

Abstract
In segments of rat vena cava preincubated with [3H]-noradrenaline and superfused with physiological salt solution, the influence of agmatine on the electrically evoked [3H]-noradrenaline release, the EP3 prostaglandin receptor-mediated and the α2D-adrenoceptor-mediated inhibition of evoked [3H]-noradrenaline release was investigated.
Agmatine (0.1–10 μM) by itself was without effect on evoked [3H]-noradrenaline release. In the presence of 10 μM agmatine, the prostaglandin E2(PGE2)-induced EP3-receptor-mediated inhibition of [3H]-noradrenaline release was not modified, whereas the α2D-adrenoceptor-mediated inhibition of [3H]-noradrenaline release induced by noradrenaline, moxonidine or clonidine was more pronounced than in the absence of agmatine. However, 1 mM agmatine antagonized the moxonidine-induced inhibition of [3H]-noradrenaline release.
Agmatine concentration-dependently inhibited the binding of [3H]-clonidine and [3H]-rauwolscine to rat brain cortex membranes (Ki values 6 μM and 12 μM, respectively). In addition, 30 and 100 μM agmatine increased the rate of association and decreased the rate of dissociation of [3H]-clonidine resulting in an increased affinity of the radioligand for the α2D-adrenoceptors.
[14C]-agmatine labelled specific binding sites on rat brain cortex membranes. In competition experiments. [14C]-agmatine was inhibited from binding to its specific recognition sites by unlabelled agmatine, but not by rauwolscine and moxonidine.
In conclusion, the present data indicate that agmatine both acts as an antagonist at the ligand recognition site of the α2D-adrenoceptor and enhances the effects of α2-adrenoceptor agonists probably by binding to an allosteric binding site of the α2D-adrenoceptor which seems to be labelled by [14C]-agmatine.
Keywords: Agmatine, rat α2-adrenoceptor, [3H]-clonidine binding, [3H]-rauwolscine binding, allosteric site, neurotransmission
Introduction
Agmatine is a polycationic guanidine derivative synthesized from L-arginine by the enzyme arginine decarboxylase. In radioligand binding studies, agmatine exhibited affinity for all four subtypes (A–D) of the α2-adrenoceptor (Ki 0.8–164 μM; Li et al., 1994; Piletz et al., 1995; Pinthong et al., 1995a,1995b; Regunathan & Reis, 1996) as well as for the non-adrenoceptor I1 (0.03–0.7 μM) and I2 (1–74 μM) imidazoline binding sites (Li et al., 1994; Piletz et al., 1995). It had no affinity for the α1- and β-adrenoceptors, 5-HT2 serotonin and D2 dopamine binding sites (Li et al., 1994), κ opiod and adenosine A1 receptors (Szabo et al., 1995). Agmatine is present in most mammalian tissues including brain and blood vessels (Raasch et al., 1995; Regunathan et al., 1996) and its distribution in the central nervous system suggests a role as a neurotransmitter (Otake et al., 1998). However, in most tissues in which functional tests have been carried out, agmatine failed to act as an agonist or antagonist at pre- and postsynaptic α2-adrenoceptors (Pinthong et al., 1995b; Gonzalez et al., 1996; Jurkiewicz et al., 1996; Pineda et al., 1996). In contrast, agmatine is able to block nicotinic and 5-HT3 receptor channels via an allosteric modulatory site (Loring 1990; Molderings et al., 1996).
In the present study, the first aim was to investigate the influence of agmatine on the α2D-autoreceptor-mediated (in comparison to the EP3-receptor-mediated) modulation of noradrenaline release from perivascular sympathetic nerves in rat vena cava. The second aim was to examine whether agmatine interacts with an allosteric site associated with α2-adrenoceptors. Since α2D-adrenoceptors on cardiovascular sympathetic nerve terminals (inhibitory presynaptic autoreceptors) and in the central nervous system are identical, the latter can be used as a model for the former. Therefore, radioligand binding experiments were performed with [3H]-rauwolscine, [3H]-clonidine and [14C]-agmatine in rat cerbral cortex membranes in which α2-adrenoceptors mainly of the α2D-subtype are known to be abundant (Greenberg et al., 1976, Bricca et al., 1989, Hussain et al., 1993).
Methods
Superfusion studies
The experiments were carried out on segments of vena cava from adult Wistar rats. The segments were incubated for 30 min in 1.5 ml physiological salt solution (37°C, composition see below) containing (−)-[2,5,6-3H]-noradrenaline 0.1 μM). Subsequently, they were mounted vertically in an organ bath (tension adjusted to 2 g) between two parallel platinum electrodes (1.5 cm long) and superfused with [3H]-noradrenaline-free physiological salt solution of 37°C at a rate of 2 ml min−1. The composition of the solution was (mM): NaCl 118, Na2HPO4 1.2, NaHCO3 25, KCl 4.7, CaCl2 1.6, MgSO4 1.2, glucose 11, ascorbic acid 0.3, Na2EDTA 0.038, aerated with 95% O2 and 5% CO2. Throughout superfusion of the blood vessels this solution contained desipramine (0.6 μM) and corticosterone (40 μM) to block neuronal and extraneuronal noradrenaline uptake.
For transmural electrical stimulation, rectangular pulses of 0.3 ms duration and 150 mA were delivered to the segments at frequencies of 2 Hz during up to five 3-min periods after 93 (S1), 117 (S2), 141 (S3), 165 (S4) and 189 (S5) min of superfusion. The superfusate was continuously collected in 3- or 6-min fractions. At the end of superfusion the blood vessels were solubilized with Soluene®. The radioactivity in the superfusate samples and blood vessels was determined by liquid scintillation counting.
