

Abstract
Replacement of the carboxylic acid group of PGF2α with the non-acidic substituents hydroxyl (-OH) or methoxy (-OCH3) resulted in an unexpected activity profile.
Although PGF2α 1-OH and PGF2α 1-OCH3 exhibited potent contractile effects similar to 17-phenyl PGF2α in the cat lung parenchymal preparation, they were approximately 1000 times less potent than 17-phenyl PGF2α in stimulating recombinant feline and human FP receptors.
In human dermal fibroblasts and Swiss 3T3 cells PGF2α 1-OH and PGF2α 1-OCH3 produced no Ca2+ signal until a 1 μM concentration was exceeded. Pretreatment of Swiss 3T3 cells with either 1 μM PGF2α 1-OH or PGF2α 1-OCH3 did not attenuate Ca2+ signal responses produced by PGF2α or fluprostenol. In the rat uterus, PGF2α 1-OH was about two orders of magnitude less potent than 17-phenyl PGF2α whereas PGF2α 1-OCH3 produced only a minimal effect.
Radioligand binding studies on cat lung parenchymal plasma membrane preparations suggested that the cat lung parenchyma does not contain a homogeneous population of receptors that equally respond to PGF2α1-OH, PGF2α1-OCH3, and classical FP receptor agonists.
Studies on smooth muscle preparations and cells containing DP, EP1, EP2, EP3, EP4, IP, and TP receptors indicated that the activity of PGF2α 1-OH and PGF2α 1-OCH3 could not be ascribed to interaction with these receptors.
The potent effects of PGF2α 1-OH and PGF2α 1-OCH3 on the cat lung parenchyma are difficult to describe in terms of interaction with the FP or any other known prostanoid receptor.
Keywords: FP receptor, prostaglandin F2α (PGF2α), calcium (Ca2+), radioligand binding, uterus, prostanoids
Introduction
Prostanoids are oxygenated fatty acids that exert a diverse and wide variety of biological activities. The pharmacology of prostanoids remained obscure and contentious for many years. The prostanoid receptor classification advanced by Kennedy et al. (1982) and Coleman et al. (1984) eventually proved to be correct and has been supported in its entirety by molecular biological studies. The key conceptual element of the prostanoid receptor classification is that each primary, natural prostanoid preferentially interacts with its own discrete population of receptors. Thus, prostaglandins D2 (PGD2), E2, (PGE2), and F2α (PGF2α) selectively interact with DP, EP and FP receptors, respectively. Similarly, prostacyclin (PGI2) preferentially interacts with IP receptors and thromboxane A2 (TxA2) and its endoperoxide precursors preferentially interact with TP receptors. There is subdivision of the EP receptor class into four subtypes; EP1, EP2, EP3, and EP4. Expansion of the existing EP receptor classification may be required to accommodate recent pharmacological findings that have suggested further heterogeneity (Lydford & McKecknie, 1994; Schaefer et al., 1998). Subdivision of IP receptors (Wise et al., 1995; Takechi et al., 1996) and DP receptors (Woodward et al., 1990; Rangachari et al, 1995; Fernandes & Crankshaw, 1995; Liu et al., 1996) has also been suggested. TP receptor heterogeneity has been widely claimed in the literature (Halushka et al., 1995) with 8-epi PGF2α providing an additional dimension in this area. The FP receptor remains the only prostanoid for which one receptor (FP) has been proposed to date, according to conventional criteria.
We report herein unexpected findings with compounds where the carboxylic acid moiety of PGF2α was replaced with certain non-acidic groups (-OH, -OCH3). These novel agents exhibited pronounced effects in certain PGF2α-sensitive preparations but lacked meaningful activity at recombinant FP receptors from the same species. The effects of the PGF2α 1-OH and PGF2α 1-OCH3 appear unique and do not seem to involve any known prostanoid receptor.
Methods
Isolated tissue studies
The smooth muscle tension of isolated tissues was measured isometrically with force displacement transducers (Grass FT-03) and recorded on a Grass Polygraph (Models 7G and 79E). The organ baths contained Krebs solution maintained at 37°C and gassed with 95% O2/5% CO2 to give a pH of 7.4. The Krebs solution had the following composition (mM): NaCl, 118.0; KCl, 4.7; KH2PO4, 1.2; CaCl2, 1.9; MgSO4, 1.18; NaHCO3, 25.0; glucose, 11.7; indomethacin, 0.001 (0.0028 for rat uterus studies).
