

Abstract
Neurally evoked contractions and release of 3H- acetylcholine (ACh) during electrical field stimulation were measured in rat urinary bladder strips.
The α1 agonist phenylephrine (PE, 2–8 μM) increased the amplitude of neurally evoked contractions, facilitated the release of ACh and increased the baseline tone of the bladder strips.
The PE-induced facilitation of the contractions did not significantly change during a prolonged exposure to PE (120 min), whereas the PE-induced rise in baseline tone gradually decreased to 65% of the initial value.
Low concentrations of specific α1A antagonists, 5-methyl urapidil (5-MU), REC15/2739 and WB-4101 competitively inhibited the facilitation of the neurally-evoked contractions (pA2: 8.77; 9.59 and 9.62, respectively), whereas higher concentrations of 5-MU (IC50: 48 nM) were required to suppress the PE-rise in baseline. WB-4101 (100 μM) inhibited the PE-induced facilitation of ACh release.
The irreversible α1B antagonist chloroethyl-clonidine (CEC, 10–50 μM) inhibited the PE-evoked rise in base line tone, but did not affect the PE-induced facilitation of the neurally evoked contractions nor the facilitation of ACh release. However, CEC increased the area and amplitude of the neurally-evoked contractions by 261±33 and 47.2±8.4%, respectively. Atropine significantly inhibited the CEC evoked increase in area and amplitude of the electrically evoked contractions (76.5±4.8 and 40.8±3%, respectively) indicating that CEC facilitated the cholinergic responses of the electrically stimulated bladder strips.
It is concluded that α1A and CEC sensitive α1B and/or α1D adrenoceptors are expressed in the rat bladder in different locations. On the cholinergic nerve terminals α1A adrenoceptors mediate prejunctional facilitation, whereas postjunctional α1B/α1D adrenoceptors mediate smooth muscle contraction.
Keywords: α1 Adrenoceptor subtypes, urinary bladder, neurally evoked contractions, ACh release
Introduction
Three subtypes of α1 adrenoceptors are present in peripheral tissues: α1A, α1B and α1D (Hieble et al., 1995). These subtypes can be identified pharmacologically with antagonists that are selective for α1A (5 methyl-urapidil;5-MU) for α1D (BMY 7378) or more effective for α1B and α1D adrenoceptors (chloroethyl-clonidine; CEC) (Han et al., 1987; Hieble et al., 1995). In the rat bladder all three subtypes of α1 adrenoceptors have been detected in the smooth muscle (Malloy et al., 1998). In addition α1 adrenoceptors are located prejunctionally on cholinergic terminals in the rat urinary bladder (Somogyi et al., 1995, de Groat et al., 1999). Activation of prejunctional α1 adrenoceptors facilitates acetylcholine (ACh) release and enhances neurogenic contractions, whereas, activation of α1 adrenoceptors in the smooth muscle increases basal tone (Ordway et al., 1986; Somogyi et al., 1995; Suzuki et al., 1999). The non-selective α1 adrenoceptor antagonist, terazosin, inhibited phenylephrine (PE)-induced facilitation of the neurally evoked contractions and facilitation of ACh release as well as the PE-evoked increase in the basal tone (Somogyi et al., 1995). The types of α1 adrenoceptors mediating the pre-and postjunctional effects of PE in the urinary bladder are not known.
In the present study we used subtype selective antagonists to examine the α1 adrenoceptor subtypes located pre- and postjunctionally in the rat bladder. A preliminary report of some of these results has been published in an abstract (Somogyi et al., 1999).
Methods
Adult female rats (at least 7 months old; 350–450 g) were used for these experiments. The bladder was removed from the abdomen following decapitation and two to four circular slices were cut from the bladder body. Bladder strips weighing 15–20 mg were mounted in a double jacketed organ bath at 37°C in Krebs solution (mM: NaCl 113, KCl 4.7, CaCl2 1.25, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, glucose 11.5) and constantly bubbled with a mixture of 95% O2 and 5% CO2.
Contractile experiments
The initial tension was set at 10 mN and isometric contractions were measured with strain-gauge transducers and recorded with a computerized data acquisition program (Windaq, DATAQ Instruments Inc, Akron, OH, U.S.A). Electrical field stimulation with a Grass 88 stimulator (Grass, ASTROMED, RI, U.S.A.) was delivered through platinum electrodes inserted from the top and bottom of the organ bath and separated by 4 cm. A stimulus intensity-response curve was constructed at the beginning of each experiment and unless otherwise stated the preparations were stimulated (20 Hz with 0.25 ms pulse duration) at a voltage producing 50% of the maximal response. Long (100 shocks) and short (10 shocks) duration trains of stimuli were used. The amplitudes and areas of the stimulation-evoked contractions were computed by the WindaqEx program (DATAQ).
Unless otherwise stated the α1-adrenoceptor agonist, PE was added to the bath at 8–10 min intervals in increasing concentrations to construct cumulative dose-response curves in the absence or presence of various concentrations of α1-adrenoceptor antagonists, which were added to the bath 20 min before each PE cumulative dose response curve.
