed two different culture systems to study eosinophil inhibition of phytohemagglutinin(PHA)-induced proliferation of these cells and our results suggest that certain adhesion molecules as well as ECP release triggered by chymotrypsin-like protease activity contributed to the mechanisms involved.
Methods
DNase, Ficoll-Hypaque, PAF, OPD(o-phenylenediamine) and PHA-p were purchased from Sigma Chemicals; chymostatin and leupeptin were obtained from The Peptide Institute (Minoh, Japan); E64-d: N-(L-3-trans ethoxycarbonyloxirane-2-carbonyl)-L-leucine-3-methylbutylamide (Hanada et al., 1978) was a gift from Taisho Pharmaceutical Co. (Tokyo, Japan); horseradish peroxide was from Wako Junyaku Co. (Osaka, Japan); purified human serum IgG was from Organon Teknica; rIL-5 was a gift from Suntory Co. (Osaka, Japan); calcium ionophore A23187 was from Calbiochem; pH probe 2′,7′ Bis (carboxyethyl) carboxyfluorescein tetraacetoxymethyl ester (BCECF-AM) was from Dojindo Laboratories (Kumamoto, Japan). PAF was dissolved in saline at a concentration of 0.1 mg ml−1 with 2.0 mg ml−1 of bovine serum albumin. Chymostatin and E64-d at 10 mM were dissolved in dimethylsulphoxide (DMSO) and kept as a stock solution. These reagents were diluted to specific concentrations with BCECF-AM loading buffer containing 153 mM NaCl, 5 mM KCl, 5 mM glucose and 20 mM HEPES (pH 7.4) without Ca2+ and Mg2+ before use. BCECF-AM was dissolved in DMSO at 1 mM and stored at −20°C until used. The monoclonal antibodies against CD3, CD4, CD8, CD9, CD20, CD11a, CD18, CD11b and CD54, and isotype-matched mouse IgG1 and IgG2a were from Becton Dickinson (CA, U.S.A). Rabbit complement for the cell depletion procedure was purchased from Hoechst, Germany. The ECP RIA-kit was purchased from Pharmacia Fine Chemicals AB (Uppsala, Sweden). Millicell-HA (0.45 μm, 12 mm diameter) was obtained from Millipore Products Division (Bedford, MA, U.S.A.), and 24-multiwell flat-bottomed tissue culture plates were purchased from Becton Dickinson. [3H]-thymidine was purchased from New England Nuclear (Boston, MA, U.S.A.).
Preparation of mononuclear cells and eosinophils from peripheral blood
Heparinized venous blood was obtained from eight patients with eosinophilia. Then, 3% gelatin was added at a ratio of 1:1 and the blood cells were allowed to sediment spontaneously for 20 min at room temperature. The buffy coat layer was collected, overlaid on 20 ml of Ficoll-Hypaque (density, 1.077) and centrifuged for 30 min at 400× g at 4°C. This layer was used as the peripheral mononuclear cell (PMNC) fraction. The bottom layer contained a mixture of eosinophils, neutrophils and erythrocytes. The cells in this bottom layer were washed and the cell pellet was suspended in 4 ml of distilled water, promptly mixed with 1 ml of a 5× concentration of HBSS and centrifuged for 10 min at 150× g to remove lysed erythrocytes. The resulting cell pellet was resuspended in 3 ml of HBSS and again overlaid on a discontinuous metrizamide gradient consisting of 2 ml each of solutions with densities of 1.120, 1.100, 1.090 and 1.080 in HBSS containing 1 mg ml−1 of DNase. The gradient preparation was centrifuged at 400× g for 30 min at 4°C and 2 ml fractions were collected from the bottom of the tube. Eosinophils were collected in fractions with a density of more than 1.090 and were washed twice with HBSS containing 10% FCS. The lymphocyte subpopulations were determined by flow cytometry using monoclonal antibody (moAb): T-cells (CD3+) and B-cells (CD20+). T-cells represented about 75% of the lymphocytes and B-cells were below 10%. We verified the eosinophil fraction by Wright-Giemsa staining and also determined the CD9+ population by flow cytometry analysis and confirmed that it was over 90%. In one experiment, we used CD4+ and CD8+ subpopulations from PMNC. Briefly, PMNC(1×107) were incubated at 4°C with either anti-CD4 or -CD8 moAbs (diluted 1×102) for 45 min and washed twice with RPMI medium.
Rabbit complement was then added to the cell suspension, and incubation was continued at 37°C for another 45 min. After two further washings, each population was depleted of either the CD4+ or CD8+ T-cell population. The viability of each population, tested by trypan blue dye exclusion, exceeded 95%.
Assay of ECP release from eosinophils
ECP release was induced by PAF, IgG-coated beads plus rIL-5, or calcium ionophore A23187 at optimal concentrations (Matsunaga et al., 1994). Eosinophils were preincubated for 15 min at 37°C with chymostatin, leupeptin or E64-d at a concentration of 1×10−6 M. The cells were washed twice to remove the inhibitors and were resuspended at 2×106 cells ml−1 in Tyrode' buffer containing 0.1% BSA. Aliquots of cell suspension (2×106 cells ml−1) were incubated for 15 min with buffer alone, PAF (1×10−7 M) or calcium ionophore A23187(1×10−7 M) or for 4 h with IgG-coated beads (2.5 mg protein ml−1 beads) plus rIL-5 (10 ng ml−1) in a final volume of 500 μl at 37°C. Reactions were stopped by adding 500 μl of ice-cold HBSS and the suspensions were centrifuged at 100× g for 5 min at 4°C. The amount of ECP released into the supernatant was measured with an ECP RIA-kit.