Cat lung parenchyma
Cat lung parenchymal tissue preparations for functional studies were prepared according to the following procedures. Adult domestic cats were euthanized by intravenous overdose of sodium pentobarbital (Anthony, Arcadia, CA, U.S.A.). The thoracic cavity was opened by ventral incision using bone scissors. The lungs were excised in their entirety by severing the trachea and all major vascular connections. The lungs were then rinsed with ice-cold Krebs buffer to remove blood. The parenchymal preparations were obtained by cutting strips of about 3 mm in width and 10 mm in length from the margins of the lower lungs. The parenchymal strips were mounted in 10 ml jacketed-organ baths under 1.0 g tension at 37°C. After equilibration, cumulative dose-response experiments were performed. All tissues were pretreated with the thromboxane receptor antagonist SQ 29548 (1 μM) 15–20 min prior to performing each dose-response experiment. PGF2α (10 μM) was used to provide a reference contraction.
Cat lung parenchymal preparations for radioligand binding studies were prepared as follows. Adult domestic cats of either sex were euthanized by intravenous Eutha 6: Tm, 1 ml heparin solution was also given to prevent blood clotting. The lungs were cleared of blood by the following procedure. The pulmonary vein was clamped and approximately 20 ml of irrigation solution was injected via the pulmonary artery. The irrigation solution was phosphate buffered saline (PBS) containing 0.1 mg ml−1 soybean trypsin inhibitor, 5 mM EDTA, 1 mM benzamidine, 0.01 mg ml−1 aprotinin, 10 μM indomethacin, and 0.5 mg ml−1 bovine serum albumin. The pulmonary vasculature was flushed by releasing the clamp on the pulmonary vein. The filling/flushing process was repeated until the lung tissue appeared rather blanched.
The lung tissue was then minced, discarding any areas that appeared unhealthy or necrotic, and placed into four 50 ml centrifuge tubes containing 40 ml of ice cold homogenization buffer (10 mM TRIS-HCl at pH 7.4, 1 mM EGTA, 0.25 M sucrose, 1 mM benzamidine, and 0.5 mg ml−1 bovine serum albumin). The tissue was homogenized with a polytron at setting 7 for about 30 s. The tubes were centrifuged at 1200 r.p.m. (400×g) for 5 min. The floating fat layer was removed and the supernatant was filtered through a triple layer of gauze. The pellet was rehomogenized and the supernatant prepared as previously described. The pooled supernatant was then centrifuged at 42,000 r.p.m. (177,000×g) for 40 min in a Beckman XL-90 ultracentrifuge. The pellet was suspended in 24 ml of homogenization buffer and layered over a 0.771/1.28 M discontinuous sucrose gradient and centrifuged at 25,000 r.p.m. (112,000× g) for 2 h using a swing-out rotor. The discontinuous gradient was selected, following several preliminary experiments. The band at the lower interface of the discontinuous gradient was removed by pipette and suspended in ‘wash' buffer which comprised 10 mM TRIS-HCl at pH 6.0, 0.5 mM EGTA, 1 mM benzamidine, and 0.5 mg ml−1 bovine serum albumin. This was finally centrifuged at 55,000 r.p.m. (304,000×g) for 40 min. The final pellet was washed with the TRIS-HCl at pH 5.75 and stored at −70°C.
Radioligand binding assays were performed in duplicate in a final volume of 200 μl for 60 min. Optimal binding conditions were determined in pilot experiments and involved the use of 1 mg ml−1 bovine serum albumin to assist in reducing the level of [3H]-17-phenyl PGF2α non-specific binding. Despite extensive preliminary efforts, the levels of specific binding ranged from 40–55%. The pH of the buffer was 5.75 and contained 5 mM MnCl2. Radioligand binding competition assays were conducted over a 60 min period. Binding reactions were started by adding plasma membrane fraction. The reaction was terminated by the addition of 4 ml ice-cold TRIS-HCl buffer at pH 5.75 and rapid filtration through Whatman GF/B filters using a Brandel cell harvester. The filters were washed three times and then oven dried for 1 h. [3H]-17-phenyl PGF2α at a 5 nM concentration, was used as the radioligand in these competition studies.
Rat uterus
Virgin, female rats weighing 240–270 g were euthanized by CO2 inhalation. The duplex uterus was excised and cleared of oviducts, excess fat, and connective tissue. Each horn was cut in half longitudinally from the top (ovarian end) to the bottom (cervical end). Two muscle strips were obtained from each uterine horn. The endometrium was removed by cotton Q-tip. Longitudinal muscle strips of approximately 8 mm in length were suspended with sutures in a 10 ml jacketed organ bath under an initial 2 g tension that was readjusted and maintained at 1 g. Tissues were washed every 15–20 min during a 45 min equilibration period. Wash-out, dose-response curves were then constructed. Only one dose-response curve was generated per tissue and PGF2α at a 10 μM concentration was used as a reference. Contractile responses were calculated as the integrated area under the tension vs time curve for 5 min after administration of each dose using DigitizeTM and a graph pad.