ACh release
Tissue slices were placed into an incubation bath with 1 ml of Krebs solution containing 5 uCi ml−1 3H-choline (80 Ci mmol−1), and constantly bubbled with a mixture of 95% O2 and 5% CO2 for 30 min at 37°C. Each slice was suspended in a separate bath after incubation and superfused at a rate of 0.3 ml min−1 with oxygenated Krebs solution. After 60 min washing (precollection period), 3 min effluents were collected with a fraction collector. All drugs except CEC were added to the perfusion solution 15 min before the stimulation and were kept in the solution until the end of the experiments. In experiments with CEC, the drug was added to the solution for 30 min, then it was washed out 15 min before the stimulation. When PE was applied to CEC treated preparations it was added to the perfusion solution after CEC was washed out. Electrical field stimulation (10 Hz, 100 shocks, 100 V, 0.25 ms stimulus duration) was delivered by a Grass S-88 stimulator through platinum plate electrodes positioned at the top and the bottom of the perfusion bath (Somogyi et al., 1995). The strips were stimulated either once (experiments with WB 4101) or twice (CEC experiments) with the above parameters. When two stimulation paradigm was used, the time interval between the two stimulations was 50 min. The radioactivity in the effluent was measured by using Optifluor scintillation reagent (Packard Industries) with a Beckman Spectrometer (Model 5801). The measured counts were corrected to absolute activity using an external standard technique. To determine the total uptake of radioactivity, the acid soluble radioactive content of the tissue slices was determined after the experiments by placing them into 1 ml of 0.1 N perchloric acid for 16 h and an aliquot of 0.1 ml was removed for counting. The released amount of radioactivity for each sample was expressed as per cent of the tissue radioactive content (fractional release). The net evoked release of ACh reflects the number of counts or fractional release contained in the efflux curve during stimulation minus baseline (non-stimulated) release. To compute net evoked release the mean of two baseline samples taken immediately before and then after the recovery from stimulation was subtracted from the total radioactivity released due to the stimulation (Somogyi & de Groat, 1992). Drug effects were calculated either as change in the fractional release or when two stimulation paradigm was used as change in S2/S1 ratio.
Drugs
Phenylephrine hydrochloride, atropine sulphate, and all constituents of the Krebs solution were purchased from Sigma Chemical Co, (St. Louis, MO, U.S.A.). 5-methyl-urapidil, 2-(2,6-dimethoxy-phenoxyethyl)aminoethyl-1,4 benzodioxane HCl (WB-4101) and chloroethylclonidine were purchased from RBI (Natick, MA, U.S.A.). Rec 15/2739 was provided by Recordati Labs. 3H choline (80 Ci mmol−1) was purchased from NEN Life Science Products (Boston MA, U.S.A.)
Statistics
The data were analysed by one way analysis of variance using the Prism Statistical Program (Graphpad, San Diego, CA, U.S.A.) using the Bonferini test as a post test. A level of P<0.05 was considered statistically significant. The data are expressed as mean±s.e.mean.
Results
Time-course of the effect of PE
To establish the validity of cumulative concentration response curves which required continuous exposure to PE for 1–2 h, it was necessary to perform preliminary studies to evaluate the effects of prolonged administration of a single concentration of PE. In these experiments the PE effects on: (1) the neurally evoked bladder contractions and (2) basal smooth muscle tone were measured. Bladder strips were stimulated by short (10 shocks) or by long trains of pulses (100 shocks) at 20 Hz every 7 min. After obtaining control responses, 8 μM PE facilitated the contractions by 30–50% and increased the basal tone in the range of 3–6 mN. The strips were further stimulated for 125 min in the presence of PE (Figure 1). The increase in basal tone gradually decreased with time; whereas the facilitation of the neurally evoked contractions evoked by 100 shocks or 10 shocks (not shown) did not change (Figure 1).
Figure 1.
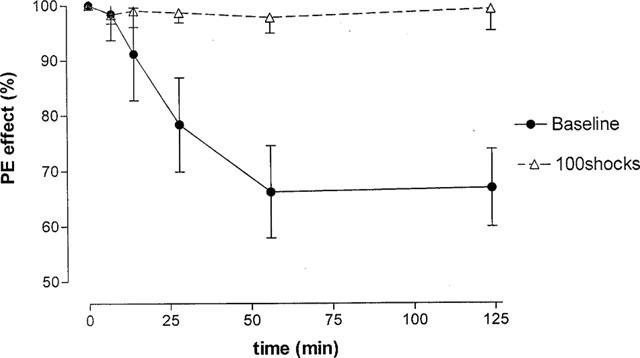
Effect of prolonged administration of phenylephrine (PE) on the amplitude of neurally evoked contractions and basal tone in bladder strips. The effect of 8 μM PE during 125 min exposure is shown as percentage of the initial response to the drug (indicated as 100%). PE was added to the strips 2–3 min before time zero. Strips were stimulated with trains of 100 shocks at 20 Hz every 7 min using submaximal stimulation. The per cent of facilitation and/or the base line tone was plotted against time (min). Note that per cent decrease in baseline and the degree of PE-evoked facilitation at 51 and 125 min after addition of PE was significantly different (P<0.05) Every point is the average of five experiments; the vertical lines denote s.e.m.