Unless stated otherwise, the receptor agonists were applied at concentrations increasing by a factor of 10 from 9 min before until 15 min after the onset of S3, S4 and S5. Separate control experiments (no agonists applied) were carried out for each series of experiments. In interaction experiments, the interacting drug (agmatine, clonidine or rauwolscine) was applied from 13 min before S1 until the end of superfusion.
Tritium efflux was calculated as the fraction of tritium present in the strip at the onset of the respective collection period. Basal tritium efflux was expressed as the ratio of the fractional efflux during the collection period immediately before S3, S4 or S5 (t3, t4, t5) over that immediately before S2 (t2); S1 (and t1 correspondingly) was not included in the evaluation, since it was applied as a conditioning stimulation period. Stimulation-evoked tritium overflow was calculated by subtraction of the basal efflux from the total efflux during the 12 min subsequent to the onset of stimulation; basal efflux was assumed to decrease linearly from the collection period before to that 12–15 min after onset of stimulation. Evoked tritium overflow was calculated as a percentage of tissue tritium at the onset of stimulation, and the ratios of the overflow evoked by S3, S4 or S5 over that evoked by S2 were determined.
Results are given as means±s.e.mean. Students' t-test for unpaired data was used for comparison of mean values. Apparent pA2 values were determined according to formula (4) of Furchgott (1972). As an estimate of potency the concentration that reduced evoked tritium overflow by 50% (IC50%) was determined by interpolation between the two nearest points of the concentration-response curve for mean values. pIC50% values are the negative logarithms of these concentrations.
Binding studies
Binding experiments were carried out in principle as described by Bylund et al. (1992) with slight modifications. Cerebral cortices obtained from male Wistar rats were homogenized (Potter-Elvehjem; 10 up and down strokes during 1 min) in 25 volumes of ice-cold Tris-HCl buffer (Tris 50 mM, pH 8.0; EDTA 5 mM; sucrose 10.27%) and centrifuged at 1000×g for 10 min (4°C). The supernatant was centrifuged at 35,000×g for 10 min and the pellet was resuspended in 10 volumes of Tris-HCl buffer and frozen at −80°C.
In the competition experiments, a 400-μl aliquot of the membranes (containing 0.2–0.3 mg protein) was incubated for 30 min with 1 nM [3H]-rauwolscine or 364 nM [14C]-agmatine and for 45 min with 5 nM [3H]-clonidine (25 μl each), respectively, at room temperature in a final volume of 0.5 ml. In the experiments with [3H]-clonidine, 1 μM cimetidine which at that concentration binds to I1 imidazoline binding sites (Ernsberger et al., 1990) was added to the assay to prevent the radioligand from binding to I1 imidazoline binding sites. The reaction was stopped by rapid vacuum filtration with a Brandel cell harvester through Whatman GF/C glass-fibre filters presoaked with polyethylenimine 0.5 M followed by rapid washing of the incubation tubes and filters with 10 ml ice-cold buffer. Filters were placed in 6 ml of scintillation fluid and shaken overnight, and the radioactivity was determined by liquid scintillation counting at 44% ([3H]) and 100% ([14C] efficiency. Non-specific binding was defined as [3H]-rauwolscine and [3H]-clonidine binding in the presence of 10 μM noradrenaline (40% and 36%, respectively). [14C]-Agmatine binding in the presence of 1 mM unlabelled agmatine revealed nonsaturable [14C]-agmatine binding sites (17%, filter binding amounted to 56% of total binding).
Kinetic experiments were carried out with 5 nM [3H]-clonidine. Association was initiated by addition of membranes and dissociation by addition of 10 μM noradrenaline after 45 min of incubation of membranes with the radioligand. Reactions were stopped at points of time between 30 s and 60 min by rapid filtration and washing with ice-cold buffer.
All experiments were carried out in triplicate. Data from the kinetic and competition experiments were analysed using the least-squares fitting program PRISM (GraphPad Software Inc., San Diego, U.S.A.). The affinity (Kd value) of agmatine in the homologous competitive binding experiments was calculated by the formulas (3) and (5) of DeBlasi et al. (1989). Results are expressed as mean values±s.e.mean. The statistical significance of differences was analysed by Dunnett' test and Bonferroni' test.
Drugs used
(−)-[2,5,6-3H]-noradrenaline, specific activity 43.7–55 Ci mmol−1; [benzene ring 3H]-clonidine, specific activity 60 Ci mmol−1 (New England Nuclear, Dreieich, Germany); [O-methyl-3H]-rauwolscine, specific activity 76 Ci mmol−1 (Amersham, Braunschweig, Germany); [14C]-agmatine, specific activity 55 mCi mmol−1 (American Radiolabeled Chem. Inc, St. Louis, U.S.A.); moxonidine (Beiersdorf, Hamburg, Germany); corticosterone, noradrenaline base, agmatine sulphate, rauwolscine hydrochloride, prostaglandin E2 (Sigma, München, Germany); clonidine hydrochloride (Boehringer, Ingelheim, Germany); desipramine hydrochloride (Ciba-Geigy, Wehr, Germany); cirazoline hydrochloride (Synthélabo, Paris, France); propranolol hydrochloride (ICI, Planckstadt, Germany). Drugs were dissolved in saline with the following exceptions: noradrenaline base was dissolved in HCl (0.01 M) and corticosterone in propandiol-1,2. The stock solutions were further diluted in saline.