Subcellular localization of BCECF-AM in eosinophils
Eosinophils were suspended in BCECF-AM loading buffer without Ca2+ and Mg2+ at 4×107 cells ml−1 and the cells were incubated for 30 min at 37°C with BCECF-AM at a final concentration of 3 μM. Incubation was carried our under a 5% CO2 in the dark. After washing twice, the cells were resuspended in RPMI medium containing 10% FCS (pH 7.4) at 1×107 cells ml−1. Each of the fractions were prepared as follows (Figure 1). The BCECF-AM-loaded eosinophils in 0.5 ml of 0.25 M sucrose were disrupted by sonication for 25 s on ice, and centrifuged at 540× g for 5 min at 4°C. The precipitate containing the nuclear fraction was designated P-1 and the post-nuclear supernatants, S-1. S-1 was further centrifuged at 12,000× g for 30 min and the precipitate containing eosinophil granules, mitochondria and lysosomes was designated P-2 and the supernatant, S-2. S-2 was further centrifuged at 100,000× g for 60 min. The resulting precipitate containing Golgi apparatus and plasma membranes was designated P-3 and the supernatant containing the cytosol fraction, S-3. Subcellular localization of BCECF-AM was measured fluorometrically with excitation and emission wavelengths of 500 and 530 nm, respectively, in a Hitachi fluorescence spectrophotometer (Model 650-10MS). The EPO contents of each fraction were also measured as a marker of eosinophil granules, as previously reported (Matsunaga et al., 1994).
Figure 1.
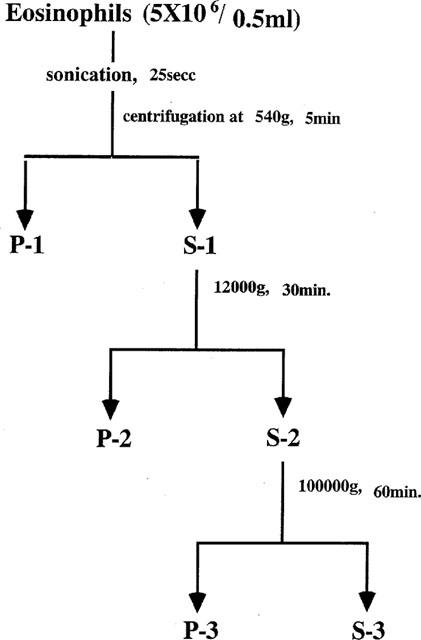
Fractionation of BCECF-AM-loaded eosinophils. BCECF-AM-loaded eosinophils were sonicated and centrifuged as described in the Methods, and the fractions were designated as follows: P-1 (nuclei and undisrupted cells), S-1 (post-nuclear fraction), P-2 (granules, mitochondria and lysosomes), S-2 (post-mitochondria and lysosome fraction), P-3 (Golgi apparatus and plasma membrane) and S-3 (cytosol).
Monitoring of eosinophil pHi changes evoked by PAF or calcium ionophore
A 100 μl suspension of BCECF-AM-loaded eosinophils was dispensed onto an albumin-coated cover slip and incubated for 30 min at 37°C under 5% CO2 in the dark. The slip was then washed twice with isotonic loading buffer and the cells adhering to the slip were used as a sample for pHi monitoring. Before stimulation by PAF or calcium ionophore A23187, the eosinophils were preincubated with protease inhibitors of chymostatin, leupeptin or E64-d at a concentration of 1×10−6 M at 37°C for 30 min. After washing twice, the sample on the cover slip was placed in a chamber kept at a temperature of 37°C under the fluorescence microscope. This chamber was perfused at 3 ml min−1 using a syringe pump. A suction apparatus was used to keep the volume of the solution in the cuvette constant at 1 ml. Fluorescence of a single cell was measured with a microfluorescence photo counter assembled for this work as described previously (Shono et al., 1993). The conditions were as follows: The microscope, an Optiphoto (Nikon, Tokyo, Japan), excitation and emission wavelengths of 360 and 470 nm, respectively; the diameter of the field diaphragm, 40 μm; the spot size, 26 μm; the measurement period, 1 s; the lamp, a high pressure 50 W lamp (Osram Gesellschaft, Berlin-Munich, Germany); PM, photomultiplier (type R649) (Hamamatsu Photonics, Hamamatsu, Japan) cooled at −20°C; photo counter (NF) (Electronic Instruments, Yokohama, Japan); and computer, PC-9801 Vm (NEC) (Tokyo, Japan). For determination of baseline pHi status, eosinophils were monitored until a steady state pHi had been established (within 300 s). DMSO at concentrations of less than 1% by volume has not effect on the pHi baseline of these cells. After achieving the steady state pHi with perfusion of BCECF-AM loading buffer containing 1 mM CaCl2 and 1 mM MgSO4, the perfusate was changed to either PAF or calcium ionophore containing BCECF-AM loading buffer with Ca2+ and Mg2+ at a concentration of 1×10−7 M, at around the 400 to 450 s time point. Then, the fluorescence intensities of the dye-loaded eosinophils on the cover slips were monitored for a period of about 250 s. Each intensity ratio was calibrated to the pHi using the standard K+-nigericin technique (Thomas et al., 1979). The patterns of intracellular pHi change were monitored at 20 s intervals for about 700 s.