Effect of specific inhibitors of α1A adrenoceptors on the PE-induced facilitation of neurogenic contractions and basal tone
After obtaining a cumulative concentration-response curve for PE (concentrations ranging from 0.5–32 μM) in preparations stimulated with trains of 10 shocks at 20 Hz, a selective antagonist for α1A adrenoceptors; 5-methyl-urapidil (5-MU); Rec15/2739 or WB-4101 was applied. The concentration-response curve of PE was repeated in the presence of different concentrations of an antagonist starting with the lowest concentration. When the maximal PE facilitation occurred, both PE and the antagonist were washed out and immediately a higher concentration of antagonist was added to the bath. As shown in Figure 2, the concentration-response curves for PE were shifted to the right in a concentration-dependent manner by 5-MU. The pA2 values for 5-MU and the other two antagonists were calculated according to Arunlakshana and Schild (1959) (Table 1). The three antagonists showed slightly different potencies; Rec15/2739 being the most potent and 5-MU the least potent. The slopes of the Schild plot were not significantly different from unity indicating that the three antagonists acted in a competitive manner. 5-MU inhibited the 8 μM PE evoked increase in basal tone with an IC50 of 48±12.5 nM (n=6), whereas the IC50 for the PE-facilitated neurally evoked contractions occurred at significantly lower concentration (10±1.5 nM; n=8; P<0.05).
Figure 2.
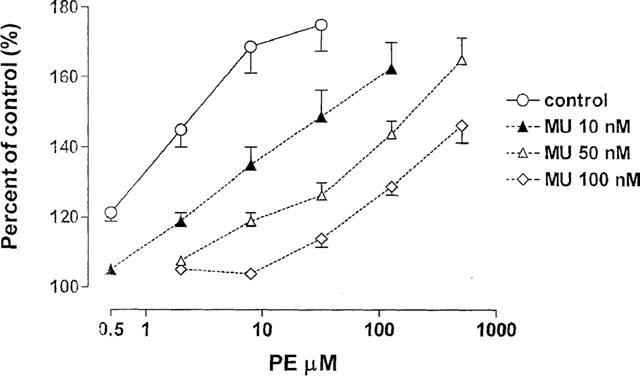
Effect of various concentrations of the α1A antagonist, 5-MU on the phenylephrine (PE)-induced facilitation of the neurally evoked contractions of rat urinary bladder strips. The strips were stimulated with trains (20 Hz, 10 shocks) at submaximal voltage. The facilitatory effect of PE expressed as a percentage of control was plotted against the concentration of PE in μM. The pA2 values calculated from the right shift of the PE concentration-response curves are shown in Table 1. Every point is an average of seven experiments.
Table 1.
Inhibition by various α1 blockers of the PE-evoked facilitation of NEC in rat urinary bladder strips

Effect of chloroethylclonidine (CEC) on the pre and postjunctional effects of PE
In another set of experiments bladder strips were exposed for 30 min to various concentrations (2.5–50 μM) of CEC, an irreversible blocker that is more effective at α1B/α1D adrenoceptors than at α1A adrenoceptors. The facilitating effect of PE on the neurally evoked contractions was not inhibited after CEC treatment (Figure 3), however, the PE-induced increase in basal tone was suppressed by 90% (Figure 4).
Figure 3.
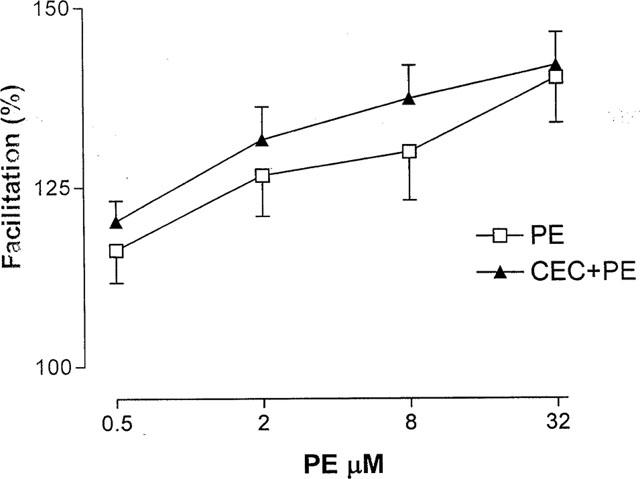
Effect of chloroethyl clonidine (CEC) on the PE evoked facilitation of the neurally evoked contractions. Contractions were evoked by a long train (100 shocks of stimulation) which is known to increase the release of ACh (Somogyi et al., 1994). After constructing a cumulative concentration-response curve for PE, 20 μM CEC was added to the bath for 30 min and then was washed out. Then the PE concentration-response curve was repeated 15 min after the washout. Note that CEC treatment did not affect the PE induced facilitation. Data represent the average of eight experiments.
Figure 4.
