

Abstract
The second messenger cyclic AMP regulates diverse biological processes such as cell morphology and cell growth. We examined the role of the second messenger cyclic AMP on rat aortic vascular smooth muscle cell (VSMC) morphology and the intracellular transduction pathway mediated by platelet-derived growth factor β-receptor (PDGF-Rβ).
The effect of PDGF-BB on VSMCs growth was assessed by [3H]-thymidine incorporation. Tyrosine phosphorylation of PDGF-Rβ, PLC-γ1, ERK1 and ERK2, p125FAK and paxillin as well as Sm α-actin was examined by the chemiluminescence Western blotting method. Actin mRNA level was quantitated by Northern blotting. Visualization of Sm α-actin filaments, paxillin and PDGF-Rβ was performed by immunfluorescence microscopy.
Cholera toxin (CTX; 10 nM) treatment lead to a large and sustained increase in the cyclic AMP concentration after 2 h which correlated with change of VSMC morphology including complete disruption of the Sm α-actin filament array and loss of focal adhesions. Treatment of VSMCs with CTX did not influence tyrosine phosphorylation of p125FAK and paxillin but decreased the content of a Sm α-actin protein. Maximal decrease of 70% was observed after 24 h of treatment. CTX also caused a 90% decrease of the actin mRNA level. CTX treatment completely abolished PDGF-BB stimulated DNA-synthesis although PDGF-Rβ level and subcellular distribution and translocation was not altered. Furthermore CTX attenuated the PDGF-BB-induced tyrosine phosphorylation of the PDGF-Rβ, PI 3′-K, PLC-γ1 and ERK1/2 indicating an action of cyclic AMP on PDGF-β receptor.
We conclude that although cyclic AMP attenuates the PDGF-Rβ mediated intracellular transduction pathway, an intact actin filament may be required for the PDGF-BB-induced DNA synthesis in VSMCs.
Keywords: PDGF, cyclic AMP, paxillin, smooth muscle, α-actin, MAP kinase, PLC-γ1, PI 3′-K
Introduction
Platelet-derived-growth factor-BB (PDGF-BB) propagates its mitogenic signals through tyrosine phosphorylation of the PDGF-Rβ receptor, thereby activating several tyrosine kinases via their phosphorylation on tyrosine residues. Binding of PDGF-BB to two PDGF-Rβ results in dimerization and autophosphorylation of tyrosine residues of the PDGF-Rβ. Upon activation of PDGF-Rβ, phospholipase C-γ1 (PLC-γ1), GTPase activating protein, phosphatidylinositol 3′ kinase (PI 3′-K) and Src tyrosine kinases bind to the autophosphorylated receptor via their Src homology 2 (SH2) domain and become tyrosine phosphorylated (Kaplan et al., 1990; Kypta et al., 1990; Pawson, 1995; Rönnstrand et al., 1992). Activation of the Ras-dependent stimulation of the mitogen-activated protein (MAP) kinases, in particular the extracellular response kinases 1 and 2 (ERK1 and ERK2) (also known as p44mapk and p42mapk) occurs by a cascade that requires the activation of the adapter protein complex Grb2/Shc/Sos, p21ras, p21ras GAP, Raf-1 kinase and MAP kinase kinase (MEK) (Marshall, 1995a,1995b). In this context, it is assumed that upon activation of PDGF-Rβ, Grb2/Shc/Sos complex is activated by the autophosphorylated PDGF-Rβ receptor resulting in activation of the p21ras signalling pathway after release of GDP by Sos, thereby allowing binding of GTP (Arvidsson et al., 1994; Boguski & McCormick, 1993). Finally, tyrosine autophosphorylated PDGF-Rβ activates the transcriptional factor Stat1 via tyrosine phosphorylation (Silvennoinen et al., 1993).
Mitogen-induced Ras signalling pathway promotes transcription of the cyclin D1 gene via a kinase cascade that depends on the sequential activities of Raf-1, mitogen-activated protein kinase kinase (MEK1 and MEK2), and ERK1/2. In this context, it is well established that progression through the G1 phase into the S phase of the cell cycle requires assembly of D-type cyclins (cyclin D1, D2, D3) with cyclin-dependent kinase (CDK) proteins (Sherr, 1993). Assembly of Cyclin D1 with cyclin-dependent kinase (CDK) proteins depends on the same cascade but the substrate(s) of the ERK1/2 phosphorylation that is mediated in this process is as yet unknown (for review see Sherr & Roberts, 1999).
It is well established that the second messenger cyclic AMP and its effector protein kinase A (PKA) regulates diverse biological processes such as cell morphology, gene transcription and cell growth (Edelman et al., 1987). Although several studies demonstrated that cyclic AMP is implicated in the regulation of different cell types such as fibroblasts (Kato et al., 1994) and VSMCs (Assender et al., 1992; Meisenhelder et al., 1989), the underlying inhibitory mechanisms are not clear. In general, a number of previous studies have favoured the concept that the anti-proliferative effect of cyclic AMP is mediated through inhibition of the growth factors-induced activation of ERK1/2 by preventing activation of the Ras-dependent activation of the serine/threonine kinase Raf-1 (Graves et al., 1993). Recent studies demonstrated that elevation of cyclic AMP in VSMCs by cyclic AMP increasing pharmacological tools significantly inhibited the PDGF-induced DNA synthesis in VSMCs probably via an inhibition of the PI 3′-K/p70 ribosomal protein S6 kinase (p70s6k) activity (Scott et al., 1996) or by an inhibition of the cyclin D1 expression (Musa et al., 1999; Vadiveloo et al., 1997).
Focal adhesion kinase (FAK) is a protein tyrosine kinase with a molecular weight of 125 kDa (p125FAK) that co-localizes with integrins in cellular focal adhesions and normally mediates signalling between the actin cytoskeleton and the integrin-regulated signalling transduction pathway (Schaller & Parsons, 1994; Zachary & Rozengurt, 1992). The p125FAK can be normally phosphorylated on tyrosine residues by extracellular adhesion matrix ligands such as fibronectin. Paxillin that is a substrate for p125FAK is tightly associated with p125FAK (Han & Rubin, 1996; Schaller & Parsons, 1994; Zachary et al., 1993). Although the cellular function of p125FAK and paxillin is not definitively elucidated, it is generally assumed that both may play an important role in the regulation of cell adhesions, morphology and motility (Han & Rubin, 1996; Schaller & Parsons, 1994; Zachary & Rozengurt, 1992; Zachary et al., 1993). Recently, there is accumulating evidence that cytoskeletal structure and cell shape may play a critical role in control of cell growth of different mammalian cells (Huang et al., 1998). Thus, VSMC morphology and cytoskeletal structure of VSMCs has been altered by increasing cyclic AMP and the effect of PDGF-BB on the DNA synthesis has been examined. Intracellular cyclic AMP was elevated by cholera toxin (CTX) that induces increase of intracellular cyclic AMP level via ADP-ribosylation of specific residues of the guanylate nucleotide-binding protein (GS) subfamily. Morphological changes were visualized by staining of Sm α-actin filament array and paxillin in focal adhesions. Moreover, Sm α-actin protein content and mRNA expression level in VSMCs has been determined in CTX-treated VSMCs. Since it has been suggested that the anti-proliferative effect of cyclic AMP might be mediated through inhibition of the growth factors-induced activation of ERK1/2 (Cook & McCormick, 1993; Graves et al., 1993; Wu et al., 1993) the PDGF-Rβ dependent intracellular signalling transduction pathway of PDGF-BB in CTX-treated cells, including tyrosine phosphorylation of PDGF-Rβ, PLC-γ1, PI 3′K and ERK1/2, has been examined.