Results
Superfusion experiments in rat vena cava
Basal tritium efflux
In control experiments in the absence of test drugs, basal tritium efflux during t2 from segments of the rat vena cava preincubated with [3H]-noradrenaline was 0.22±0.03 nCi min−1 (n=7; in a representative control series), corresponding to a fractional rate of efflux of 0.00059±0.00002 min−1. It did not significantly differ from that in the presence of 10–1000 μM agmatine and/or 0.3 μM rauwolscine from 13 min before S1 until the end of the experiments. Basal efflux decreased with time, as reflected by tn/t2 ratios which, in the control experiments, declined from t3/t2 (0.86±0.04) to t5/t2 (0.77±0.02). The tn/t2 values were not altered by 10 to 1000 μM agmatine, 10 μM clonidine and/or by 0.3 μM rauwolscine present from 13 min before S1 until the end of the experiments (controls for the interaction experiments of these drugs with other compounds) nor by noradrenaline, moxonidine, clonidine and PGE2 (see Figure 1 for concentrations and time schedule).
Figure 1.
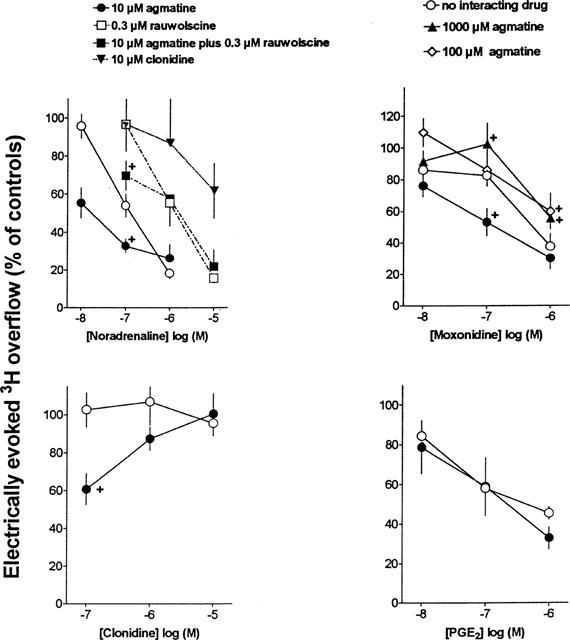
Effects of noradrenaline, moxonidine, clonidine and PGE2 on electrically evoked tritium overflow from segments of the rat vena cava preincubated with [3H]-noradrenaline and interaction with agmatine, clonidine and/or rauwolscine. Interacting drugs were present from 13 min before S1 until the end of the experiments. The experiments with noradrenaline and the corresponding controls were carried out in the presence of 1 μM propranolol throughout superfusion. Ordinate, S3/S2, S4/S2 and S5/S2 overflow ratios, expressed as percentage of ratios in corresponding control experiments without noradrenaline, moxonidine, clonidine or PGE2. Means±s.e.mean from 5–12 tissue segments. All Sn/S2 ratios below 75% of the corresponding Sn/S2 ratios in the respective controls were significantly different from these control ratios (i.e. in the absence of the respective agonist).+P<0.05, compared with the effect of the corresponding agonist concentration in the absence of agmatine.
Evoked tritium overflow under control conditions
In a representative series of control experiments in the absence of test drugs from 13 min before S1 until the end of the experiments, the tritium overflow evoked at 2 Hz by S2 amounted to 7.77±0.38 nCi (corresponding to 1.45±0.30% of tissue tritium; n=7); evoked tritium overflow remained constant (S3/S2: 0.90±0.04; S4/S2: 0.90±0.05; S5/S2: 0.86±0.05). These findings were not significantly different from the values determined in control experiments in which 10–1000 μM agmatine, 10 μM clonidine and/or 0.3 μM rauwolscine was/were present from 13 min before S1 until the end of the experiments.
Actions of α2-adrenoceptor agonists, agmatine and PGE2
Agmatine in concentrations up to 1 mM tended to increase the electrically-evoked tritium overflow (0.1 μM: 93.5±7.2%; 1 μM: 88.6±5.8%; 10 μM: 84.6±9.3%; 100 μM: 134.9± 14.9%; 300 μM: 121.1±15.5%; 1000 μM: 112.3±9.9%; n=6–7; values represent percentages of the overflow at S3, S4 and S5, respectively, in control experiments in the absence of agmatine); however, due to a high variance of the data the facilitatory effect did not reach the level of significance.
In the absence of agmatine, the full α2-adrenoceptor agonists noradrenaline and moxonidine and the EP3 receptor agonist PGE2 inhibited the electrically evoked tritium overflow (Figure 1, open circles; pIC50% values of noradrenaline, moxonidine and PGE2: 6.89, 6.44, and 6.37, respectively). In contrast, clonidine which is a partial agonist with low intrinsic activity at α2-adrenoceptors (Jasper et al., 1998) failed to inhibit evoked tritium overflow (Figure 1, open circles).
In the presence of 10 μM agmatine from 13 min before S1 until the end of the experiments, the concentration-response curves of noradrenaline and moxonidine for their inhibitory effect on evoked tritium overflow were shifted to the left (Figure 1, closed circles; pIC50% values of noradrenaline and moxonidine: 7.76 and 6.86, respectively). However, in the presence of 100 μM agmatine, the concentration response-curve of moxonidine tended to be shifted to the right (Figure 1). A more clear-cut rightward shift of the concentration-response curve of moxonidine was determined in the presence of 1000 μM agmatine (Figure 1, closed triangles up).
In the presence of 10 μM agmatine, clonidine 0.1 μM significantly inhibited evoked tritium overflow, whereas higher concentrations did not (Figure 1, closed circles). The concentration-response curve of PGE2 was not altered in the presence of 10 μM agmatine (Figure 1, closed circles).