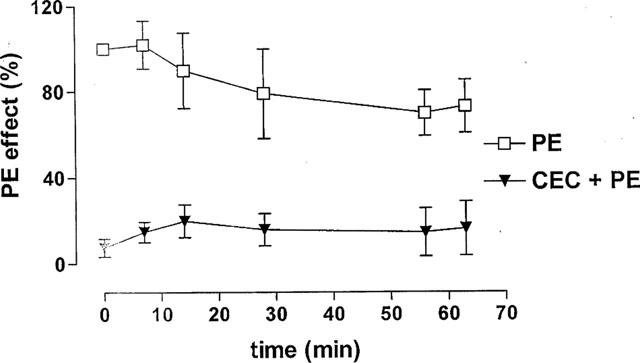
Effect of CEC on PE-evoked increase in basal tone. PE (8 μM) was applied for 65 min before and after exposure to CEC (20 μM). CEC was applied for 20 min between the two applications of PE and washed out before the second application. Vertical axis: increase in basal tone expressed as a percentage of the initial effect of PE (indicated as 100%) during the first application. Note that CEC significantly inhibited the effect of PE. Also there was a significant rundown of the PE induced increase in tone in controls; whereas there was a slight increase in baseline tone in the first 10 min of the PE effect in CEC treated preparations.
CEC in concentrations of 2.5–20 μM also increased the amplitude (by 47.2±8.4 %; n=8 in a concentration of 20 μM) (Figure 5) and increased the duration and area of the contractile curves (by 261±33%; n=8 in a concentration of 20 μM) (Figure 5). Atropine (1 μM) suppressed both the amplitude and area of contractile curves in CEC-treated preparations (Figure 6, Table 2). The effect of atropine was more prominent in CEC-treated than in untreated preparations (Table 2). 5-MU (10–100 nM) administered 15 min before CEC did not reduce the CEC-increased contractions (data not shown) indicating that the increase in contractile amplitude by CEC is not due to activation of prejunctional α1A receptors.
Figure 5.
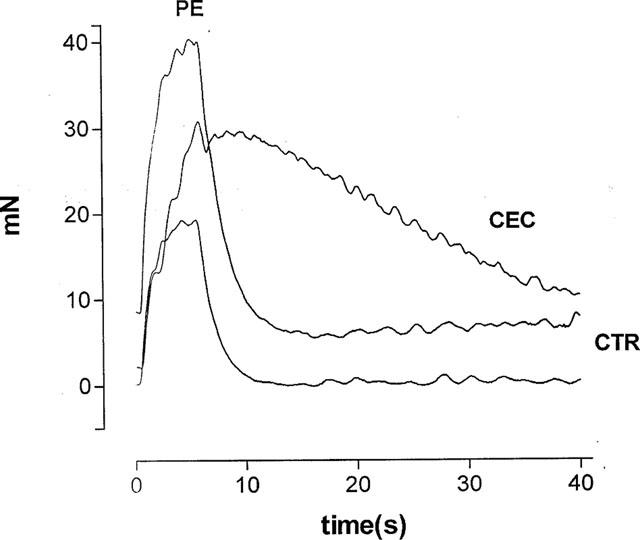
Effect of CEC (20 μM) on the contractions of the bladder strips. Individual contractions of a bladder strip were evoked by trains of 100 shocks at 20 Hz at submaximal voltage. CTR=control contraction, PE=contraction in the presence of 8 μM PE, and ‘CEC' denotes a contraction after exposing the strip 20 μM CEC for 30 min and then washing out CEC.
Figure 6.
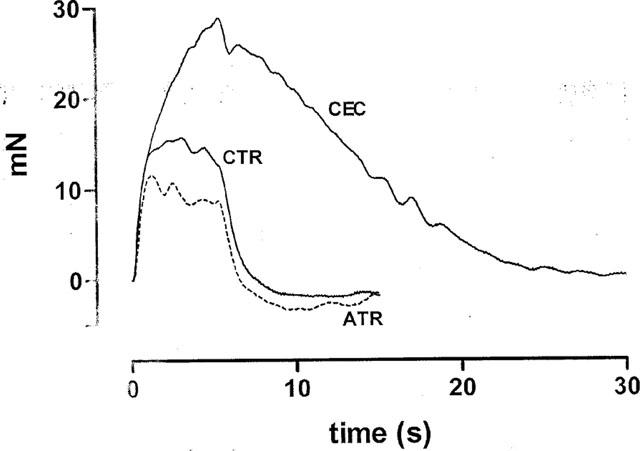
Effect of atropine (1 μM) on the contractions of the bladder strips in the presence and absence of CEC (20 μM). Individual contractions of a bladder strip were evoked by trains of 100 shocks at 20 Hz at submaximal voltage. CTR=control contraction, ATR=atropine after 15 min exposure to the drug after CEC; ‘CEC' denotes a contraction after exposing the strip to 20 μM CEC for 30 min and then washing out CEC.
Table 2.
Effect of atropine (1 μM) on contractile response of intact and CEC treated bladder strips

Effect of α1A and α1B/α1D adrenoceptor blockade on the PE-evoked facilitation of ACh release
PE-induced facilitation of ACh release was examined in the presence and absence of CEC (20 μM) and WB-4101 (100 nM). For practical reasons the two stimulation paradigm was used in the CEC experiments. With this stimulation pattern both the effect of PE and CEC could be tested with a smaller number of experiments. The bladder strips were stimulated twice (S1 and S2) and CEC was added to the perfusion solution before S2 for a period of 30 min. CEC was washed out before the addition of PE (2 μM) to the perfusion solution. As shown in Figure 7, ACh release was facilitated by PE (2 μM) to the same extent in the presence and absence of CEC. However, ACh release in the absence of PE was significantly increased by CEC itself (P<0.05), a finding which is consistent with that obtained in the contractile experiments. On the other hand, WB-4101 (100 nM), an α1A adrenoceptor antagonist, which alone did not alter the evoked release of ACh, inhibited the facilitatory effect of PE on the electrically evoked release of 3H-ACh (Figure 8).