Methods
Isolation and culture of vascular smooth muscle cells
Rat aortic VSMCs were isolated from the thoracic aorta of Wistar-Kyoto rats (6–8 weeks old, Charles River Wiga GmbH) by enzymatic dispersion using a slight modification of the method as described previously (Sachinidis et al., 1995). Cells were cultured in DMEM supplemented with 10% FCS (v v−1, nonessential amino acids, penicillin 100 IU ml−1 and streptomycin 100 μg ml−1 at 37°C in the Steri-cult incubator (Forma Scientific, Göttingen, Germany) in a humidified atmosphere of 95% air and 5% CO2. Cells were grown in 75 cm2 flasks to confluence over 4–5 days.
Measurement of cyclic AMP content
VSMCs were seeded on petri dishes (3 cm diameter) and cultivated in culture medium until confluence. Cells were washed with 1 ml PBS and medium was replaced with 0.5 ml serum-free medium consisting of a mixture of DMEM and Ham' F-10 medium (1 : 1). Following a 24 h incubation, 10 nM CTX was added to the serum-free medium for various times. Experiments were terminated by adding small amounts of 2 N HCl at 4°C until it had a HCl concentration of 0.01 N. Cells were scraped and centrifuged for 5 min at 10,000×g. Supernatants of 25 μl were used for cyclic AMP determination according to instructions of cyclic AMP[125I]-assay system (single range, code RPA508) from Amersham Buchler, Braunschweig, Germany. Cell pellet was dissolved in 0.5 ml 0.5 N NaOH and aliquots of 25 μl were used for protein determinations using the Bio-Rad (Munich, Germany) protein assay as described previously (Bradford, 1976).
Gel electrophoresis and immunostaining
Determination of the tyrosine phosphorylated ERK1/2, PLC-γ1, PI 3′-K, PDGF-Rβ, p125FAK and paxillin were performed after precipitation of tyrosine phosphorylated proteins using sepharose-coupled anti-phosphotyrosine antibodies. Briefly, confluent cells in 10 cm (diameter) culture dishes were incubated in serum-free medium consisting of a mixture of DMEM and Ham' F-10 medium (1 : 1) for 24 h before addition of CTX. After cell treatment with 10 nM CTX for 24 h, cells were stimulated for different time periods with 50 ng ml−1 PDGF-BB. After removing the medium cells were lysed with a 1 ml RIPA buffer (in mM): NaCl 50, Tris-HCl 20, NaF 50, EDTA 10, Na4P2O7 10H2O 20, Triton X-100 1%, pH 7.4 containing 1 mM Na3VO4, 1 mM phenylmethylsulphoxide (PMSF), 10 μg ml−1 leupeptin, 10 μg ml−1 antipain, and 0.023 TIU ml−1 aprotinin. After 10 min at 0°C, cell lysates were centrifuged at 14,000×g for 2 min. Then supernatant was mixed with 90 μl sepharose-coupled anti-phosphotyrosine antibody and shaken for minimum 2 h at 4°C. After elution of tyrosine phosphorylated proteins with 100 μl the lysis buffer containing 5 mM phenylphosphate protein determination was performed using the Bio-Rad DC Protein Assay. For determination of Sm α-actin protein, confluent cells in 3 cm (diameter) culture dishes were incubated in serum-free medium consisting of a mixture of DMEM and Ham' F-10 medium (1 : 1, v v−1) for 24 h before addition of the CTX. AFter 24 h cultivation, cells were lyzed and protein determination was performed with the Bio-Rad DC Protein Assay. Proteins were separated in a 7.5% SDS polyacrylamide gel (SDS–PAGE). Proteins were transferred to a PVDF membrane overnight at 100 mA with a buffer containing 25 mM Tris-HCl, 192 mM glycin and 20% methanol, pH 8.3. Enhanced chemiluminescence Western blotting analysis was performed with the chemiluminescence Western blotting system from NEN Life Science Products, Inc. (Boston, MA, U.S.A.), using primary monoclonal anti-PI 3′-K (1 : 5000, anti-PLC-γ1 (1 : 1000), anti-p125FAK (1 : 1000), anti-paxillin IgG (1 : 10,000), and anti-α-Sm (1 : 5000) IgGs or polyclonal anti-PDGF-Rβ (1 : 500) and phospho-specific polyclonal anti-ERK1/ERK2 (1 : 1000) IgGs. The phospho-specific primary polyclonal anti-ERK1/ERK2 IgG recognizes phosphorylated (activated) ERK1/2 on the tyrosine 204 residue. Phosphorylation of the ERK1/2 on tyrosine 204 residue is essential for the activation of ERK1/2 (Marshall, 1995a). Chemiluminescence detection of ERK1/2 and PDGF-Rβ was performed using secondary horseradish peroxidase-labelled anti-rabbit IgG (1 : 5000). Detection of PI 3′-K, PLC-γ1, paxillin, Sm α-actin and FAK was performed using secondary horseradish peroxidase-labelled anti-mouse IgG (1 : 5000).
Visualization of Sm α-actin filaments, paxillin and PDGF-Rβ in VSMCs
VSMCs were cultured on round glass cover slips in 24-well culture plates (Falcon, Heidelberg, Germany) and cultivated until they reached about 50% confluence. Cells were then preincubated for 24 h in serum-free medium before addition of 10 nM CTX. Following different time incubation, cells were washed with PBS, fixed with 2% formaldehyde (v v−1) for 5 min and permeabilized with PBS containing 0.2% Triton X-100 (v : v) for 5 min. Following treatment of the cells with PBS containing 2% BSA (w v−1) for 1 h, cells were incubated with a monoclonal antibody to Sm α-actin (mouse IgG2a isotype) (10 μl per slide of the working dilution 1 : 100), anti-paxillin antibody (10 μl per slide of the working solution 1 : 500) or anti-PDGF-Rβ (10 μl per slide of the working solution 1 : 50) for 1 h. Cells were washed with PBS containing 0.15% Tween 20 (v : v) and were incubated with fluoresceinisothiocyanate (FITC)-conjugated swine anti-rabbit IgG for 1 h (10 μl per slide of the working dilution 1 : 200) and visualized using an immunoflourescence microscope (Leitz DMRB, Leica, Köln, Germany). All experimental steps were performed at room temperature.
RNA extraction and analysis
The expression of actin mRNA was studied after preincubation of confluent cells (75 cm2 culture flasks) in serum-free medium for 24 h before addition of 10 nM CTX. After 24 h incubation, medium was aspirated and cells were lysed with 1 ml TRI reagent (Sigma) and total RNA was extracted according to manufacturer' protocol as described previously (Chomczynski & Sacchi, 1987). Ten μg of total RNA were separated by electrophoresis in a 6% formaldehyde/1.2% agarose gel, blotted on Hybond N+ membranes (Amersham, Little Chalfont, U.K.), washed at room temperature in 5×SSC (1×=0.15 M NaCl, 0.015 M sodium citrate) for 5 min, and fixed with u.v. irradiation. After fixing, the blots were washed at 60°C in 0.1×SSC, 0.1% SDS for 5 min. Prehybridization and hybridization were performed overnight at 60°C in 5×SSC, 0.2% SDS (w v−1), 50 mM sodium phosphate, 10×Denhardt' solution (Sigma Chemical, Deisenhofen, Germany) 200 μg ml−1 salmon spern DNA. The DNA probes were labelled with [32P]-deoxycytidine triphosphate ([32P]-dCTP) by the random oligonucleotide priming method (Amersham Buchler, Braunschweig, Germany). The stringency of the final wash was 0.2×SSC containing 0.1% SDS at 65°C for 2×45 min. A 0.77 kg cDNA probe for actin mRNA were used as probes. Blots were exposed to Kodak films (Kodak X-OMAT, 8×10 inch, Rochester, U.S.A.) for 1–7 days at −70°C. The size in kilobases (kb) of the detected mRNA was estimated by the 18S (1.8 kb) and 28S (4.6 kb) ribosomal RNA migration from the gel wells.