Proliferation of PMNC in response to PHA with or without eosinophils
PMNC (5×105 cells ml−1) in complete medium (RPMI 1640+10% FCS) were incubated with PHA-p (2 μg ml−1) for 72 h in the presence or absence of eosinophils (1×105 cells ml−1). We employed two different culture systems in the present study: a coculture and a separated culture. In the coculture, PMNC and eosinophils were cultured together on them same plate, and in the separated culture, eosinophils were plated in culture plate inserts manufactured by Transwell™ (Millicell-HA, 0.45 μm pore size) and incubated in 24-well culture plates with PMNC, so that only soluble factors from the eosinophils, but not cell components, could pass through the membrane filter. We checked for the presence of eosinophils in the lymphocyte population after co-culture using the Transwell™ inserts. We used FACS analysis, in view of the FSc and SSc, to determine the size and characteristics of the cells within the 24-well plate and confirmed at 72 h post-incubation that no migration of eosinophils into the lymphocyte population had occurred. In both culture systems, we added the protease inhibitors chymostatin, leupeptin and E64-d to the cultures at a concentration of 1×10−6 M and incubated these for 72 h. The cultures were pulsed with 1 μCi of [3H]-thymidine for the final 8 h and the incorporation of [3H]-thymidine was determined using a liquid scintillation counter. Cell proliferation, in terms of [3H]-thymidine incorporation, was expressed as counts per minute (c.p.m.). In the coculture system, we also used monoclonal antibodies against adhesion molecules. PMNC and eosinophils were cocultured in the same wells with anti-CD11a, -CD18, -CD11b, -CD54 moAbs and isotype-matched mouse IgG1 and IgG2a. In one experiment, double or triple combinations of anti-adhesion molecule moAbs were added in the coculture system, and these were anti-CD11a+anti-CD18, anti-CD11a+anti-CD54, anti-CD18+anti-CD54 and anti-CD11a+anti-CD18+anti-CD54. These combinations were added at a 1:1000 dilution. [3H]-thymidine incorporation assays were performed as described above. We checked the viability of the eosinophils at the 72 h time point by FACS analysis using FITC-annexin V binding according to the methods of Van Engeland et al. (1998). Briefly, at 72 h post-incubation, eosinophils were resuspended in annexin V buffer (Clontech, Palo Alto, CA, U.S.A.). Propidium iodide (2.5 μg ml−1) and annexin V-fluorescein isothiocyanate at 1 μg ml−1 (Clontech) were added, and the cells were allowed at room temperature for 15 min before flow cytometric analysis. Results showed that at the 72 h time point, no significant cell death had occurred.
T-cell proliferative response to immobilized anti-CD3 in the presence or absence of eosinophils
PMNC were passed through a nylon wool column, and the resulting cells were used as the T-cell population. To determine the proliferative response to immobilized anti-CD3, 1 μg ml−1 of anti-CD3 moAb in Tris buffer was added and the plates were incubated overnight at room temperature. After washing with HBSS, T-cells (5×105 cells) were cultured on the plates with or without eosinophils (1×105 cells) for 72 h under 5% CO2 at 37°C, and the proliferation assay was performed as described above.
Results
Effects of protease inhibitors on ECP release by activated eosinophils
We examined the inhibition of ECP release using a variety of protease inhibitors; chymostatin, an inhibitor of chymotrypsin-type serine proteases, leupeptin, an inhibitor of trypsin-type and E64-d, an inhibitor of cysteine proteases. For these experiments, eosinophils were activated with IgG-coated beads plus rIL-5, PAF or calcium ionophore A23187. Basal ECP release was 8–12 ng ml−1 which corresponds to about 2% of the total cellular ECP content. Stimulation of eosinophils with rIL-5 (10 ng ml−1)+IgG beads (2.5 mg ml−1), PAF (1×10−7 M) or calcium ionophore (1×10−7 M) caused release into the medium of 48.5, 68 and 75 ng ml−1 of ECP per 1×106 eosinophils, corresponding to around 12, 17 and 19%, respectively, of the total cellular ECP content. However, preincubation of eosinophils with chymostatin (1×10−6 M) for 15 min significantly inhibited the ECP release induced with the IgG-coated beads plus rIL-5 (43% decrease) as well as that induced with PAF (47% decrease) (P <0.01) but had no effect the ECP release induced with the calcium ionophore A23187. Leupeptin and E64-d also had a slight effect on ECP release induced with the IgG-coated beads plus rIL-5 or PAF, but in neither case was this significant, and the release induced with the calcium ionophore was unaffected by any of the three inhibitors (Figure 2a,b,c).
Figure 2.