Figure 7.
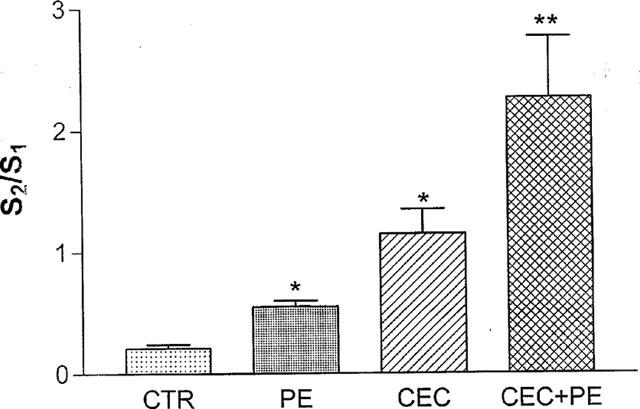
Effect of CEC (20 μM) on the PE-evoked facilitation of ACh release. The effect of drugs is expressed as change in S2/S1 ratio. CEC was added before S2 for 30 min and then was washed out. PE (2 μM) was applied before or after CEC was added to the perfusion solution (see methods for details). The vertical bars denote s.e.m., n=9 for CTR (control); 7 for PE (phenylephrine); 10 for CEC (chloroethylclonidine) and 10 for CEC+PE. * or ** denote significant difference (P<0.05 and P<0.01, respectively, one way analysis of variance, Bonferini test was used as a post test).
Figure 8.
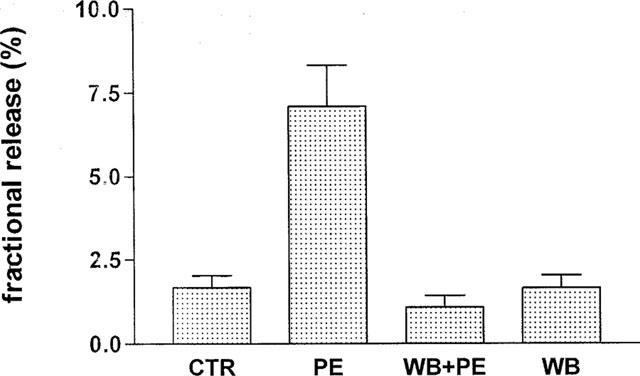
Effect of WB 4101 (100 nM) on the PE-induced facilitation of ACh release. The release of ACh was expressed as fractional release. Bladder strips were stimulated at 20 Hz with 100 shocks starting from the 6th min of the collection period. Drugs were added 15 min before S1. Vertical bars denote s.e.m; n=8 for each group. Abbreviations: CTR=control, PE=phenylephrine, WB=WB 4101.
Discussion
The present experiments revealed that PE, an α1 adrenoceptor agonist has both pre- and postjunctional actions on strips removed from the bladder body of the female rat. PE enhanced neurally evoked contractions (prejunctional action) and also increased basal tone of the bladder strips (postjunctional action). Using subtype selective antagonists we obtained evidence that different subtypes of α1 adrenoceptors are involved in the pre- and postjunctional actions of PE.
In our earlier work (Somogyi et al. 1995) we showed that both the pre- and postjunctional effects of PE could be inhibited by the non-selective α1 adrenoceptor antagonist, terazosin. However, the subtypes of α1 receptors mediating the pre- and postjunctional effects of PE were not identified. In the present study the PE-induced facilitation of neurally evoked contractions was suppressed by the specific α1A adrenoceptor antagonist, 5-MU (Gross et al., 1988). 5-MU shifted the PE dose response curve to the right in a parallel fashion and the Schild analysis indicated that the antagonism was competitive (Figure 1). Similarly, other α1A selective antagonists, WB-4101 (Minneman et al., 1998) or Rec15/2739 (Testa et al., 1997; Leonardi et al., 1997) were also competitive antagonists under our experimental conditions (Table 1). On the other hand CEC which is more effective at the α1B/α1D adrenoceptors, (Bylund et al. 1994; Hieble et al., 1995) did not alter the PE-evoked increase of the neurally evoked contractions indicating the involvement of α1A but not α1B/α1D adrenoceptors in the facilitation.
The α1A adrenoceptor antagonist WB-4101 inhibited the PE-evoked facilitation of ACh release indicating that facilitatory α1A adrenoceptors operate in the bladder postganglionic parasympathetic terminals. Although the concentration of WB-4101 (100 nM) used in these experiments was above the range of α1A selective concentrations reported in other experiments (Hieble et al., 1995), the fact that CEC did not inhibit the PE-facilitated ACh release confirmed that α1B/α1D adrenoceptors did not participate in the presynaptic facilitation. Thus it seems likely that increased release of ACh after PE is the principal mechanism underlying the facilitation of the neurally evoked contractions.