Determination of cell counts and cell diameter
For cell counting, VSMCs were cultured in DMEM, supplemented with 10% FCS (v v−1), non essential amino acids, penicillin 100 IU ml−1 and streptomycin 100 μg ml−1 at 37°C for 24 h until a cell confluence of approximately 70% was reached. The medium was then replaced by serum-free medium consisting of DMEM and Ham' F-10 (1 : 1, v v−1). After 24 h VSMCs were treated with 10 nM CTX. After 24 h the cells were trypsinized and cell counting as well as determination of cell diameter was performed using the CASY-1 system based on the coulter counter principle (Schärfe, Reutlingen, Germany).
Determination of DNA Synthesis
The effect of PDGF-BB on [3H]-thymidine incorporation into cell DNA was assessed as previously described (Sachinidis et al., 1995). VSMCs were cultured until approximately 70% confluence. Then, the medium was replaced by serum-free medium. After 24 h, 10 nM CTX was added to the medium. After 24 h, cultures were exposed to 50 ng ml−1 PDGF-BB and 3 μCi ml−1 [3H]-thymidine for 12, 16, 18, 24 and 36 h. Experiments at the different time periods were terminated by aspirating the medium and subjecting the cultures to sequential washes with PBS containing 1 mM CaCl2, 1 mM MgCl2, then with 10% trichloroacetic acid and finally with ethanol : ether (2 : 1, v v−1). Acid-insoluble [3H]-thymidine was extracted into 0.5 M NaOH and [3H]-thymidine incorporation was quantified by liquid scintillation counting (Beckman LS 3801, Düsseldorf, Germany).
Materials
Cholera toxin was obtained from Calbiochem (Bad Soden, Germany). Dulbecco' modified Eagles medium (DMEM), Ham' F-10 and Dulbecco' phosphate-buffered saline (PBS) were obtained from Gibco BRL (Eggenstein, Germany). PDGF-BB was prepared as described previously (Hoppe et al., 1989). Monoclonal mouse anti-PI 3′ K, mouse anti-PLC-γ1, mouse anti-paxillin IgG, and mouse anti-FAK antibodies were obtained from Transduction Laboratories (Lexington, Kentucky, U.S.A.). Monoclonal mouse anti-Sm α-actin antibodies (mouse IgG2a) and sepharose-coupled anti-phosphotyrosine antibodies were obtained from Sigma (St. Louis, MO, U.S.A.). The anti-PDGF-Rβ rabbit polyclonal antibodies were obtained from Santa Cruz (Heidelberg, Germany). The phospho-specific ERK1/ERK2 rabbit polyclonal antibodies were obtained from New England BioLabs (Beverly, MA, U.S.A). The horseradish peroxidase-labelled anti-rabbit and anti-mouse antibodies were obtained from Amersham Life Sciences (Little Chalfont, U.K.). The 0.77 kb actin cDNA probe was obtained from Dianova/Oncor Science (Hamburg, Germany). FITC-conjugated anti-mouse and FITC-conjugated anti-rabbit antibodies were obtained from DAKO Corporation (Carpinteria, CA, U.S.A.). Hybond N+ membranes, [32P]-deoxycytidine triphosphate ([32P]-dCTP), [Methyl-3H]-thymidine were obtained from Amersham (Little Chalfont, U.K.).
Statistics
Values are expressed as means±s.e.mean. Statistical analysis of the data was performed using the one factor Anova-Scheffe F-test. P<0.05 was considered to be statistically significant.
Results
Figure 1 shows the time course of the CTX-induced intracellular formation of cyclic AMP. A first increase from 0.03 nmol g−1 protein to 3.4 nmol g−1 protein cyclic AMP was observed after 30 min. Treatment of the cells for 2, 4, 8, and 24 h caused an increase of cyclic AMP from 0.03 (untreated cells) to 34.2, 40.9, 42.9, and 42.6 nmol g−1, respectively.
Figure 1.
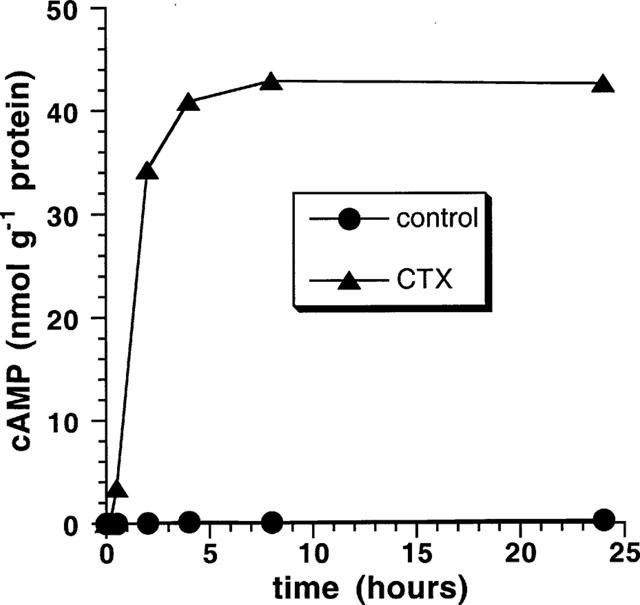
CTX evokes a large and sustained increase in cyclic AMP concentration. Confluent VSMCs in 3 cm (diameter) culture dishes were cultured in serum-free medium for 24 h. Cells were then incubated with CTX 10 nM for various times. Aliquots of the cell lysate were used for the determination of cyclic AMP by using the cyclic AMP[125I] radioimmunoassay.
CTX caused time-dependent drastic changes in cell morphology as inspected by phase contrast microscopy (Figure 2a). Change of the cell morphology correlates well with the time kinetics of the CTX-induced formation of cyclic AMP in VSMCs (see Figure 1). After 30 min some of the VSMCs began to transit their shape to a rounder shape. Almost all of the cells exhibited the ‘rounder shape' after 2 h and this morphology remained stable within 24 h treatment. Sm α-actin filament array and focal adhesions (stained with antibodies directed against paxillin) in control and CTX-treated cells are shown in Figure 2b,c, respectively. Changes in VSMCs morphology correlate with disruption of the Sm α-actin filament array and loss of focal adhesions. Disruption of the filament array and loss of focal adhesions started after 30 min treatment with CTX. Treatment of the cells for longer than 2 h caused a complete disruption of the Sm α-actin filament array and a complete loss of focal adhesions (representative graphs in Figure 2b,c show the effect of CTX after 24 h.
Figure 2.
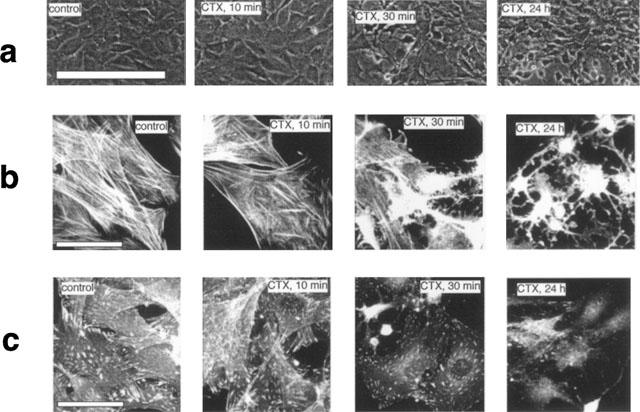
Morphological changes of VSMCs after treatment with CTX. (a) Confluent VSMCs in 3 cm (diameter) culture dishes were cultured in serum-free medium for 24 h. Then cells were treated with 10 nM CTX for different time periods. Cells were photographed by phase-contrast light microscope. Calibration bar represents 200 μm. Visualization of Sm α-actin filaments (b) and focal adhesions in VSMCs. (c) VSMCs were cultured on round glass cover slips (1 cm diameter) and treated as above. After fixing of the cells, Sm α-actin filaments and focal adhesions were visualized using monoclonal antibodies against Sm α-actin or paxillin, respectively. The secondary antibody was fluoresceinisothiocyanate (FITC)-conjugated anti-mouse IgG. Bar represents 20 μm.