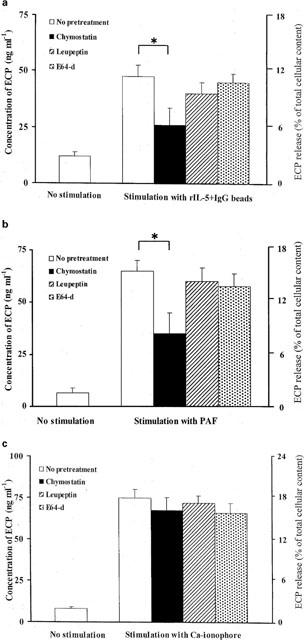
Effects of protease inhibitors on EPC release from activated eosinophils. Eosinophils (1×106 cells ml−1) were preincubated with medium, chymostatin, leupeptin or E64-d at 1×10−6 M for 15 min at 37°C and ECP release was induced with (a) IgG beads (2.5 mg ml−1 beads) plus rIL-5 (10 ng ml−1) after a 4 h incubation or with (b) PAF (1×10−7 M) or (c) calcium ionophore A23187 (1×10−7 M) after a 15 min incubation. The amount of ECP released into the medium was assayed with an ECP RIA kit. Values are expressed as means±s.d. of triplicate samples per assay from eight different donors. The total cellular ECP in eosinophils (405 ng per 1×106 cells) was determined and values were expressed as percentages of the total cellular ECP concentration. Chymostatin produced a significant reduction in ECP release (*P<0.01). Data were analysed by Student' t-test and the range of basal ECP release into the medium under conditions of no pretreatment and no stimulation was 8–12 ng l−1. (n=8).
Determination of the subcellular distribution of BCECF-AM in eosinophils
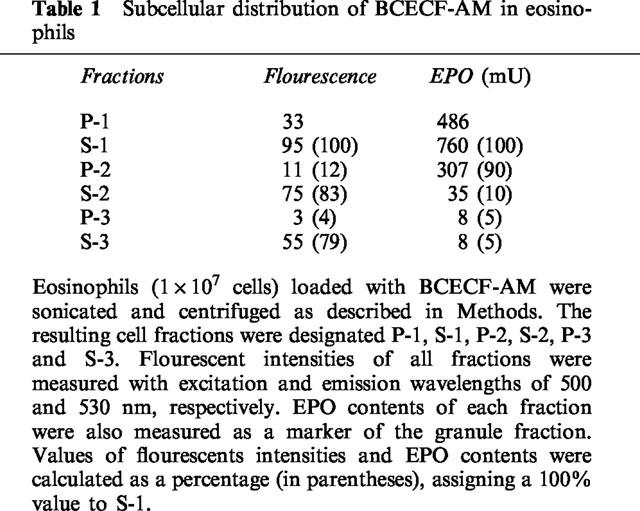
The distribution of fluorescence intensities and EPO contents of each fraction was determined as described in the Methods. Since BCECF-AM is a light-sensitive probe and since the intensity gradually decreased with time after separation, the intensity of each fraction was corrected according to the order of the recovery of the samples and these values are indicated in the parentheses in Table 1. The corrected values clearly indicated that about 80% of the BCECF-AM probe was distributed in the S-3 (cytosol) fraction. Distribution data indicated that about 90% of the EPO in the post-nuclear supernatant was recovered in the P-2 (granules, mitochondria and lysosomes) fraction. These results indicated that the pH probe was distributed mainly in the cytosol and that the cellular fluorescence intensity principally reflected this cytosol pHi change (Table 1).
Table 1.
Subcellular distribution of BCECF-AM in eosinophils
Induction of intracellular pH changes by PAF or calcium ionophore, and the inhibitory effects of protease inhibitors on these changes
For pHi measurement in our present system, alkalization of eosinophils was carried out by exposure to 10 mM NH4Cl and a rise in pHi from 6.94 and 7.25 was noted (Figure 3a). The eosinophil pHi baseline was at about pH 6.94. PAF (1×10−7 M) was found to induce remarkable pHi changes, raising the pH to about 7.3 within 20 s after stimulation without any discernible plateau phase (Figure 3b). Pretreatment of eosinophils with chymostatin at a concentration of 1×10−6 M completely inhibited the pHi changes evoked by PAF, however, leupeptin and E-64d at that same concentration had no effect (Figure 3c,e). Calcium ionophore A23187 (1×10−7 M) also induced a remarkable pHi change, and shifted the pHi from around 6.95 to 7.28 within 20 s, however, chymostatin (1×10−6 M) did not inhibit this pHi shift (Figure 3f,g).
Figure 3.
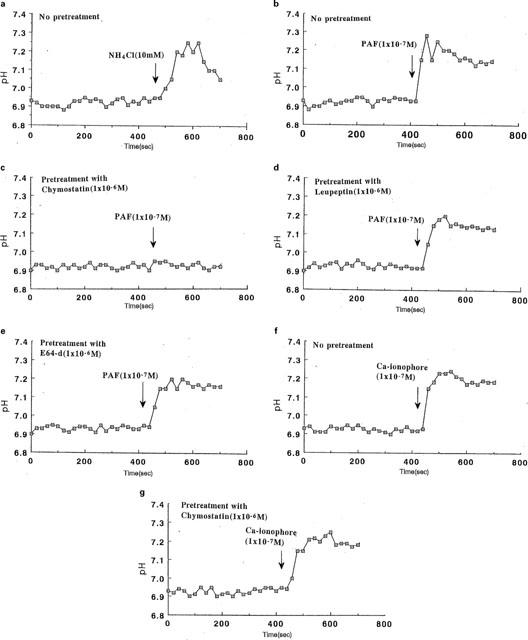
Effects of protease inhibitors on pHi changes in activated eosinophils. Eosinophils were loaded with BCECF-AM as described in the Methods and pHi was monitored at 20 s intervals. Cells were perfused with loading buffer (pH 7.4) containing Ca2+ and Mg2+, and after a steady state was achieved, the perfusate was changed to (a) NH4 Cl(10 mM) or (b) PAF(1×10−7 M) containing buffer. Cells were preincubated with the following protease inhibitors at a concentration of 1×10−6 M for 30 min; (c) chymostatin, (d) leupeptin or (e) E-64d and they were then exposed to PAF-containing perfusate. Cells were exposed to calcium ionophore without pretreatment (f) or with pretreatment with chymostatin (g).