Rauwolscine 0.3 μM present in the superfusion fluid from 13 min before S1 until the end of the experiments, shifted the concentration-response curve of noradrenaline to the right yielding an apparent pA2 value of 7.31 (Figure 1, open squares). In the presence of both 0.3 μM rauwolscine and 10 μM agmatine, 0.1 μM noradrenaline inhibited the evoked tritium overflow (Figure 1, closed squares), whereas in the presence of rauwolscine alone (i.e. in the absence of agmatine) it was without effect (Figure 1, open squares). The apparent pA2 value for rauwolscine determined in the presence of 10 μM agmatine amounted to 8.49. Clonidine 10 μM present in the superfusion fluid from 13 min before S1 until the end of the experiments, also shifted the concentration-response curve of noradrenaline to the right (Figure 1, closed triangles) yielding an apparent pA2 value of 7.05 (determined at the level of IC30%).
Radioligand binding experiments
The specific binding of [3H]-clonidine (5 nM) increased with time and reached equilibrium already after about 20 min (not shown). The time t1/2 (0.693/observed rate of association) at which half of the equilibrium specific binding of [3H]-clonidine was reached amounted to 2.15 min (Table 1). Agmatine 30 μM did not significantly change the association of the radioligand, whereas association was significantly enhanced in the presence of 100 μM agmatine (t1/2=0.85 min; Table 1).
Table 1.
Half life (t1/2) of dissociation and association and Kd of [3H]-clonidine derived from the respective kinetic data in the absence and presence of agmatine in the incubation buffer. Number of experiments in parentheses
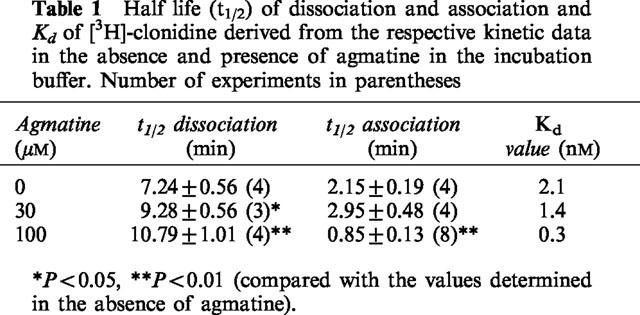
The dissociation of [3H]-clonidine from its binding sites (induced by 10 μM noradrenaline) was monoexponential with a dissociation rate constant of 0.084±0.011 min−1 (t1/2= 7.24 min; see Table 1). In the presence of 30 and 100 μM agmatine the dissociation of the radioligand from its specific binding sites was significantly delayed (Table 1). The Kd values calculated from the kinetic experiments decreased with increasing agmatine concentration in the incubation buffer (Table 1). Higher agmatine concentrations could not be investigated, since at those concentrations agmatine inhibited [3H]-clonidine binding nearly complete (see Figure 2); hence, no specific binding could be reliably detected in respective kinetic experiments.
Figure 2.
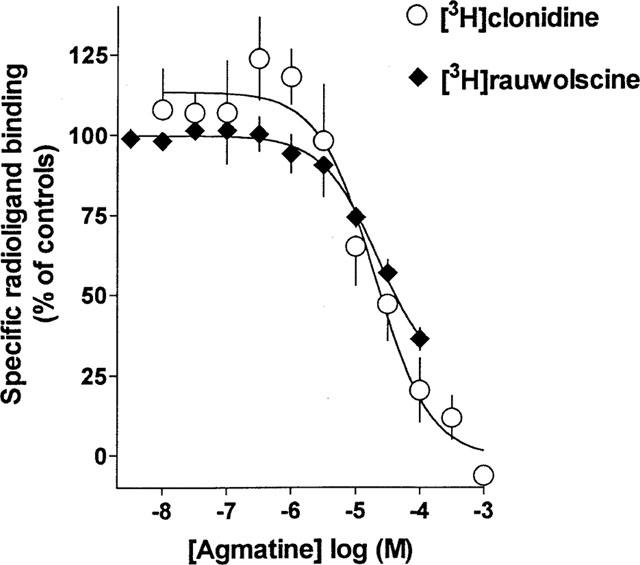
Competition of agmatine with 5 nM [3H]-clonidine or 1 nM [3H]-rauwolscine for their specific binding sites in rat cerebral cortex membranes. Each point is the mean of 3–4 experiments performed in triplicate.
In competition experiments of agmatine with [3H]-clonidine at 5 nM, agmatine induced a monophasic inhibition (nHill=−0.97, not significantly different from unity; Figure 2). The Ki-value of agmatine amounted to 5.8±1.7 μM.
In competition experiments of agmatine with 1 nM [3H]-rauwolscine, agmatine also produced a monophasic inhibition (nHill=−0.95, not significantly different from unity; Figure 2). The Ki-value of agmatine amounted to 12.1±2.5 μM.
To rule out that [14C]-agmatine retained in the brain membranes after the incubation period was due to an incorporation into vesicles by an agmatine transporter (recently described by Sastre et al., 1997), the time course of dissociation of [14C]-agmatine was studied. The dissociation of [14C]-agmatine from its binding sites (induced by 1 mM agmatine) was monoexponential with a dissociation rate constant k−1=0.107±0.101 min−1 (n=4). Hence, it appears justified to assume that [14C]-agmatine labelled specific binding sites in the membranes rather than being transported into the vesicles. Homologous competition experiments of [14C]-agmatine with unlabelled agmatine revealed a reaction with one binding site with a Kd value of 158.7±100.5 μM (Figure 3). Cirazoline also weakly inhibited specific binding of [14C]-agmatine in a concentration-dependent manner. Cirazoline at 100 μM (i.e. the highest concentration tested) produced an inhibition by 33% (extrapolated Ki value 199 μM; Figure 3). Rauwolscine and moxonidine failed to inhibit specific [14C]-agmatine binding at concentrations up to 100 μM (not shown).