In order to assess the viability, the number of intact cells after treatment with CTX for 2 and 24 h was determined by the CASY cell culture counter. There was a slight reduction in cell number of about 10% after 2 h. Less than 20% of the cells were lost after 24 h treatment, indicating that the drastic morphological changes are not paralleled by a loss in cell viability. Furthermore the cell diameter did not change during CTX treatment and remained constant at 15 μM. Thus, both the viability and the cell volume were not altered.
In the search for possible signalling pathway that might be involved in the actin remodelling the phosphorylation status of the p125FAK and paxillin was studied. As demonstrated in Figure 3a, treatment of VSMCs with 10 nM CTX for various times had no significant effects on p125FAK and paxillin (Figure 3b) tyrosine phosphorylation.
Figure 3.
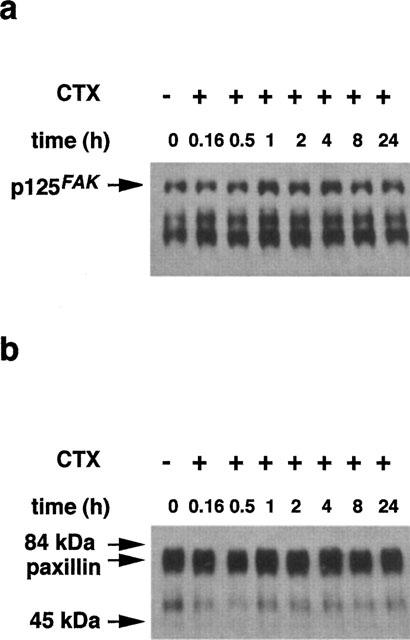
CTX has no effect on p125FAK and paxillin tyrosine phosphorylation. Confluent cells in 75 cm2 culture flasks were preincubated in serum-free medium for 24 h. Then cells were incubated with 10 nM CTX for various times. Then cells were lysed and tyrosine-phosphorylated proteins were immunoprecipitated using an anti-phosphotyrosine antibody coupled to sepharose. Proteins (5 μg) were analysed by 7.5% SDS–PAGE. Tyrosine phosphorylated p125FAK (a) and paxillin (b) were detected by the enhanced chemiluminescence method using the respective monoclonal antibodies.
We next investigated the cellular content of Sm α-actin. As demonstrated in Figure 4a, treatment of VSMCs with 10 nM CTX caused a time-dependent decrease of the Sm α-actin content. CTX-treatment for 4, 8 and 24 h resulted in a 22±8 (n=3), 36±6 (n=3) and 68±2.4 (n=14) inhibition of the Sm α-actin content in VSMCs (Figure 4b).
Figure 4.
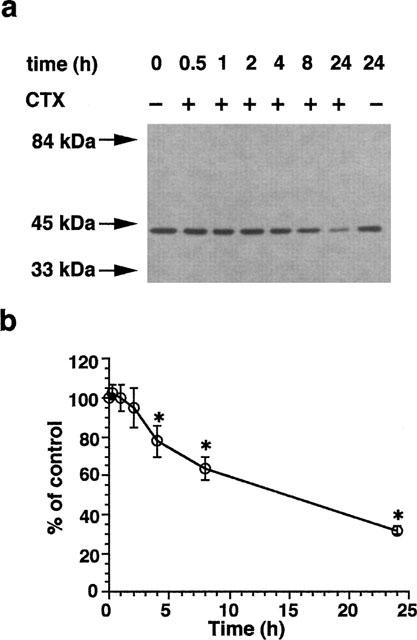
Reduction of Sm α-actin level in VSMCs after CTX treatment. Confluent VSMCs in 3 cm (diameter) culture dishes were precultured in serum-free medium for 24 h. Cells were then incubated in the presence and absence of 10 nM CTX for different time. Cells were then lysed and 30 μg proteins were analysed by SDS–PAGE. Sm α-actin was detected after blotting on PVDF membranes by a specific antibody that recognizes the Sm α-actin isoform (a). (b) Densitometric analysis of blots derived form different experiments (0.5, 1, 2, 4 and 8 h, n=3, 24 h, n=14. *P<0.05).
The reduced Sm α-actin content after CTX treatment was apparently due to a suppression of actin mRNA transcription level in VSMCs. As demonstrated in Figure 5a, pretreatment of the cells with CTX for 24 h caused a 85±10 (mean±s.e.mean, n=3) reduction of the actin mRNA level.
Figure 5.
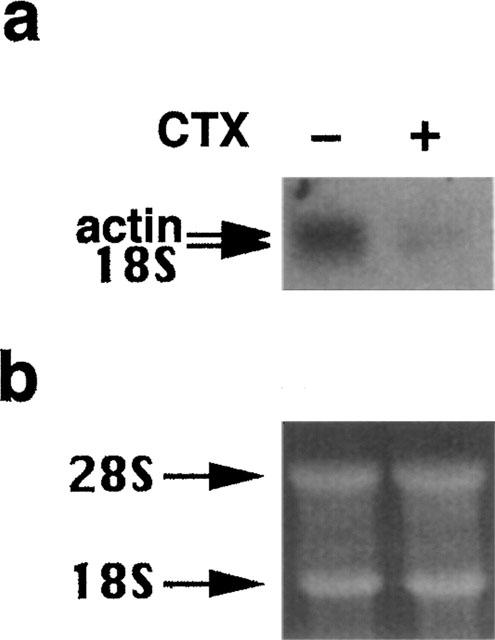
Inhibition of the action mRNA expression in VSMCs after treatment with CTX. (a) Confluent cells in 75 cm2 culture flasks were preincubated in serum-free medium for 24 h. Then cells were incubated with 10 nM CTX for 24 h. After RNA extraction 10 μg of total RNA was separated on a formaldehyde-agarose gel, blotted on Hybond N+ membranes and probed with a [32P]-labelled 0.77 kb cDNA probe for actin). Arrows indicate the actin mRNA (2.0 kb) and 18 S (1.8 kb) ribosomal RNA. (b) Equal loading was demonstrated by showing the ethidium bromide gel.
When control cells were treated with 50 ng ml−1 PDGF-BB for 16, 18, 24 and 36 h, a significant 2.2, 3.7, 4.2 and 6.3 fold increase in DNA synthesis, respectively, was observed (Figure 6). DNA synthesis started after 16 h and continued for at least 36 h. Interestingly, a complete inhibition of the PDGF-BB-induced DNA synthesis was observed in CTX-treated cells. To exclude the possibility that the unresponsiveness was simply due to a receptor downregulation the overall content of PDGF-Rβ was determined by Western blotting. As shown in (Figure 7a) treatment of the cells for 24 h with CTX had no effects on the PDGF-Rβ content. To further analyse the impaired function of the PDGF-Rβ the subcellular distribution and translocation after PDGF-BB was studied by cellular immunohistochemical methods. In control cells, the PDGF-Rβ is evenly distributed over the entire cells membrane (Figure 7b) and also detected in the endoplasmic reticulum around the periphery of the nuclei (Simm et al., 1998). Stimulation of the cells with 50 ng ml−1 PDGF-BB resulted in the formation of highly fluorescing clusters which disappeared after 1 h stimulation with PDGF-BB. The major fluorescence due to PDGF-Rβ being is predominantly located in the endoplasmic reticulum. Pretreatment of the cells with CTX for 24 h did not alter subcellular localization of the PDGF-Rβ in unstimulated and PDGF-BB-stimulated cells.
Figure 6.