Rat aorta
The rat aorta was used as a smooth muscle TP receptor preparation. Adult Sprague Dawley rats weighing 180–220 g were anaesthetized by CO2 inhalation and then decapitated and exsanguinated. The thoracic aorta was removed and cleaned of any adhering tissue. The lumen was flushed to remove any blood. The aorta was dissected into three small segments of 5–8 mm length. Each segment was mounted under 2.0 g of tension in a jacketed 10 ml organ bath with the aid of two wire hooks placed through the lumen of the vessel. This arrangement allowed for the measurement of contractile forces developed by the circular smooth muscle. The tissues were allowed to equilibrate for 1 h before compounds were added cumulatively to the organ baths. The response to 100 nM U-46619 was determined at the beginning and end of each experiment as a reference. A 30–45 min recovery period was allowed after complete wash-out of drug.
Guinea-pig ileum/chick ileum
Activity at prostaglandin-sensitive EP1- and EP3-receptor subtypes was determined by the ability of each compound to contract the guinea-pig or chick ileum, respectively. Portions of the guinea-pig or chick ileum 1.5 cm in length were suspended under 1.0 g tension. After a 1 h equilibration period, a standard dose-response curve to PGE2 was obtained in a non-cumulative fashion with 30 min wash-out periods between individual doses. Subsequently, test compound was added non-cumulatively. A maximal dose of PGE2 (1 μM) was given at the end to serve as a second reference standard. Contractile activity at each concentration was then calculated as per cent of the maximal PGE2 response reached at a 1 μM concentration.
Rabbit jugular vein
Activity at DP, EP2, and EP4 receptor subtypes was determined by examining relaxant effects on endothelium-denuded rabbit jugular vein segments pre-contracted with histamine as previously described (Lawrence & Jones, 1992; Chen et al., 1995). Briefly, the external jugular vein was excised from New Zealand white rabbits. Vascular segments 4–5 mm in length were suspended in organ baths and equilibrated under 0.5–0.75 g tension for 60 min. The tissue was then pre-contracted with histamine for 30 min and thereafter cumulative dose-response curves were constructed.
Cell studies
Human platelets
Activity at the DP-, TP- and IP- receptor types was determined by an ability to cause aggregation (TP-receptor activity) or inhibit ADP-induced aggregation of human platelets (DP- and IP-receptor activity). Fresh whole blood was obtained from consenting healthy human volunteers and mixed with acid citrate-dextrose (ACD). The blood was centrifuged at 1000 r.p.m. for 15–20 min to obtain platelet rich plasma (PRP). Compound solution or vehicle (4.5 μl volume) was added to 450 μl of PRP and incubated for 2 min at 37°C in a Payton aggregometer to record any aggregatory activity. ADP (final concentration 20 μM) was then added to induce full aggregation. Inhibition of aggregation was calculated as the percentage difference between aggregation evoked by 20 μM ADP in the absence and presence of drug. Aggregatory activity was calculated as the per cent aggregation induced by the compound relative to the aggregation induced by 20 μM ADP. Standard aggregatory responses to 20 μM ADP along were performed at the beginning and end of each experiment.
Human dermal fibroblasts and Swiss mouse 3T3 cells: Ca2+ signalling
Human CRL 1497 cells or Swiss 3T3 cells were plated in culture flasks and fed Dulbecco's modified Eagle's medium (DMEM) containing 10% foetal calf serum, 2 mM L-glutamate, and 0.05 mg ml−1 gentacin (all purchased from Gibco, Grand Island, NY, U.S.A.). Cell cultures were maintained in a humidified atmosphere of 95% air, 5% CO2 and grown to confluency.
Cells were removed from the culture flasks by approximately 1 min treatment with trypsin 0.05%/0.53 mM EDTA in HBSS (Gibco, Grand Island, NY, U.S.A.) at 37°C. Proteolytic activity was arrested by adding 5 ml of 10% foetal bovine serum in DMEM. The cells were consecutively washed in Hank's BSS and medium containing (mM): NaCl 140, KCl 50, MgCl2 1, CaCl2 1.5 HEPES: TRIS 10, glucose 5, Na pyruvate 5, 0.1% bovine serum albumin at pH 7.4: centrifugation for the washes was performed for 15 min at 200×g at room temperature. Cells were counted, resuspended in the above medium and incubated with 2 μM Fura 2/acetoxymethyl ester in a shaking water bath for 30 min at 37°C. The cells were subsequently washed in medium as above and resuspended at a concentration of 2×106 cells ml−1. Aliquots of 0.5 ml cell suspension were then added to autocap microtubes to provide 106 cells per experimental determination of intracellular free Ca2+ concentration ([Ca2+]i).