Figure 3.
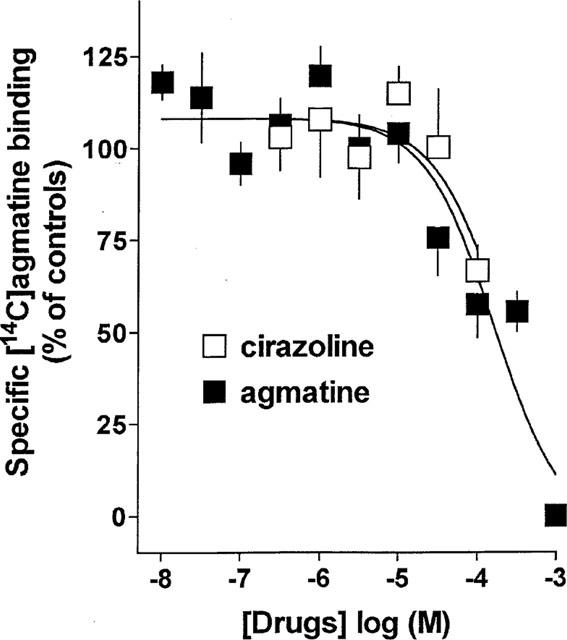
Competition of unlabelled agmatine and cirazoline with [14C]-agmatine (364 nM) for its specific binding sites in rat cerebral cortex membranes. Each point is the mean of 5–7 experiments performed in triplicate.
Discussion
One of the aims of the present study was to investigate the influence of agmatine on presynaptic α2D-autoreceptors of the sympathetic nerves in the rat vena cava (Göthert et al., 1986; subclassification by Trendelenburg et al., 1997). For this purpose, the effects of the full α2-adrenoceptor agonists noradrenaline and moxonidine and of the partial α2-adrenoceptor agonist clonidine on the electrically evoked tritium overflow from the rat vena cava preincubated with [3H]-noradenaline was investigated in the absence and presence of agmatine. Under the conditions applied (i.e. presence of neuronal and extraneuronal uptake inhibitors), evoked tritium overflow reflects action-potential-induced release of tritiated and unlabelled noradrenaline from the sympathetic axon terminals contained in the blood vessel wall (for further details, see Göthert et al., 1986).
The main aim of this investigation is based on the suggestion that agmatine is a novel neurotransmitter capable of recognizing both α2-adrenoceptors and non-adrenoceptor imidazoline binding sites (Reis & Regunathan, 1999). Agmatine binds to imidazoline sites with high to moderate affinity in radioligand binding studies (Li et al., 1994; Piletz et al., 1995) and exhibits intrinsic activity in functional assays for imidazoline recognition sites in the brain and periphery (for review, see Regunathan & Reis, 1996; Molderings 1997). However, no α2-adrenoceptor-mediated function could be reliably ascribed to agmatine in several α2-adrenoceptor containing preparations of various species (for references, see Introduction). In accordance with those findings, agmatine failed to significantly modulate the electrically evoked noradrenaline release in the present experiments on the rat vena cava, only 100 μM agmatine tended to facilitate evoked noradrenaline release, whereas at higher concentrations no enhancement was observed. The same result has been obtained in the rabbit pulmonary artery and aorta (unpublished observations). Obviously, agmatine influences sympathetic neurotransmission in the rat vena cava in a more complex manner. The modification of the concentration-response curves for the full α2-adrenoceptor agonists noradrenaline and moxonidine by the relatively low agmatine concentration of 10 μM basically differs from the change of the moxonidine concentration-response curve produced by 1 mM agmatine. Whereas the latter concentration produced a rightward shift of the concentration-response curve, which can be most plausibly explained by a weak competitive antagonism, application of the lower agmatine concentration resulted in a higher potency of the α2-adrenoceptor agonists (leftward shift by 0.87 log unit). In the presence of 100 μM agmatine the facilitatory and inhibitory effects of agmatine seemed to be balanced, since the rightward shift of concentration-response curve of moxonidine was not as clear-cut as that in the presence of 1 mM agmatine. The potentiating effect of agmatine appears to be due to an influence on α2-adrenoceptors because the concentration-response curve of PGE2 which inhibited evoked noradrenaline release from the sympathetic nerves via presynaptic EP3 receptors (Molderings et al., 1992) to the same extent as noradrenaline and moxonidine, was not altered by agmatine (Figure 1). The results of the interaction experiments of 10 μM agmatine with noradrenaline and moxonidine suggest that agmatine may be a positive allosteric modulator at the presynaptic α2D-adrenoceptor in rat vena cava leading to an increased affinity of α2-adrenoceptor agonists for the α2-adrenoceptor. It is compatible with the assumption of an allosteric modulation that also the potency of rauwolscine to antagonize the noradrenaline-induced inhibition of noradrenaline release was higher in the presence than in the absence of agmatine (difference in apparent pA2 value: 1.18 log unit).