Eosinophils inhibited PHA-induced PMNC proliferation, but not that induced by anti-CD3
To explore the possible mechanisms whereby eosinophils inhibit PMNC proliferation in response to mitogens, we employed two culture systems. In the cell-separated culture system, eosinophils in the upper chamber of the two chamber arrangement inhibited PMNC proliferation induced by PHA by about 40%, but proliferation was restored to 90% of its original level when the former cells were pretreated with chymostatin (1×10−6 M), suggesting that chymostatin-sensitive soluble factors from the eosinophils had contributed to the mechanisms involved in inhibition. However, in the direct coculture system, eosinophils also exhibited a high level of suppression, reducing PMNC proliferation by about 60%, and this effect was not abrogated by the chymostatin pretreatment (Figure 4). We attempted to identify the T lymphocyte subpopulation targeted by eosinophils using the direct coculture system. Both the CD4+ depleted and CD8+ T cell subpopulations were highly proliferative in response to PHA, reaching levels of about 3.3×104 c.p.m. Eosinophils exhibited an equal level of suppression (66% inhibition) with both types of T cells, reducing proliferation to 1.1×104 c.p.m., and chymostatin had no effect on this suppression whatever (data not shown). We concluded that eosinophils targeted both CD4+ and CD8+ populations equally and that these cells inhibit proliferation by mechanisms other than just the ECP pathway. We also examined the ability of eosinophils to suppress T-cell proliferation in response to anti-CD3 since anti-CD3 stimulation involves a different stimulatory pathway from that of PHA. Interestingly, eosinophils exhibited no suppression of PMNC proliferation in response to anti-CD3 stimulation in either culture system (Figure 5).
Figure 4.
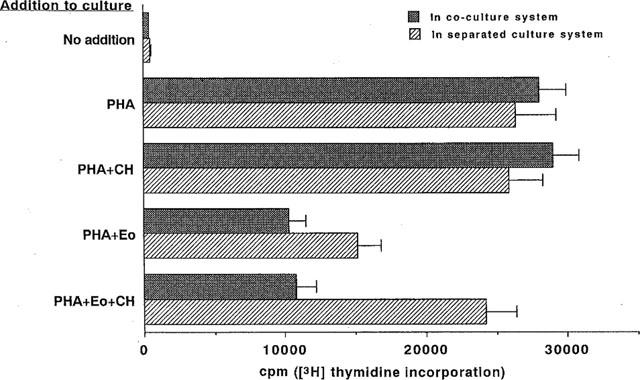
Chymostatin abrogated eosinophil suppression of PMNC proliferation in response to PHA in the separated culture system. PMNC (5×105 cells ml−1) were incubated with PHA-p (2 μg ml−1) for 72 h in the presence or absence of autologous eosinophils (1×105 cells ml−1) with or without chymostatin (CH) at 1×10−6 M in the coculture or separated culture systems. The cultures were pulsed with 1 μCi of [3H]-thymidine for the final 8 h and the incorporation of [3H]-thymidine was assayed using a liquid scintillation counter. [3H]-thymidine incorporation was expressed as counts per minute (c.p.m.). (n=8).
Figure 5.
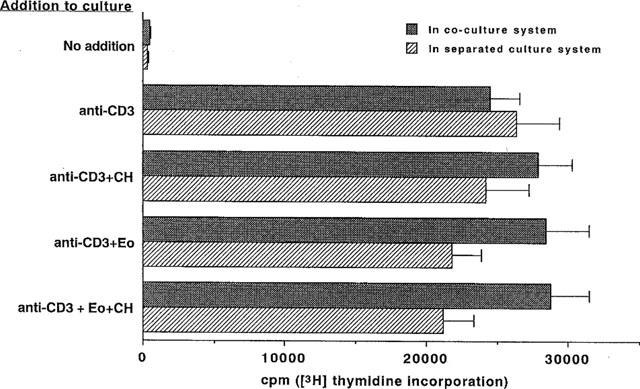
Eosinophils exhibited no suppression of the PMNC proliferative response to anti-CD3 stimulation. PMNC (5×105 cells ml−1) were cultured in immobilized anti-CD3-coated wells for 72 h in the presence or absence of eosinophils (1×105 cells ml−1) in the coculture or separated culture system. The cultures were pulsed with 1 μCi of [3H]-thymidine and the incorporation was assayed as described above. Data were expressed as counts per minute. (n=6).
Adhesion molecules restore eosinophil inhibition of PMNC proliferation in the coculture system
Since chymostatin could not abrogate the ability of eosinophils to suppress PMNC proliferation in the coculture system, we explored other possible mechanisms by which eosinophils suppress proliferation in response to PHA. We first tested anti-adhesion molecule moAbs in coculture and found that addition of either anti-CD11a, -CD18 or -CD54 moAbs could restore the suppressed PMNC proliferation by around 60%, although anti-CD11b moAB could not, suggesting that CD11a, CD18 and CD54 might be involved in the inhibition mechanisms (Figure 6a). On using double or triple combinations of anti-adhesion molecule moAbs, as described in the Methods, we did not observe complete reversal although up to 70% restoration with the triple combination (Figure 6b).