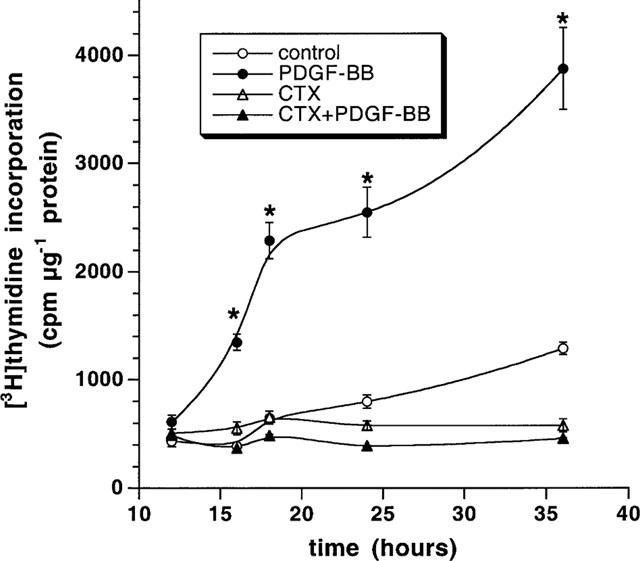
Effect of CTX on VSMCs DNA synthesis. Confluent cells (24-well plates) were precultured for 24 h in serum-free medium. Cells were then treated with 10 nM CTX 24 h. Cells were then stimulated with 50 ng ml−1 PDGF-BB in the presence of 2 μCi ml−1 [3H]-thymidine. [3H]-thymidine incorporation into cell DNA was quantified at 12, 16, 18, 24 and 36 h. *P<0.05 for PDGF-BB effect vs control.
Figure 7.
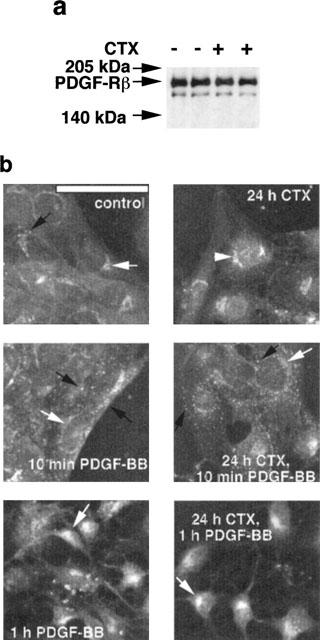
CTX treatment has no effect on PDGF-Rβ content and subcellular localization in VSMCs. (a) Confluent VSMCs in 3 cm (diameter) culture dishes were precultured in serum-free medium for 24 h. Cells were then incubated in the presence and absence of 10 nM CTX for 24 h. Cells were then lysed and 30 μg proteins were analysed by SDS–PAGE. PDGF-Rβ was detected by the enhanced chemiluminescence method and anti-PDGF-Rβ. (b) VSMCs were cultured on round glass cover slips (1 cm diameter) and cultivated in 10% FCS medium. Following 24 h in serum-free medium, cells were treated with 10 nM CTX for 24 h and then stimulated with PDGF-BB for different time periods. After fixing of the cells, subcellular localization of PDGF-Rβ was visualized using monoclonal anti-PDGF-Rβ and fluoresceinisothiocyanate (FITC)-conjugated swine anti-rabbit IgG for 1 h. Bar represents 20 μm.
Since both receptor number and subcellular distribution were not altered after CTX treatment, we next studied several downstream signals from the receptor i.e. PDGF-BB-induced tyrosine phosphorylation of PDGF-Rβ, ERK1/2, PI 3′-K, and PLC-γ1. After preincubation of VSMCs with 10 nM CTX for 24 h, VCMCs were stimulated for different time periods with PDGF-BB. After immunoprecipitation of tyrosine phosphorylated proteins with anti-tyrosine sepharose, specific proteins were detected simultaneously using the appropriate antibodies. PDGF-BB caused in untreated VSMCs a time-dependent phosphorylation of PDGF-Rβ, ERK1/2, PI 3′-K and PLC-γ1 with a maximum at 15, 3, 5 and 5 min, respectively. In general, PDGF-BB also stimulated phosphorylation of PDGF-Rβ, ERK1/2, PI 3-′K and PLC-γ1 in CTX-treated VSMCs with similar time-course kinetic but the amount of the phosphorylated kinases was attenuated in CTX treated VSMCs compared with untreated cells (Figure 8). Results derived from three independent experiments show that phosphorylation of PDGF-BB-induced tyrosine phosphorylation of ERK1/2, PLC-γ1, PI 3′-K and PDGF-Rβ occurring at 5 min was inhibited by 42±5, 48±7, 46±8 and 32±5%, respectively.
Figure 8.
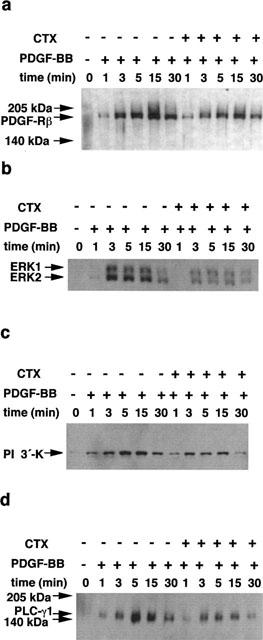
Effect of PDGF-BB on tyrosine phosphorylation of PDGF-Rβ, ERK1/2, PI 3′-K and PLC-γ1 in CTX-treated VSMCs. Confluent cells in 75 cm2 were preincubated in serum-free medium for 24 h before addition of 10 nM CTX. After 24 h VSMCs were stimulated with 50 ng ml−1 PDGF-BB for various time periods. Then cells were lysed and tyrosine-phosphorylated proteins were immunoprecipitated using an anti-phosphotyrosine antibody coupled to sepharose. Immunoprecipitated proteins (5 μg) were analysed by SDS–PAGE. Tyrosine phosphorylated PDGF-Rβ (a), ERK1/2 (b), PI 3′-K (c) and PLC-γ1 (d) was detected by the enhanced chemiluminescence method using the respective monoclonal antibodies.
To examine whether PDGF-BB may prevent the CTX-induced disruption of the Sm α-actin filament array or Sm α-actin expression, VSMCs were simultaneously treated with PDGF-BB and CTX or first with PDGF-BB for 30 min (ERK1/2 and PI 3′-K signals returned to basal values) and then with CTX for 24 h. As shown in Figure 9 treatment of the cells with PDGF-BB for 24 h had no effect on the Sm α-actin protein (Figure 9a) or on Sm α-actin filament array (Figure 9b) in comparison to unstimulated control cells. Also, simultaneous addition of PDGF-BB with CTX or addition of PDGF-BB 30 min before addition of CTX did not prevent disruption and inhibition of the expression of Sm α-actin induced by CTX (Figure 9a,b).
Figure 9.
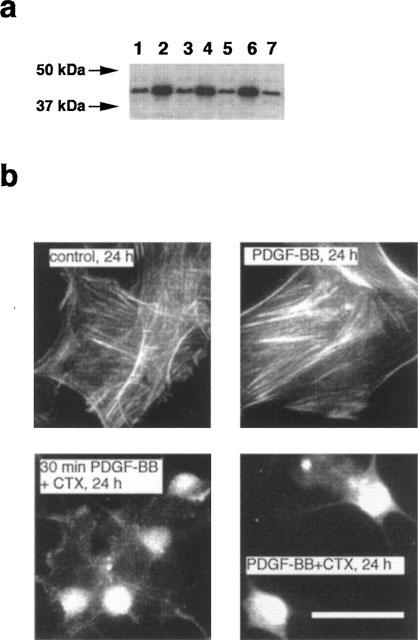
PDGF-BB cannot prevent the CTX-induced reduction of Sm α-actin level and disruption of Sm α-actin filaments. (a) Confluent VSMCs in 3 cm (diameter) culture dishes were precultured in serum-free medium for 24 h. Cells were then stimulated with 50 ng ml−1 PDGF-BB for 30 min and then incubated with 10 nM CTX or PDGF-BB was simultaneously added to the cells with CTX. After 24 h cells were lysed and 30 μg proteins were analysed by SDS–PAGE. After blotting on PVDF membranes Sm α-actin was detected (lane 1: CTX, 24 h; lane 2: PDGF-BB, 24 h; lane 3: 30 min PDGF-BB+CTX, 24 h; lane 4: control, 24 h; lane 5: CTX, 24 h; lane 6: PDGF-BB, 24 h; lane 7: PDGF-BB+CTX, 24 h (simultaneous addition). (b) VSMCs were cultured on round glass cover slips (1 cm diameter) and treated with PDGF-BB and CTX as above. After 24 h cells were fixed and Sm α-actin filaments were visualized. Bar represents 20 μm.