The agmatine-induced modification of the effect of clonidine can also be explained on the basis of a positive allosteric modulation by the polyamine. Clonidine has been reported to be a partial agonist with low intrinsic activity at α2D-adrenoceptors (Jasper et al., 1998). In accordance with this finding, clonidine markedly shifted the concentration-response curve of noradrenaline to the right suggesting an estimate of affinity for the presynaptic α2D-autoreceptors (apparent pA2 value) of about 7.00. Since the intrinsic activity of the partial agonist clonidine is relatively small, the error factor introduced into the estimate because of its agonistic effect is also small. In the absence of agmatine, clonidine at all concentrations investigated did not modify noradrenaline release; this can be explained by the suggestion that an inhibitory effect of clonidine due to its agonistic property is compensated by its antagonism against endogenous noradrenaline released by electrical stimulation. However, when 100 nM clonidine was applied in the presence of 10 μM agmatine, the positive allosteric effect of agmatine appears to result in a shift in the equilibrium between agonistic and antagonistic properties of clonidine towards agonism, leading to receptor activation. At the higher clonidine concentrations, the agonism unmasked by the positive allosteric effect of agmatine is re-balanced by the increased ability of clonidine to antagonize the effect of endogenous noradrenaline. Finally, a simultaneous action of clonidine at presynaptic imidazoline recognition sites present in the rat vena cava (Molderings & Göthert 1998) at which the drug is an agonist, might also influence the effect of clonidine.
Taken together, the data from our superfusion experiments suggest that agmatine enhances α2-adrenoceptor-mediated inhibition of noradrenaline release from presynaptic nerve terminals via an allosteric binding site and that at higher concentration the compound acts as an antagonist at the ligand recognition site of the α2-adrenoceptor. Since the presynaptic α2D-autoreceptors in the cardiovascular system are identical with the postsynaptic α2D-adrenoceptors in the brain cortex, the latter can be used as a model for the former. Therefore, radioligand binding experiments were carried out with the radiolabelled partial α2D-adrenoceptor agonist [3H]-clonidine (in the presence of 1 μM cimetidine to mask I1 imidazoline binding sites; Ernsberger et al., 1990), the non-selective α2-adrenoceptor antagonist [3H]-rauwolscine and with [14C]-agmatine on rat cerebral cortex membranes. An interference of [3H]-clonidine with presynaptic imidazoline recognition sites is not to be expected because we previously failed to demonstrate the presence of presynaptic imidazoline recognition sites in the rat brain cortex (Schlicker et al., 1997).
In competition experiments agmatine inhibited [3H]-clonidine and [3H]-rauwolscine binding to sites, previously shown to be α2-adrenoceptors (Greenberg et al., 1976; Bricca et al., 1989; Hussain et al., 1993), with a potency (Ki values 6 μM and 12 μM, respectively) comparable to that reported in the literature (4 μM, Li et al., 1994; 8–17 μM, Pinthong et al., 1995a,1995b, 26–164 μM, Piletz et al., 1995). It can be concluded from this finding that agmatine binds with μmolar affinity to the ligand recognition site of α2-adrenoceptor. This inhibition of radioligand binding to the α2-adrenoceptor may be assumed to reflect the antagonistic property of agmatine at these receptors in the superfusion experiments (see above).
To identify the postulated allosteric modulatory site for agmatine at α2-adrenoceptors in radioligand binding experiments, association and dissociation kinetics of [3H]-clonidine in the absence and presence of increasing concentrations of agmatine were determined. These experiments revealed that agmatine concentration-dependently accelerates association of the radioligand to and delayed dissociation from the specific binding sites (Table 1). Accordingly, the affinity of [3H]-clonidine for the α2-adrenoceptors was 7 fold increased by 100 μM agmatine. These findings strongly supported the suggestion that agmatine acts at an allosteric site of the α2D-adrenoceptor. This allosteric site may be assumed to represent the allosteric site of action of agmatine in the superfusion experiments. Yet it should be noted that agmatine concentrations as high as 100 μM were necessary to markedly increase the affinity of clonidine for the α2-adrenoceptor in the radioligand binding experiments, whereas the increase in potency of the ligands in the functional experiments was detected at 10 times lower agmatine concentration. However, this quantitative difference may be related to differences in the experimental procedures: a drop in potency is not unusual when results of experiments obtained in isolated organs are compared with those observed in more artificial systems such as radioligand binding experiments. In contrast to the latter experiments, the physiological readout in isolated veins involves a whole sequence of events from activation of the α2D-adrenoceptors, Ca2+ influx and modulation of adenylate cyclase. Thus, the differences in potency between these bioassay systems probably reflect, among others, differences in signal amplification.
Finally an attempt was made to identify the allosteric modulatory site for agmatine in binding experiments with [14C]-agmatine. Due to the expected low affinity of agmatine for this allosteric site, we used a relatively high concentration of the radioligand. Since specific binding in absolute terms was low, the data can be interpreted in qualitative but not in quantitative terms. It can be derived from the data shown in Figure 2 that at this radioligand concentration only 3–6% of the ligand recognition sites of the α2-adrenoceptors are labelled by [14C]-agmatine. Hence, specific binding of [14C]-agmatine must represent another ligand binding site. Accordingly, neither rauwolscine (α2-adrenoceptor antagonist) nor moxonidine (α2-adrenoceptor/I1-imidazoline recognition site agonist) inhibited [14C]-agmatine binding, indicating that under the present conditions [14C]-agmatine did indeed not label α2-adrenoceptors or I1-imidazoline binding sites. In addition, cirazoline which is a high affinity ligand at α1-adrenoceptors and I2-imidazoline binding sites, up to 100 μM only produced a very weak inhibition of [14C]-agmatine binding, ruling out substantial binding to α1-adrenoceptors and I2-imidazoline binding sites. Hence, it seems justified to assume that [14C]-agmatine bound to the allosteric modulatory site at α2-adrenoceptors postulated on the basis of the superfusion experiments. Whether or not the clearly lower affinity of agmatine for the [14C]-agmatine binding sites found in the homologous displacement experiments than its potency to enhance the inhibitory effect of the α2-adrenoceptor agonists in the superfusion experiments reflects the true affinity of agmatine for the modulatory site remains to be established in future experiments.