Figure 6.
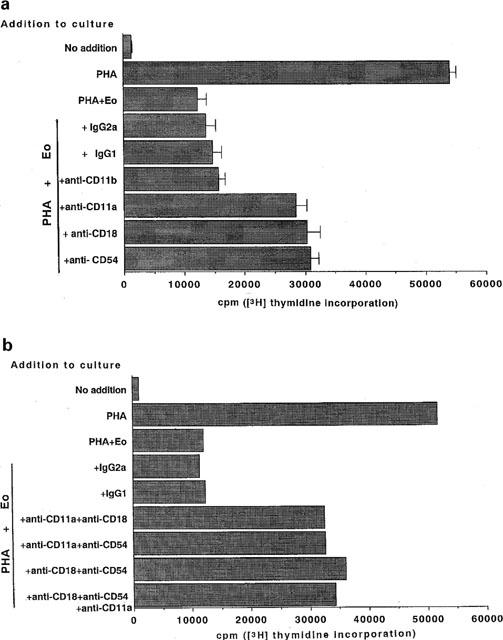
Anti-adhesion molecule moAbs partially restored eosinophil inhibition of PMNC proliferation in the coculture. (a) Anti-CD11a, -CD11b, -CD18, or -CD54 moAB was added at a dilution of 1:1000 to the coculture system of PMNC (5×105 cells ml−1) with PHA-p (2 μg ml−1) and incubated for 72 h in the presence of autologous eosinophils (1×105 cells ml−1). The cultures were pulsed with 1 μCi of [3H]-thymidine and the incorporation of [3H]-thymidine was assayed using a liquid scintillation counter. Cell proliferation was expressed as counts per minute (c.p.m.) (n=6). (b) Double or triple combinations of anti-CD11a, -CD18, and -CD54 moAbs were added to the coculture at a dilution of 1:1000, as described in Methods, and the incorporation of [3H]-thymidine was assayed. Each value indicates a mean of two donors.
Discussion
Cytoplasmic pH and intracellular calcium play important roles in the regulation of a variety of cell functions. Furthermore, an association between calcium ion influx (Cai2+) and pHi has been reported in number of cell systems. As an example, cytoplasmic alkalization has been shown to increase Cai2+ in rat lymphocytes (Grinstein & Goetz, 1985). In the present study, we investigated the effects of various types of protease inhibitors on eosinophil pHi changes evoked by PAF or calcium ionophore. PAF is produced by many types of cells involved in inflammation, including neutrophils, monocytes, alveolar macrophages, platelets and eosinophils (Kroegel et al., 1989a). It has been reported that PAF induces a rise in Cai2+ in eosinophils (Kroegel et al., 1989b), and our present data indicate that PAF also induced pHi alkalization in these cells. These findings support a link between this alkalizing change in pHi and the rise in Cai2+ that has been seen in a variety of cell systems (Tasaka et al., 1987). In the present study, we demonstrated that chymostatin completely inhibited the alkalizing change in eosinophil pHi evoked by PAF, whereas, at the same concentrations, leupeptin or E64-d did not. These results were compatible with the chymostatin inhibition of ECP release, suggesting that alkalization in eosinophils might be closely associated with the degranulation process. It has been reported that PAF induced Ca2+ release from intracellular Ca2+ stores via the IP3 pathway (Berridge et al., 1987). Calcium ionophore induced the same rise in eosinophil pHi that was seen with PAF and this effect was resistant to chymostatin, however, in the case of PAF, the pHi changes were inhibited by chymostatin, suggesting that this inhibitor might suppress the release of Ca2+ from the intracellular Ca2+ stores.