Discussion
We found that treatment of VSMCs with CTX for 24 h resulted in a profound increase in intracellular cyclic AMP. Morphological changes induced by elevated cyclic AMP concentration were not accompanied by a loss of PDGF-Rβ protein or by a distinct subcellular localization of PDGF-Rβ in comparison to unstimulated VSMCs. Furthermore, similar subcellular distribution of PDGF-Rβ was observed in CTX-treated and untreated cells after stimulation with PDGF-BB. Morphological changes of VSMC induced by elevated cyclic AMP concentration were accompanied by disruption of the Sm α-actin filament array and loss of focal adhesions. Increasing levels of cyclic AMP did not elicit the dephosphorylation of p125FAK and paxillin. Similar studies demonstrated that increased cyclic AMP causes a reorganization of the actin cytoskeleton in REF52 fibroblasts (Lamb et al., 1988) and adrenal cortex-derived Y1 cells (Han & Rubin, 1996) resulting in a conversion of the flat cell shape into rounded (Han & Rubin, 1996). The cyclic AMP-induced morphological changes of Y1 cells were associated with a depletion of paxilin from focal adhesions and a rearrangement of the actin cytoskeleton. However, a tyrosine dephosphorylation of paxillin has been observed (Han & Rubin, 1996). Authors postulated that tyrosine dephosphorylation of paxillin is not dependent on the level of tyrosine kinases but cyclic AMP probably activates a protein-tyrosine-phosphatase (PTP) (Han & Rubin, 1996). The inconsistencies among our findings and findings reported in Han & Rubin (1996) most likely reflect differences in the origin of the cells used.
Besides disruption of Sm α-actin filament array, the Sm α-actin protein was significantly lowered in CTX-treated cells. A marked decrease of the Sm α-actin protein in CTX-treated cells strongly correlates with its effects on cyclic AMP level. Moreover, we found that actin mRNA transcript level was dramatically lowered in CTX-treated cells. Therefore, we may suggest that reduction of the Sm α-actin protein by CTX occurs on transcriptional level. Since a decrease of the Sm α-actin was first detectable after 4 h treatment, whereas a complete disruption occurred after 2 h treatment, we may assume that disruption of the Sm α-actin filaments and decrease of Sm α-actin by cyclic AMP are induced through different cellular mechanisms. In this context, we may speculate that disruption of Sm α-actin and loss of focal adhesions may occur through reduction or loss of integrins by elevation of intracellular cyclic AMP. This hypothesis is supported by findings showing that an elevation of cyclic AMP on glomerular mesangial cells (smooth muscle-like cells) resulted in disruption of actin filaments and loss of focal adhesions that was associated with a loss of vitronectin receptor (α5β1 integrin) (Glass & Kreisberg, 1993).
The question whether cyclic AMP-increasing agents inhibit the growth promoting effects of several growth factors by inhibition of the Ras-dependent activation of ERK1/2 is controversially discussed. In this context, it has been shown that cyclic AMP-increasing agents inhibit the PDGF-BB- and the EGF-induced activation of ERK1/2 in VSMCs (Graves et al., 1993) and Rat-1 fibroblasts (Wu et al., 1993), respectively (Cook & McCormick, 1993). We also demonstrated that CTX caused a significant inhibition of the PDGF-BB-induced phosphorylation of the ERK1/2. However, we found that not only ERK1/2 but also the PDGF-BB-induced tyrosine phosphorylation of PDGF-Rβ, PI 3′-K, PLC-γ1 were significantly attenuated. Our findings suggest that cyclic AMP attenuates the tyrosine phosphorylation of PDGF-Rβ resulting in an attenuation of the downstream early signalling of PDGF-Rβ. Other authors have demonstrated that although cyclic AMP-raising agents slightly modify the time course of MAP kinase activation in response to mitogens in CCL39 fibroblasts, the cyclic AMP-induced inhibition of cell growth occurred through a ERK1/2 independent pathway (McKenzie & Pouysségur, 1996). Furthermore, elevation of cyclic AMP caused an inhibition of the Angiotensin II-induced growth without affecting ERK1/2 activation in VSMCs (Giasson et al., 1997; Takahashi et al., 1996). We found that although CTX attenuated the PDGF-BB-induced tyrosine phosphorylation of the PDGF-Rβ, PI 3′-K, PLC-γ1 and ERK1/2 inhibited completely the PDGF-BB-induced DNA synthesis. Therefore, the profound inhibitory effect of CTX on DNA synthesis can not only be attributed to its ability to attenuate the PDGF-BB-induced tyrosine phosphorylation of the PDGF-Rβ, PI 3′-K, PLC-γ1 and ERK1/2.
CTX completely inhibited the PDGF-BB-induced DNA synthesis started after 16 h stimulation. These results demonstrate that treatment of VSMCs with CTX prevents progression of cells from G1 to the S of cell cycle. This observation is in agreement with studies with other cell types demonstrating that cyclic AMP increasing agents prevent cells to enter from the G1 to the S phase (Johnson et al., 1988; Kato et al., 1994; L'Allemain et al., 1997; Magnaldo et al., 1989; McKenzie & Pouysségur, 1996). It is well established that progress through the G1 phase into the S phase of the cell cycle requires assembly of D-type cyclins (cyclin D1, D2, D3) with cyclin-dependent kinase (CDK) proteins (Sherr, 1993). In this context, it has been shown that cyclic AMP-induced G1 phase arrest is mediated by a blockade of the growth factor-induced increase in cyclin D1 protein expression (Kato et al., 1994; L'Allemain et al., 1997).