Because field stimulation releases NA as well as ACh (Somogyi et al., 1995; 1996) in the rat urinary bladder it might be expected that endogenous NE released during electrical stimulation would activate a portion of the presynaptic α1A adrenoceptors and that ACh release would be facilitated. However, in the absence of PE WB-4101 did not inhibit the electrical stimulation-evoked ACh release suggesting that the endogenous NA does not activate presynaptic α1A adrenoceptors. These results are consistent with our previous findings which showed that terazosin, a non-selective α1 antagonist did not alter ACh release or the neurally evoked contractions of the bladder (Somogyi et al., 1995). On the other hand, ACh released from bladder cholinergic nerves can modulate NA release (Mattiasson et al., 1987; Somogyi & de Groat, 1990; 1999), indicating that cholinergic and adrenergic varicosities are located close enough to allow heterosynaptic interactions. Thus, the distance between the two types of varicosities cannot explain the lack of effect of endogenous NA on ACh release. The most probable explanation is that the concentration of NA is not sufficient to activate the α1A adrenoceptors in the cholinergic terminals. Although PE does act postjunctionally to increase basal tone, postjunctional mechanisms must be relatively unimportant for the PE-facilitation of the neurally evoked contractions because contractile responses to exogenous ACh were not facilitated by PE (Somogyi et al., 1995).
Previous studies conducted in our laboratory on young mature rats (3 months old; 250–300 g) (Somogyi et al., 1995) did not reveal a consistent postjunctional effect after PE administration. PE-induced contractions occurred in less than 10% of bladder strips indicating that α1 adrenergic excitatory mechanisms are relatively weak in the bladder smooth muscle of young animals. However, similar to the findings of Ordway et al. (1986) the present experiments revealed that bladder strips from older rats (7 months or older) consistently exhibited an increase in basal tone when exposed to PE. In contrast to our findings (Somogyi et al., 1995) and those of Ordway et al., (1986) a recently published paper by Suzuki et al. (1999) reported that PE consistently increased smooth muscle tone in bladder strips from young adult Fisher rats (11 weeks old). The explanation for this difference may be that Fisher rats can express α1 adrenoceptors in bladder smooth muscle at younger age than the Sprague Dawley rats used in our experiments.
The PE-evoked contraction of the bladder strips was inhibited by various concentrations of CEC (5–20 μM) which is an irreversible inhibitor of the α1B adrenoceptors (Han et al., 1987), and a weak inhibitor of α1D adrenoceptors (Bylund et al., 1994; Hieble et al., 1995), whereas, the PE-evoked facilitation of neurally evoked contractions was not inhibited by CEC suggesting that the postjunctional α1 adrenoceptors are of the α1B subtype. 5-MU inhibited the PE-evoked increases in baseline tone only in concentrations (IC50=48 nM) higher than those (IC50=10 nM) which suppressed the PE induced facilitation of the neurally evoked contractions and which are presumably selective for α1A adrenoceptors.
When the time course of the facilitatory effect of PE on the neurally evoked contraction was compared with that of the direct PE contractile effect, there was a significant run down over time in the latter responses, while former effect was unchanged suggesting that the prejunctional and postjunctional receptors are significantly different. The run down is probably the result of the internalization of the α1 receptors in the smooth muscle membrane after activation by PE (Wang et al., 1997).
Because rat bladders exhibit all three subtypes of the α1 adrenoceptors (Malloy et al., 1998) both the α1B and α1D adrenoceptor are possible mediators of the postjunctional action of PE. However, the above mentioned desensitization which has been described for α1B and not for α1D receptors suggests that the postjunctional α1B adrenoceptors may have a more important role in the action of PE.
One problem encountered in the use of CEC was that low concentrations (2.5–20 μM) of the drug increased the amplitude and duration of the neurally evoked contractions. In addition CEC by itself enhanced the release of ACh. 5-MU did not antagonize this effect of CEC; therefore it is unlikely that CEC acts by stimulating prejunctional α1A adrenoceptors. CEC could act on prejunctional α2 adrenoceptors, since Bultmann & Starke (1993) reported that CEC activates presynaptic α2 adrenoceptors in the rat vas deferens. However, in our studies (Somogyi, unpublished) the α2 adrenoceptor agonist, xylazine (0.5–1 μM), had only a slight inhibitory effect on the neurally evoked contractions and had no effect on ACh release in the rat urinary bladder. Since CEC increased both the amplitude of the neurally evoked contractions and the release of ACh, the opposite effect of the α2 agonist, it is unlikely that α2 adrenoceptors are involved in the action of CEC in the rat bladder. On the other hand, atropine significantly inhibited the CEC-induced enhancement of the area and amplitude of the neurally evoked contractions. In addition the inhibitory effect of atropine was more prominent on the area than on the amplitude of the contractions. Larger inhibitory effects of atropine on area versus amplitude of the neurally evoked contractions were also observed in normal and spinal cord transected rats (Somogyi et al., 1998). It is known that the bladder contractions in the rat are composed of a cholinergic and a purinergic component, and that the cholinergic component is longer lasting (MacKenzie & Burnstock, 1984; Maggi et al., 1985; Brading & Mostwin, 1989). Thus, area is a better estimate of the cholinergic part of the contractile curves (Somogyi et al., 1998). The more prominent inhibitory effect of atropine on the area of the contractile curves in CEC-treated versus CEC-non-treated bladder strips suggests that the cholinergic response was increased by CEC. A direct effect on postjunctional muscarinic or α1 receptors seems unlikely because the drug did not increase basal tone of bladder strips. Eserine, an anticholinesterase agent can produce a similar enhancement of the neurally evoked contraction (Somogyi, unpublished) and of ACh release (Somogyi & de Groat, 1992; Somogyi et al., 1994). Thus it seems reasonable to propose that CEC may inhibit acetylcholinesterase in addition to irreversibly blocking postjunctional α1 receptors.