In the present study, we focused on the mechanisms whereby eosinophils inhibit lymphocyte proliferation in response to PHA. As expected from our previous study of EPO release (Matsunaga et al., 1994), chymostatin was also found to inhibit ECP release from eosinophils activated by PAF- or IgG plus rIL5. Using the separated culture system, we confirmed that these cells suppress PMNC proliferation via ECP release, and that chymostatin can abrogate this inhibitory activity, suggesting that chymotrypsin-like serine proteases in eosinophil granules also regulate ECP release from the activated forms of these cells. However, in the coculture system, chymostatin was unable to reverse the suppression of PMNC proliferative response to PHA, suggesting that eosinophils utilize inhibitory mechanisms other than solely that of ECP release. We explored the possible involvement of adhesion molecules in these mechanisms. Leukocyte function-associated antigen-1 (LFA-1), namely CD11a (LFA-1a) and CD18(LFA-1b), belongs to the β2 integrin family (Rothlein et al., 1988a) and its expression is limited to leucocytes, including lymphocytes and eosinophils (Patarroyo et al., 1989). LFA-1 mediates a wide range of adhesion-dependent functions and anti-LFA-1 moAb can inhibit virtually every immune phenomenon that involves T cells (Larson & Springer, 1990). CD11b(Mac-1) also belongs to the β2 integrin family (Diamond et al., 1991), and is expressed on the surfaces of neutrophils, eosinophils, monocytes, and approximately 30% of peripheral blood lymphocytes (Patarroyo & Makgoba, 1989). CD54 (ICAM-1) is a member of the immunoglobulin gene family (Staunton et al., 1988) and consists of five extracellular immunoglobulin-like-domains (Staunton et al., 1989). It is widely expressed on several cell types, including lymphocytes (Rothlein et al., 1988b). Although CD54 can act as a counterstructure for LFA-1 and CD11b, the binding sites for CD11b and LFA-1 on CD54 are different; CD11b is mapped to the third NH2-terminal immunoglobulin domain (Marlin et al., 1987), whereas LFA-1 is mapped to both the first and second NH2-terminal immunoglobulin domains (Staunton et al., 1990). In the present study, antibodies against CD11a, CD18 and CD54 had almost the same ability to reduce the inhibitory effect of eosinophils, but anti-CD11b was unable to reduce it at all, indicating that the first and second, but not the third NH2-terminal immunoglobulin domain on CD54 may be involved, and that eosinophil interaction with LFA-1 and CD54 is essential for eosinophil inhibition of PMNC proliferation in response to PHA. However, various combinations of anti-adhesion molecule moAbs could not completely restore eosinophil inhibition of PMNC proliferation. For a complete reversal, we speculate that other mechanisms may be involved, including eosinophil production of eicosanoids through oxidative metabolism of arachidonic acid via the cyclooxygenase and lipoxygenase pathways and that these mechanisms may act in modulating lymphocyte proliferation (Shapiro & Meydani, 1993). Further studies are required to completely elucidate the mechanisms underlying this phenomenon. We further investigated the T-cell subpopulations targeted by eosinophils in our coculture system, and found that subpopulations of CD8+ and CD4+ were both equally affected by these cells. We also investigated the anti-CD3 stimulation pathway in the coculture system since this pathway does not require antigen presenting cells (APC) for T-cell activation. Our present data suggest that eosinophils failed to inhibit PMNC proliferation via anti-CD3 stimulation, and that the means by which eosinophils suppress PMNC proliferation might be limited to stimulation mediated by APC.
Accordingly, we suggest at least two mechanisms whereby eosinophils exert inhibitory effects on lymphocyte proliferation in response to PHA: one involves interactions between CD11a, CD18 and CD54 and, the second involves ECP release, accompanied by cytoplasmic [pHi] changes, which is regulated by chymotrypsin-like serine proteases.
Abbreviations
- BCECF-AM
2′,7′ Bis (carboxyethyl) carboxy-fluorescein tetra-acetoxymethyl ester
- DMSO
dimethylsulphoxide
- ECP
eosinophil cationic protein
- EDN
eosinophil-derived neurotoxin
- EPO
eosinophil peroxidase
- ICAM-1
intercellular adhesion molecule-1
- LFA-1
leukocyte function-associated antigen-1
- Mac-1
macrophage-1 antigen
- MBP
major basic protein
- PAF
platelet-activating factor
- PHA
phytohaemagglutinin
- pHi
intra-cellular pH
- PMNC
peripheral mononuclear cell
- rIL-5
recombinant interleukin-5
References
- BERRIDGE M.J. Inositol trisphosphate and diacylglycerol: two interacting second messengers. Annu. Rev. Biochem. 1987;56:159–193. doi: 10.1146/annurev.bi.56.070187.001111. [DOI] [PubMed] [Google Scholar]
- CARLSON M.G.Ch., PETERSON C.G.B., VENGE P. Human eosinophil peroxidase: Purification and characterization. J. Immunol. 1985;134:1875–1879. [PubMed] [Google Scholar]
- DAHL R., VENGE P., OLSSON I. The content of eosinophil cationic protein in eosinophil leukocytes. Study on normal controls and patients with bronchial asthma. Allergy. 1978;33:152–154. doi: 10.1111/j.1398-9995.1978.tb01526.x. [DOI] [PubMed] [Google Scholar]