In conclusion, our data suggest that agmatine is a positive modulator at an allosteric site of α2-adrenoceptors and an antagonist at the ligand recognition site of the α2-adrenoceptor. This pattern of effects may explain why agmatine either failed to act at α2-adrenoceptors or yielded inconsistent results in the respective previous studies.
Acknowledgments
The technical assistance of Mrs M. Hartwig and Mrs D. Funccius is gratefully acknowledged. The authors wish to thank Prof K. Mohr (Department of Pharmacology and Toxicology, Institute of Pharmacy, Bonn, Germany) for critically reading the manuscript and for his helpful comments and suggestions.
Abbreviations
- PGE2
prostaglandin E2
- S1–S5
stimulation periods 1–5
- t2–t5
collection period immediately before S1–S5, respectively
References
- BRICCA G., DONTENWILL M., LONIES A., FELDMAN J., BELCOURT A., BOUSQUET P. The imidazoline preferring receptor: binding studies in bovine, rat and human brainstem. Eur. J. Pharmacol. 1989;162:1–9. doi: 10.1016/0014-2999(89)90597-9. [DOI] [PubMed] [Google Scholar]
- BYLUND D.B., BLAXALL H.S., IVERSEN L.J., CARON M.G., LEFKOWITZ R.J., LOMASNEY J.W. Pharmacological characteristics of α2-adrenergic receptors: comparison of pharmacologically defined subtypes with subtypes identified by molecular cloning. Mol. Pharmacol. 1992;42:1–5. [PubMed] [Google Scholar]
- DE BLASI A., O'REILLY K., MOTULSKY H.J. Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol. Sci. 1989;10:227–229. doi: 10.1016/0165-6147(89)90266-6. [DOI] [PubMed] [Google Scholar]
- ERNSBERGER P., GIULIANO R., WILLETTE R.N., REIS D.J. Role of imidazole receptors in the vasodepressor response to clonidine analogs in the rostral ventrolateral medulla. J. Pharmacol. Exp. Ther. 1990;253:408–418. [PubMed] [Google Scholar]
- FURCHGOTT R.F.The classification of adrenoceptors (adrenergic receptors). An evaluation from the standpoint of receptor theory Handbook of experimental pharmacology. Catecholamines 1972XXXIIIBerlin, Heidelberg, New York: Springer; 283–335.ed. Blaschko H, Muscholl E. pp [Google Scholar]
- GONZALEZ C., REGUNATHAN S., REIS D.J., ESTRADA C. Agmatine, an endogenous modulator of noradrenergic neurotransmission in the rat tail artery. Br. J. Pharmacol. 1996;119:677–684. doi: 10.1111/j.1476-5381.1996.tb15726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GÖTHERT M., SCHLICKER E., KOLLECKER P. Receptor-mediated effects of serotonin and 5-methoxytryptamine on noradrenaline release in the rat vena cava and in the heart of the pithed rat. Naunyn-Schmiedeberg' Arch. Pharmacol. 1986;332:124–130. doi: 10.1007/BF00511401. [DOI] [PubMed] [Google Scholar]
- GREENBERG D.A., U'PRICHARD D.C., SNYDER S.H. α-Noradrenergic receptor binding in mammalian brain: differential labelling of agonist and antagonist states. Life Sci. 1976;19:67–76. doi: 10.1016/0024-3205(76)90375-1. [DOI] [PubMed] [Google Scholar]
- HUSSAIN J.F., KENDALL D.A., WILSON V.G. Species selective binding of [3H]-idazoxan to α2-adrenoceptor and non-adrenoceptor imidazoline binding sites in the central nervous system. Br. J. Pharmacol. 1993;109:831–837. doi: 10.1111/j.1476-5381.1993.tb13650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JASPER J.R., LESNICK J.D., CHANG L.K., YAMANISHI S.S., CHANG T.K., HSU S.A.O., DAUN D.A., BONHAUS D.W., EGLEN R.M. Ligand efficacy and potency at recombinant α2 adrenergic receptors. Biochem. Pharmacol. 1998;55:1035–1043. doi: 10.1016/s0006-2952(97)00631-x. [DOI] [PubMed] [Google Scholar]
- JURKIEWICZ N.H., DO CARMO L.G., HIRATA H., DA COSTA SANTOS W., JURKIEWICZ A. Functional properties of agmatine in rat vas deferens. Eur. J. Pharmacol. 1996;307:299–304. doi: 10.1016/0014-2999(96)00274-9. [DOI] [PubMed] [Google Scholar]
- LI G., REGUNATHAN S., BARROW C.J., ESHRAGI J., COOPER R., REIS D.J. Agmatine: an endogenous clonidine-displacing substance in the brain. Science. 1994;263:966–969. doi: 10.1126/science.7906055. [DOI] [PubMed] [Google Scholar]
- LORING R.H. Agmatine acts as an antagonist of neuronal nicotinic receptors. Br. J. Pharmacol. 1990;99:207–211. doi: 10.1111/j.1476-5381.1990.tb14680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLDERINGS G.J. Imidazoline receptors: basic knowledge, recent advances and future prospects for therapy and diagnosis. Drugs Future. 1997;22:757–772. [Google Scholar]