Recently, it has been demonstrated that cytoskeletal structure and cell shape may play a critical role in control of cell cycle progression (Huang et al., 1998). In this context, it has been reported that ERK1/2 were normally stimulated in capillary endothelial cells that were prevented from spreading in medium containing growth factors. However, in contrast to spread cells, these non-spread cells failed to progress through G1 and enter S phase (Huang et al., 1998). As described by authors, this shape-dependent block in cell cycle progression correlated with the failure to increase cyclin D1 protein levels. Furthermore, a similar block in cell cycle progression was induced by disruption of the actin array using cytochalasin or by inhibiting of cytoskeletal tension generation (Huang et al., 1998). Authors also supposed that the early intracellular signalling events alone are not sufficient for growth. Subsequent cell cycle progression and, hence, cell proliferation are controlled on cell shape and cytoskeletal structure (Huang et al., 1998). From our findings, we may postulate that PDGF-BB failed to stimulate DNA synthesis in morphologically changed VSMCs. Besides induction of morphological changes, CTX attenuates the PDGF-Rβ mediated early signals that might cause reduction of the D-type cyclins thereby additionally contributing to the inhibitory effects of cyclic AMP on VSMCs growth. Recently, there is accumulating evidence that the Rho small Gi proteins consisting of the Rho, Rac and Cdc42 subfamilies are involved in the regulation of actin cytoskeleton and cell shape organization in response to growth factor signalling (Schoenwaelder & Burridge, 1999; Small et al., 1999) and may play a critical role in control of cell cycle progression (Olson et al., 1995). In this context, it has been demonstrated that short time stimulation (15–30 min) of different cell types with growth factors like PDGF (Eriksson et al., 1992; Nobes et al., 1995; Ridley et al., 1992) and insulin (Nobes et al., 1995) stimulate polymerization of actin at the plasma membrane resulting in membrane ruffling. Furthermore, it has been shown that activation of the PI 3′-K is essential for the PDGF- and insulin-induced cell membrane ruffling (Nobes et al., 1995; Wennstrom et al., 1994a, 1994b). Other growth factors like lysophosphatidic (LPA) and bombesin induces a PI 3′-K-independent actin stress fibre formation (Ridley et al., 1992). We demonstrated that long term treatment of VSMCs (24 h) with PDGF-BB has no effects on Sm α-actin filament array and Sm α-actin protein content. We further demonstrated that addition of PDGF-BB for 30 min before addition of CTX or simultaneous addition of PDGF-BB with CTX did not prevent the CTX-induced disruption of the Sm α-actin filament array. These findings demonstrated that activation of ERK1/2 and PI 3′-K by PDGF-BB cannot prevent the CTX-induced disruption of Sm α-actin filament array or the inhibition of Sm α-actin protein and provide further evidence that inhibition of the PDGF-BB-induced stimulation of ERK1/2 and PI 3′-K by CTX is probably not essential to the effects of cyclic AMP on DNA synthesis.
In summary, we found that elevated intracellular levels of cyclic AMP attenuate the PDGF-Rβ-mediated signalling transduction pathway including tyrosine phosphorylation of PDGF-Rβ, PI 3′-K, PLC-γ1 and ERK1/2. But most importantly, cyclic AMP-mediated inhibition of the transition of the cells to the S phase after stimulation with PDGF-BB correlates well with the ability of cyclic AMP to cause morphological changes of VSMCs via disruption of Sm α-actin filament array and loss of focal adhesions. These findings demonstrate that full mitogenic activity of PDGF-BB on VSMCs may only be exerted if they possess an intact Sm α-actin filament array. These findings may provide new molecular mechanism for the growth inhibitory action of cyclic AMP obtained in VSMCs.
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Sa568/3-1) and by a grant from Sonderforschungsbereich 176 TPA-10.
Abbreviations
- CDK
cyclin-dependent kinase
- CTX
cholera toxin
- DMEM
Dulbecco' modified Eagle' medium
- ERKs
extracellular response kinases
- FAK
focal adhesion kinase
- FCS
foetal calf serum
- GAP
GTPase activating protein
- GTP
guanosine triphosphate
- MAP
mitogenic-activated protein
- PDGF
platelet-derived growth factor
- PI 3′
phosphatidylinositol 3′
- PLC-γ1
phospholipase C-γ1
- Raf-1
a 74 kDa protein kinase encoded by the proto-oncogene raf-1
- SDS–PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- VSMCs
vascular smooth muscle cells
References
- ARVIDSSON A.K., RUPP E., NÅNBERG E., DOWNWARD J., RÖNNSTRAND L., WENNSTRÖM S., SCHLESSINGER J., HELDIN C.H., CLAESSON-WELSH L. Tyr-716 in the platelet-derived growth factor beta-receptor kinase insert is involved in GRB2 binding and Ras activation. Mol. Biol. Cell. 1994;14:6715–6726. doi: 10.1128/mcb.14.10.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASSENDER J.W., SOUTHGATE K.M., HALLETT M.B., NEWBY A.C. Inhibition of proliferation, but not of Ca2+ mobilization, by cyclic AMP and GMP in rabbit aortic smooth-muscle cells. Biochem. J. 1992;288:527–532. doi: 10.1042/bj2880527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOGUSKI M.S., MCCORMICK F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- BRADFORD M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- COOK S.J., MCCORMICK F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993;262:1069–1072. doi: 10.1126/science.7694367. [DOI] [PubMed] [Google Scholar]
- EDELMAN A.M., BLUMENTHAL D.K., KREBS E.G. Protein serine/threonine kinases. Annu. Rev. Biochem. 1987;56:567–613. doi: 10.1146/annurev.bi.56.070187.003031. [DOI] [PubMed] [Google Scholar]
- ERIKSSON A., SIEGBAHN A., WESTERMARK B., HELDIN C.H., CLAESSON-WELSH L. PDGF alpha- and beta-receptors activate unique and common signal transduction pathways. EMBO J. 1992;11:543–550. doi: 10.1002/j.1460-2075.1992.tb05085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIASSON E., SERVANT M.J., MELOCHE S. Cyclic AMP-mediated inhibition of angiotensin II-induced protein synthesis is associated with suppression of tyrosine phosphorylation signaling in vascular smooth muscle cells. J. Biol. Chem. 1997;272:26879–26886. doi: 10.1074/jbc.272.43.26879. [DOI] [PubMed] [Google Scholar]
- GLASS W.F., KREISBERG J.I. Regulation of integrin-mediated adhesion at focal contact by cyclic AMP. J. Cell. Physiol. 1993;157:296–306. doi: 10.1002/jcp.1041570212. [DOI] [PubMed] [Google Scholar]
- GRAVES L.M., BORNFELDT K.E., RAINES E.W., POTTS B.C., MACDONALD S.G., ROSS R., KREBS E.G. Protein kinase A antagonizes platelet-derived growth factor-induced signaling by mitogenic-activated protein kinase in human arterial smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10300–10304. [Google Scholar]
- HAN J.-D., RUBIN C.S. Regulation of cytoskeleton organization and paxillin dephosphorylation by cAMP. J. Biol. Chem. 1996;271:29211–29215. doi: 10.1074/jbc.271.46.29211. [DOI] [PubMed] [Google Scholar]
- HOPPE J., WEICH H.A., EICHNER W. Preparation of biologically active PDGF type BB from a fusion protein expressed in Escherichia Coli. Biochemistry. 1989;28:2956–2960. doi: 10.1021/bi00433a032. [DOI] [PubMed] [Google Scholar]