In summary α1A and α1B/α1D adrenoceptors are localized at different sites in the rat bladder. The α1A adrenoceptors mediate prejunctional facilitation at the cholinergic nerve terminals, whereas, α1B or α1D adrenoceptors mediate smooth muscle contractions. The latter response is more prominent in bladders of older animals. In human bladder body α1A and α1D adrenoceptors have also been identified (Malloy et al., 1998). Because α1 adrenoceptors may play a significant role in the hyperactivity of obstructed (Lepor et al., 1990) and neurogenic bladders (Swierzewski et al., 1994), a more detailed analysis of the relative contribution of prejunctional and postjunctional α1 adrenoceptor mechanisms is warranted. Adrenoceptor antagonists targeting α1A prejunctional and α1B or α1D postjunctional adrenoceptors might have different effects on bladder activity and have different clinical uses.
Acknowledgments
This work was supported by the NIH grant RO1 DK-45741.
Abbreviations
- ACh
acetylcholine
- CEC
chlorethylclonidine
- 5-MU
5-methyl-urapidil
- PE
phenylephrine
- NA
noradrenaline
References
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRADING A.F., MOSTWIN J.L. Electrical and mechanical responses of guinea-pig bladder muscle to nerve stimulation. Br. J. Pharmacol. 1989;98:1083–1090. doi: 10.1111/j.1476-5381.1989.tb12651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BULTMANN R., STARKE K. Chlorethylclonidine: an irreversible agonist at prejunctional α2 adrenoceptors in rat vas deferens. Br. J. Pharmacol. 1993;108:336–341. doi: 10.1111/j.1476-5381.1993.tb12806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE GROAT W.C., YOSHIYAMA M., RAMAGE A.G., YAMAMOTO T., SOMOGYI G.T. Modulation of voiding and storage reflexes by activation of α1 adrenoceptors. Eur. Urol. 1999;36 suppl. 1:68–73. doi: 10.1159/000052324. [DOI] [PubMed] [Google Scholar]
- BYLUND D.B., EIKENBERG D.C., HIEBLE P.J., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., MOLINOFF P.R., RUFFOLO R.R. , JR, TRENDELENBURG U. IV. International Union of Pharmacology nomenclature of adrenoceptors. Pharm. Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- GROSS G., HANFT G., RUGEVICS C. 5-methyl-urapidil discriminates between subtypes of the α1-adrenoceptor. Eur. J. Pharmacol. 1988;151:333–335. doi: 10.1016/0014-2999(88)90819-9. [DOI] [PubMed] [Google Scholar]
- HAN C., ABEL P.W., MINNEMAN K.P. Heterogeneity of α1 receptors revealed by chloroethylclonidine. Mol. Pharmacol. 1987;32:505–510. [PubMed] [Google Scholar]
- HIEBLE J.P., BYLUND D.B., CLARKE D.E., EIKENBURG D.C., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., RUFFOLO R.R, , JR International Union of Pharmacology X. Recommendation for nomenclature of α1 adrenoceptors: Consensus update. Pharm. Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- LEONARDI A., HIEBLE J.P., GUARNERI L., NASELSKY D.P., POGGESI E., SIRONI G., SULPIZIO A.C., TESTA R. Pharmacological characterization of the uroselective alpha-1 antagonist Rec 15/2739 (SB216469): Role of the αIL adrenoceptor in tissue selectivity, part1. J. Pharmacol. Exp. Ther. 1997;281:1272–1283. [PubMed] [Google Scholar]
- LEPOR H., KNAP-MALONEY G., SUNSHINE H. A dose titration study evaluating terazosin, a selective, once-a-day α1 blocker for the treatment of symptomatic benign prostatic hyperplasia. J. Urol. 1990;144:1393–1398. doi: 10.1016/s0022-5347(17)39751-3. [DOI] [PubMed] [Google Scholar]
- MACKENZIE I., BURNSTOCK G. Neuropeptide action on the guinea-pig urinary bladder; a comparison with the effect of field stimulation and ATP. Eur. J. Pharmacol. 1984;105:85–94. doi: 10.1016/0014-2999(84)90651-4. [DOI] [PubMed] [Google Scholar]
- MAGGI C.A., SANTICIOLI P., MELI A. Pharmacological evidence for existence of two components in twitch response to field stimulation of detrusor strips from the rat urinary bladder. J. Auton. Pharmacol. 1985;5:221–230. doi: 10.1111/j.1474-8673.1985.tb00123.x. [DOI] [PubMed] [Google Scholar]
- MALLOY B.J., PRICE D.T., PRICE R.R., BIENSTOCK A.M., DOLE M.K., FUNK B.L., RUDNER X.L., RICHARDSON C.D., DONATUCCI C.F., SCHWINN D.A. α1-adrenergic receptor subtypes in human detrusor. J. Urol. 1998;160:937–943. doi: 10.1016/S0022-5347(01)62836-2. [DOI] [PubMed] [Google Scholar]