- MOLDERINGS G.J., GÖTHERT M. Presynaptic imidazoline receptors mediate inhibition of noradrenaline release from sympathetic nerves in rat blood vessels. Fundam. Clin. Pharmacol. 1998;12:388–397. doi: 10.1111/j.1472-8206.1998.tb00962.x. [DOI] [PubMed] [Google Scholar]
- MOLDERINGS G.J., MALINOWSKA B., SCHLICKER E. Inhibition of noradrenaline release in the rat vena cava via prostanoid receptors of the EP3 subtype. Br. J. Pharmacol. 1992;107:352–355. doi: 10.1111/j.1476-5381.1992.tb12750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLDERINGS G.J., SCHMIDT K., BÖNISCH H., GÖTHERT M. Inhibition of 5-HT3 receptor function by imidazolines in mouse neuroblastoma cells: potential involvement of σ2 binding sites. Naunyn-Schmiedeberg' Arch. Pharmacol. 1996;354:245–252. doi: 10.1007/BF00171054. [DOI] [PubMed] [Google Scholar]
- OTAKE K., RUGGIERO D.A., WANG H., MILNER T.A., REIS D.J. Regional localization of agmatine in the rat brain: an immunocytochemical study. Brain Res. 1998;787:1–14. doi: 10.1016/s0006-8993(97)01200-6. [DOI] [PubMed] [Google Scholar]
- PILETZ J.E., CHIKKALA D.N., ERNSBERGER P. Comparison of the properties of agmatine and endogenous clonidine-displacing substance at imidazoline and alpha-2 adrenergic receptors. J. Pharmacol. Exp. Ther. 1995;272:581–587. [PubMed] [Google Scholar]
- PINEDA J., RUIZ-ORTEGA J.A., MARTIN-RUIZ R., UGEDO L. Agmatine does not have activity at α2-adrenoceptors which modulate the firing rat of locus coeruleus neurones: an electrophysiological study in rat. Neuroscience Letters. 1996;219:103–106. doi: 10.1016/s0304-3940(96)13180-3. [DOI] [PubMed] [Google Scholar]
- PINTHONG D., HUSSAIN J.F., KENDALL D.A., WILSON V.G. Comparison of the interaction of agmatine and crude methanolic extracts of bovine lung and brain with α2-adrenoceptor binding sites. Br. J. Pharmacol. 1995a;115:689–695. doi: 10.1111/j.1476-5381.1995.tb14988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINTHONG D., WRIGHT I.K., HANMER C., MILLNS P., MASON R., KENDALL D.A., WILSON V.G. Agmatine recognizes α2-adrenoceptor binding sites but neither activates nor inhibits α2-adrenoceptors. Naunyn-Schmiedeberg' Arch. Pharmacol. 1995b;351:10–16. doi: 10.1007/BF00169058. [DOI] [PubMed] [Google Scholar]
- RAASCH W, REGUNATHAN S., LI G., REIS D.J. Agmatine, the bacterial amine, is widely distributed in mammalian tissues. Life Sci. 1995;56:2319–2330. doi: 10.1016/0024-3205(95)00226-v. [DOI] [PubMed] [Google Scholar]
- REGUNATHAN S., FEINSTEIN D.L., RAASCH W., REIS D.J. Agmatine, the decarboxylated arginine is localized and synthesized in glial cells. Neuroreport. 1996;6:1897–1900. doi: 10.1097/00001756-199510020-00018. [DOI] [PubMed] [Google Scholar]
- REGUNATHAN S., REIS D.J. Imidazoline receptors and their endogenous ligands. Annu. Rev. Pharmacol. Toxicol. 1996;36:511–544. doi: 10.1146/annurev.pa.36.040196.002455. [DOI] [PubMed] [Google Scholar]
- REIS D.J., REGUNATHAN S. Agmatine: an endogenous ligand at imidazoline receptors is a novel neurotransmitter. Ann. New Yord Acad. Sci. 1999;881:65–80. doi: 10.1111/j.1749-6632.1999.tb09343.x. [DOI] [PubMed] [Google Scholar]
- SASTRE M., REGUNATHAN S., REIS D.J. Uptake of agmatine into rat brain synaptosomes: possible role of cation channels. J. Neurochem. 1997;69:2421–2426. doi: 10.1046/j.1471-4159.1997.69062421.x. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., FINK K., KATHMANN M., MOLDERINGS G.J., GÖTHERT M. Effects of imidazolines on noradrenaline release in brain: an investigation into their relationship to imidazoline, α2 and H3 receptors. Neurochem. Int. 1997;30:73–83. doi: 10.1016/s0197-0186(96)00045-9. [DOI] [PubMed] [Google Scholar]
- SZABO B., URBAN R., LIMBERGER N., STARKE K. Cardiovascular effects of agmatine, a “clonidine-displacing substance”, in conscious rabbits. Naunyn-Schmiedeberg' Arch. Pharmacol. 1995;351:268–273. doi: 10.1007/BF00233246. [DOI] [PubMed] [Google Scholar]
- TRENDELENBURG A.U., SUTEJ I., WAHL C.A., MOLDERINGS G.J., RUMP C.L., STARKE K. A re-investigation of questionable subclassifications of presynaptic α2-autoreceptors: rat vena cava, rat atria, human kidney and guinea-pig urethra. Naunyn-Schmiedeberg' Arch. Pharmacol. 1997;356:721–737. doi: 10.1007/pl00005111. [DOI] [PubMed] [Google Scholar]