- HUANG S., CHEN C.S., INGBER D.E. Control of cyclin D1, p27 (Kip1), and cell cycle progression in human capillary endothelial cells by cell shape and cytoskeletal tension. Mol. Biol. Cell. 1998;9:3179–3193. doi: 10.1091/mbc.9.11.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON K.W., DAVIS B.H., SMITH K.A. cAMP antagonizes interleukin 2-promoted T-cell cycle progression at a discrete point in early G1. Proc. Natl. Acad. Sci. U.S.A. 1988;85:6072–6076. doi: 10.1073/pnas.85.16.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAPLAN D.R., MORRISON D.K., WONG G., MCCORMICK F., WILLIAMS L. PDGF β-receptor stimulates tyrosine phosphorylation of GAP and association of GAP with a signaling complex. Cell. 1990;61:125–133. doi: 10.1016/0092-8674(90)90220-9. [DOI] [PubMed] [Google Scholar]
- KATO J.Y., MATSUOKA M., POLYAK K., MASSAGUÉ J., SHERR C.J. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell. 1994;79:487–496. doi: 10.1016/0092-8674(94)90257-7. [DOI] [PubMed] [Google Scholar]
- KYPTA R.M., GOLDBERG Y., ULUG E.T., COURTNEIDGE S.A. Association between the PDGF receptor and members of the src family of tyrosine kinases. Cell. 1990;62:481–492. doi: 10.1016/0092-8674(90)90013-5. [DOI] [PubMed] [Google Scholar]
- L'ALLEMAIN G., LAVOIE J.N., RIVARD N., BALDIN V., POUYSSEGUR J. Cyclin D1 expression is a major target of the cAMP-induced inhibition of cell cycle entry in fibroblasts. Oncogene. 1997;14:1981–1990. doi: 10.1038/sj.onc.1201038. [DOI] [PubMed] [Google Scholar]
- LAMB N.J., FERNADEZ A., CONTI M.A., ADELSTEIN R., GLASS D.B., WELCH W.J., FERAMISCO J.R. Regulation of actin microfilament integrity in living nonmuscle cells by the cAMP-dependent protein kinase and the myosin light chain kinase. J. Cell Biol. 1988;106:1955–1971. doi: 10.1083/jcb.106.6.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGNALDO I., POUYSSÉGUR, PARIS S. Cyclic AMP inhibits mitogen-induced DNA synthesis in hamster fibroblasts, regardless of the signalling pathway involved. FEBS Lett. 1989;245:65–69. doi: 10.1016/0014-5793(89)80193-0. [DOI] [PubMed] [Google Scholar]
- MARSHALL C.J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995a;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- MARSHALL M.S. Ras target proteins in eukaryotic cells. FASEB J. 1995b;9:1311–1318. doi: 10.1096/fasebj.9.13.7557021. [DOI] [PubMed] [Google Scholar]
- MCKENZIE F.R., POUYSSÉGUR J. cAMP-mediated growth inhibition in fibroblasts is not mediated via mitogen-activated protein (MAP) kinase (ERK) inhibition. cAMP-dependent protein kinase induces a temporal shift in growth factor-stimulated MAP kinases. J. Biol. Chem. 1996;271:13476–13483. doi: 10.1074/jbc.271.23.13476. [DOI] [PubMed] [Google Scholar]
- MEISENHELDER J., SUH P.A., RHEE S.G., HUNTER T. Phospholipase C-γ is a substrate for the PDGF and EGF receptor protein-tyrosine kinases in vivo and in vitro. Cell. 1989;57:1109–1122. doi: 10.1016/0092-8674(89)90048-2. [DOI] [PubMed] [Google Scholar]
- MUSA N.L., RAMAKRISHNAN M., LI J., KARTHA S., LIU P., PESTELL R.G., HERSHENSON M.B. Forskolin inhibits cyclin D1 expression in cultured airway smooth-muscle cells. Am. J. Respir. Cell Mol. Biol. 1999;20:352–358. doi: 10.1165/ajrcmb.20.2.3160. [DOI] [PubMed] [Google Scholar]
- NOBES C.D., HAWKINS P., STEPHENS L., HALL A. Activation of the small gtp-binding proteins rho and rac by growth factor receptors. J. Cell Sci. 1995;108:225–233. doi: 10.1242/jcs.108.1.225. [DOI] [PubMed] [Google Scholar]
- OLSON M.F., ASHWORTH A., HALL A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- PAWSON T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- RIDLEY A.J., PATERSON H.F., JOHNSTON C., DIEKMANN D., HALL A. The small gtp-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- RÖNNSTRAND L., MORI S., ARRIDSON A.K., ERIKSSEN A., WERNSTEDT C., HELLMAN U., CLAESSON-WELSH L., HELDIN C.H. Identification of two C-terminal autophosphorylation sites in the PDGF-β-receptor: involvement in the interaction with phospholipase C-γ. EMBO J. 1992;11:3911–3919. doi: 10.1002/j.1460-2075.1992.tb05484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SACHINIDIS A., FLESCH M., KO Y., SCHRÖR K., BÖHM M., DÜSING R., VETTER H. Thromboxane A2 and vascular smooth muscle cell proliferation. Hypertension. 1995;26:771–780. doi: 10.1161/01.hyp.26.5.771. [DOI] [PubMed] [Google Scholar]
- SCHALLER M.D., PARSONS J.T. Focal adhesion kinase and associated proteins. Curr. Opin. Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- SCHOENWAELDER S.M., BURRIDGE K. Bidrectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 1999;11:274–286. doi: 10.1016/s0955-0674(99)80037-4. [DOI] [PubMed] [Google Scholar]
- SCOTT P.H., BELHAM C.M., AL-HAFIDH J., CHILVERS E.R., PEACOCK A.J., GOULD G.W., PLEVIN R. A regulatory role for cAMP in phosphatidylinositol 3-kinase/p70 ribosomal S6 kinase-mediated DNA synthesis in platelet-derived-growth-factor-stimulated bovine airway smooth-muscle cells. Biochem. J. 1996;318 (Pt 3):965–971. doi: 10.1042/bj3180965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHERR C.J. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- SHERR C.J., ROBERTS J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- SILVENNOINEN O., SCHINDLER C., SCHLESSINGER J., LEVY D.E. Ras-independent growth factor signaling by transcription factor tyrosine phosphorylation. Science. 1993;261:1736–1739. doi: 10.1126/science.8378775. [DOI] [PubMed] [Google Scholar]
- SIMM A., HOPPE V., KARBACH D., LEICHT M., FENN A., HOPPE J. Late signals from the PDGF receptors leading to the activation of the p70S6-kinase are necessary for the transition from G1 to S phase in AKR-2B cells. Exp. Cell Res. 1998;244:379–393. doi: 10.1006/excr.1998.4200. [DOI] [PubMed] [Google Scholar]
- SMALL J.V., ROTTNER K., KAVERINA I. Functional design in the actin cytoskeleton. Curr. Opin. Cell Biol. 1999;11:54–60. doi: 10.1016/s0955-0674(99)80007-6. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI T., KAWAHARA Y., OKUDA M., YOKOYAMA M. Increasing cAMP antagonizes hypertrophic response to angiotensin II without affecting Ras and MAP kinase activation in vascular smooth muscle cells. FEBS Lett. 1996;397:89–92. doi: 10.1016/s0014-5793(96)01145-3. [DOI] [PubMed] [Google Scholar]
- VADIVELOO P.K., FILONZI E.L., STANTON H.R., HAMILTON J.A. G1 phase arrest of human smooth muscle cells by heparin, IL-4 and cAMP is linked to repression of cyclin D1 and cdk2. Atherosclerosis. 1997;133:61–69. doi: 10.1016/s0021-9150(97)00116-0. [DOI] [PubMed] [Google Scholar]
- WENNSTROM S., HAWKINS P., COOKE F., HARA K., YONEZAWA K., KASUGA M., JACKSON T., CLAESSON-WELSH L., STEPHENS L. Activation of phosphoinositide 3-kinase is required for pdgf-stimulated membrane ruffling. Curr. Biol. 1994a;4:385–393. doi: 10.1016/s0960-9822(00)00087-7. [DOI] [PubMed] [Google Scholar]
- WENNSTROM S., SIEGBAHN A., YOKOTE K., ARVIDSSON A.K., HELDIN C.H., MORI S., CLAESSON-WELSH L. Membrane ruffling and chemotaxis transduced by the pdgf beta-receptor require the binding site for phosphtidylinositol 3′ kinase. Oncogene. 1994b;9:651–660. [PubMed] [Google Scholar]
- WU J., DENT P., JELINEK T., WOLFMAN A., WEBER M., STURGILL T.W. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′, monophosphate. Science. 1993;262:1065–1068. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- ZACHARY I., ROZENGURT E. Focal adhesion kinase (p125FAK): a point of convergence in the action of neuropeptides, integrins, and oncogenes. Cell. 1992;11:891–894. doi: 10.1016/0092-8674(92)90385-p. [DOI] [PubMed] [Google Scholar]
- ZACHARY I., SINNETT-SMITH J., TURNER C.E., ROZENGURT E. Bombesin, vasopressin, and endothelin rapidly stimulate tyrosine phosphorylation of the focal adhesion-associated protein paxillin in Swiss 3T3 cells. J. Biol. Chem. 1993;268:22060–22065. [PubMed] [Google Scholar]