- MATTIASSON A., ANDERSSON K-E., ELBADAWI A., MORGAN E., SJÖGREN C. Interaction between adrenergic and cholinergic nerve terminals in the urinary bladder of rabbit, cat and man. J. Urol. 1987;137:1017–1019. doi: 10.1016/s0022-5347(17)44350-3. [DOI] [PubMed] [Google Scholar]
- MINNEMAN K.P., HAN C., ABEL P.W. Comparison of α1-adrenergic receptor subtypes distinguished by chlorethylclonidine and WB 4101. Mol. Pharmacol. 1988;33:509–514. [PubMed] [Google Scholar]
- ORDWAY G.A., KOLTA M.G., GERALD M.C., WALLACE L.J. Age related change in alpha-adrenergic responsiveness of the urinary bladder of the rat is regionally specific. Neuropharmacol. 1986;25:1335–1340. doi: 10.1016/0028-3908(86)90105-x. [DOI] [PubMed] [Google Scholar]
- SOMOGYI G.T., DE GROAT W.C. Evidence for inhibitory nicotinic and facilitatory muscarinic receptors on cholinergic nerve terminals of the rat urinary bladder. J. Auton. Nerv. Syst. 1992;37:89–98. doi: 10.1016/0165-1838(92)90237-b. [DOI] [PubMed] [Google Scholar]
- SOMOGYI G.T., DE GROAT W.C. Modulation of the release of 3H-norepinephrine from the base and body of the rat urinary bladder by endogenous adrenergic and cholinergic mechanisms. J. Pharmacol. Exp. Ther. 1990;255:204–210. [PubMed] [Google Scholar]
- SOMOGYI G.T., DE GROAT W.C. Function, signal transduction mechanisms and plasticity of presynaptic muscarinic receptors in the urinary bladder. Life Sci. 1999;64:411–418. doi: 10.1016/s0024-3205(98)00580-3. [DOI] [PubMed] [Google Scholar]
- SOMOGYI G.T., TANOWITZ M., DE GROAT W.C. M-1 muscarinic receptor mediated facilitation of acetylcholine release in the rat urinary bladder. J. Physiol. (Lond.) 1994;480:81–89. doi: 10.1113/jphysiol.1994.sp020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOMOGYI G.T., TANOWITZ M., DE GROAT W.C. Prejunctional facilitatory alphal-adrenoceptors in the rat urinary bladder. Br. J. Pharmacol. 1995;114:1710–1716. doi: 10.1111/j.1476-5381.1995.tb14961.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOMOGYI G.T., TANOWITZ M., ZERNOVA G.V., DE GROAT W.C. M1 muscarinic receptor facilitation of ACh release in rat bladder mediated by protein kinase C. J. Physiol. (Lond.) 1996;496:245–254. doi: 10.1113/jphysiol.1996.sp021681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOMOGYI G.T., YAMAMOTO T., SZÉLL E.A., DE GROAT W.C. Smooth muscle and parasympathetic nerve terminals have different subtypes of α1 adrenoceptors in the rat urinary bladder. Abstracts, Soc. for Neurosci. 1999;25:1170. doi: 10.1038/sj.bjp.0703475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOMOGYI G.T., ZERNOVA G.V., YOSHIYAMA M., YAMAMOTO T., DE GROAT W.C. Frequency dependence of muscarinic facilitation of transmitter release in the urinary bladder strips from neurally intact or chronic spinal cord transected rats. Br. J. Pharmacol. 1998;125:241–246. doi: 10.1038/sj.bjp.0702041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI Y., MORIYAMA N., KANADA A., OKAYA Y., KAWABE K., AISAKA K. The role of αIL -adrenoceptor in rat urinary bladder: Comparison between young adult and aged rats. Life Sci. 1999;65:2553–2559. doi: 10.1016/s0024-3205(99)00524-x. [DOI] [PubMed] [Google Scholar]
- SWIERZEWSKI S.J., GORMLEY E.A., BELVILLE W.D., SWEETSER P.M., WAN J., MCGUIRE E.J. The effect of terazosin on bladder function in the spinal cord injured patients. J. Urol. 1994;151:2951–2954. doi: 10.1016/s0022-5347(17)35132-7. [DOI] [PubMed] [Google Scholar]
- TESTA R., GUARNERI L., ANGELICO P., POGGESI E., TADDEI C., SIRONI G., COLOMBO D., NASELSKY J.P., HIEBLE J.P., LEONARDI A. Pharmacological characterization of the uroselective α1 antagonist Rec 15/2739 (SB216469): Role of the αIL adrenoceptor in tissue selectivity, part II. J. Pharmacol. Exp. Ther. 1997;281:1284–1293. [PubMed] [Google Scholar]
- WANG J., ZHENG J., ANDERSON J.L., TOEWS M.L. A mutation in the hamster αIB-adrenergic receptor that differentiates two steps in the pathway of receptor internalization. Mol. Pharmacol. 1997;52:306–313. doi: 10.1124/mol.52.2.306. [DOI] [PubMed] [Google Scholar]