- DIAMOND M.S., STAUNTON D.E., MARLIN S.D., SPINGER T.A. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglubin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- DURACK D.T., ACKERMAN S.J., LOEGERING D.A., GLEICH G.J. Purification of human eosinophil-derived neurotoxin. Proc. Natl. Acad. Sci. U.S.A. 1981;78:5165–5169. doi: 10.1073/pnas.78.8.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLEICH G.J., FRIGAS E., LOEGERING D.A., WASSOM D.L., STEINMULLER D. Cytotoxic properties of the eosinophil major basic protein. J. Immunol. 1979;123:2925–2927. [PubMed] [Google Scholar]
- GRINSTEIN S., GOETZ J.D. Control of free cytoplasmic calcium by intracellular pH in rat lymphocytes. Biochem. Biophys Acta. 1985;819:267–270. doi: 10.1016/0005-2736(85)90183-x. [DOI] [PubMed] [Google Scholar]
- HANADA K., TAMAI M., OHMURA S., SAWADA J., SEKI T., TANAKA I. Structure and synthesis of E-64, a new thiol protease inhibitor. Agric. Biol. Chem. 1978;42:529–536. [Google Scholar]
- ISHIZAKA T., STERK A.D., DAERON M., BECKER E.L., ISHIZAKA K. Biochemical analysis of desensitization of mouse mast cells. J. Immunol. 1985;135:492–501. [PubMed] [Google Scholar]
- KITA H., ABU-GHAZALEH R.I., GLEICH G.J., ABRAHAM R.T. Regulation of Ig-induced eosinophil degranulation by adenosine 3′,5′-cyclic monophosphate. J. Immunol. 1991;146:2712–2718. [PubMed] [Google Scholar]
- KROEGEL C., PLEASS R., YUKAWA T., CHUNG K.F., WESTWICK J., BARNES P.J. Characterization of platelet-activating factor-induced elevation of cytosolic free calcium concentration of eosinophils. FEBS Lett. 1989b;243:41–46. doi: 10.1016/0014-5793(89)81214-1. [DOI] [PubMed] [Google Scholar]
- KROEGEL C., YUKAWA T., DENT G., VENGE P., CHUNG K.F., BARNES P.J. Stimulation of degranulation from human eosinophils by platelet-activating factor. J. Immunol. 1989a;142:3518–3526. [PubMed] [Google Scholar]
- LARSON R.S., SPRINGER T.A. Structure and function of leukocyte integrins. Immunol. Rev. 1990;114:181–217. doi: 10.1111/j.1600-065x.1990.tb00565.x. [DOI] [PubMed] [Google Scholar]
- LAWSON D., RAFF M.C., GOMPERTS B., FEWTRELL C., GILULA N.B. Molecular events during membrane fusion. A study of exocytosis in rat peritoneal mast cells. J. Cell Biology. 1977;72:242–259. doi: 10.1083/jcb.72.2.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARLIN S.D., SPRINGER T.A. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–819. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- MATSUNAGA Y., KIDO H., KAWAJI K., KAMOSHITA K., KATUNUMA N., OGURA T. Inhibitors of chymotrypsin-like protease inhibit eosinophil peroxidase release from activated human eosinophils. Arch. Biochem. Biophys. 1994;321:67–74. doi: 10.1006/abbi.1994.1281. [DOI] [PubMed] [Google Scholar]
- PATARROYO M., MAKGOBA M.W. Leucocyte adhesion to cells: molecular basis, physiological relevance, and abnormalities. Scand. J. Immunol. 1989;30:129–164. doi: 10.1111/j.1365-3083.1989.tb01197.x. [DOI] [PubMed] [Google Scholar]
- PETERSON C.G.B., SKOOG V., VENGE P. Human eosinophil cationic proteins (ECP and EPX) and their suppressive effects on lymphocyte proliferation. Immunobiology. 1986;171:1–13. doi: 10.1016/S0171-2985(86)80013-4. [DOI] [PubMed] [Google Scholar]
- ROTHLEIN R., CZAJKOWSKI M., O'NEILL M.M., MARLIN E.M., MERLUZZI V.J. Induction of intercellular adhesion molecule 1 on primary and continuous cell lines by pro-inflammatory cytokines. J. Immunol. 1988a;141:1665–1669. [PubMed] [Google Scholar]
- ROTHLEIN R., DUSTIN M.L., MARLIN S.D., SPRINGER T.A. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J. Immunol. 1988b;137:1270–1274. [PubMed] [Google Scholar]
- SHAPIRO A.C., MEYDANI S.N. Eicosanoids derived from arachidonic and eicosapentaenic acids inhibit T cell proliferative response. Prostaglandins. 1993;45:229–240. doi: 10.1016/0090-6980(93)90049-d. [DOI] [PubMed] [Google Scholar]
- SHONO M., YAMADA M., YAMAGUCHI H., MIYAMOTO H. Aerobic responses of mouse macrophages to phagocytosis against Escherichia coli as revealed by microphotometry. Cell. Mol. Biol. 1993;39:361–370. [PubMed] [Google Scholar]
- SMITH H., COOK R.M. The Handbook of Immunopharmacology. CA. Academic Press; 1993. Immunophalmacology of eosinophils; pp. 43–55. [Google Scholar]
- STAUNTON D.E., DUSTIN M.L., SPRINGER T.A. Functional cloning of CAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature. 1989;339:61–64. doi: 10.1038/339061a0. [DOI] [PubMed] [Google Scholar]
- STAUNTON D.E., DUSTIN M.L., ERICKSON H.P., SPRINGER T.A. The arrangement of the immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinovirus. Cell. 1990;61:243–254. doi: 10.1016/0092-8674(90)90805-o. [DOI] [PubMed] [Google Scholar]
- STAUNTON D.E., MARLIN S.D., STRATOWA C., DUSTIN M.L., SPRINGER T.A. Primary structure of intercellular adhesion molecule 1 (ICAM-1) demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell. 1988;52:925–933. doi: 10.1016/0092-8674(88)90434-5. [DOI] [PubMed] [Google Scholar]
- TASAKA K., MIO M., OKAMOTO M. The role of intracellular Ca2+ in the degranulation of skinned mast cells. Agents. Actions. 1987;20:157–160. doi: 10.1007/BF02074656. [DOI] [PubMed] [Google Scholar]
- THOMAS J.A., BUCHSBAUM R.N., ZIMNIAK A., RACKER E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probe generated in situ. Biochemistry. 1979;18:2210–2218. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- VAN ENGELAND M., NIELAND L.J.W., RAMAEKERS F.C.S., SCHUTTE B., REUTELING SPERGER C.P.M. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry. 1998;31:1–9. doi: 10.1002/(sici)1097-0320(19980101)31:1<1::aid-cyto1>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]