Fluorescence was measured in a Perkin-Elmer LS-5 fluorescence spectrophotometer at excitation and emission wavelengths of 340 and 492 nm, respectively, with both slits at 10 nm. For each experimental determination 106 cells were washed (200×g for 5 min) and suspended in a 2 ml cuvette with buffer containing (mM): NaCl 120, KCl 6, MgSO4 1, CaCl2 1.5, HEPES 20, 1 mg ml−1 glucose and 1 mg ml−1 Na pyruvate. Stirring was achieved by an overhead-mounted, paddle stirrer with the temperature maintained at 37°C. Calibration of the Fura 2 signal was as previously described for UMR-106 cells (Yamaguchi et al., 1988). The cells were lysed with digitonin (10 μl of 100 mg ml−1 DMSO) to obtain fmax. EGTA (100 mM) and sufficient 10 N NaOH to adjust the pH to 8.5 were then successively added to obtain Fmin. For functional antagonism studies in Swiss 3T3 cells, cells were treated for 10 min in the cuvette with PGF2α 1-OH or PGF2α 1-OCH3 before addition of the selected FP receptor agonist.
HEL cells: Ca2+ signalling
HEL cells were grown in suspension in 10% foetal calf serum RPMI and antibiotic/antimycotic (Gibco). The cells were harvested and pelleted by centrifugation at 1200 r.p.m. for 5 min. Cells were resuspended in medium and loaded with Fura 2-AM (1 μM for 1 h at 37°C. Cells were then washed and resuspended at 2×106 ml−1. All other conditions are identical to the above, except 0.5×106 cells were used for experimental determinations.
Swiss 3T3 cells: radioligand binding
A number of preliminary experiments were needed to optimize experimental conditions for [3H]-PGF2α and [3H]-17-phenyl PGF2α binding as previously described (Woodward & Lawrence, 1994; Woodward et al., 1995). Plasma membrane fractions were obtained as follows. Cells were removed from culture flasks by scraping, suspended in 50 ml Hanks' medium, and centrifuged at 6500×g for 10 min. The pellet was suspended in 50 ml of ice-cold 0.32 M sucrose-50 mM Tris buffer at pH 7.4 and centrifuged as above. This pellet was then resuspended in 40 ml of sucrose-Tris buffer and homogenized with a Polytron homogenizer for 1 s at setting 8. The homogenate was centrifuged at 400×g for 5 min, and the supernatant was removed with a Pasteur pipette and saved. The residue was resuspended in 25 ml of buffer, homogenized and centrifuged as before. The resulting pooled supernatant was then centrifuged at 177,000×g for 40 min. This crude membrane pellet was suspended over a sucrose cushion, 1.06 M providing optimal specific binding. Four millilitres of homogenate were layered on top of 4 ml of 1.06 M sucrose contained in six (10 ml) Beckman polycarbonate centrifuge tubes, and centrifuged at 112,000×g for 2 h. The top band separating the interface of the 1.06 M sucrose cushion and the subcellular suspension was enriched with plasma membranes. The bands from each of the tubes were aspirated with a Pasteur pipette, combined and diluted 1:2 with 50 mM Tris buffer. They were then centrifuged at 304,000×g for 40 min. The final pellet was washed once with Tris-HCl buffer and stored at −70°C.
Radioligand binding competition studies were as described for cat lung parenchymal tissue plasma membrane fractions. The radioligands were 6.1 nM [3H]-PGF2α and 5 nM [3H]-17-phenyl PGF2α. Specific binding exceeded 70% for [3H]-17-phenyl PGF2α but was only 36% for [3H]PGF2α.
PI turnover assay
Receptor-mediated phosphoinositide (PI) hydrolysis was determined by measuring the accumulation of total inositol phosphates (IP) in cells preincubated with [3H]-myoinositol. Stable cell lines of HEK 293 cells expressing the human and cat FP receptors were plated in 10 cm dishes (106 cells per dish in DMEM with 10% FBS). The following day, the cells were incubated with 18 μCi myo-[2-3H]-inositol (Amersham; 10–20 μCi mmol−1) in 6 ml of DMEM with 10% FBS for 24 h at 37°C. The cells were then rinsed twice with PBS, incubated for 5 min with 1 ml of trypsin-EDTA and suspended in 10 ml DMEM containing 25 mM HEPES buffer. Cells were pelleted at 1000 r.p.m. and resuspended in DMEM/25 mM HEPES containing 10 mM LiCl for 10 min. Aliquots of 200 μl cell suspension were incubated with 50 μl drug for 30 min at 37°C (duplicate determination), and agonist stimulation was terminated with a 750 μl mixture of chloroform:methanol:4 N HCl (100:200:2), followed by addition of 250 μl of chloroform and 0.5 N HCl for extraction of inositol phosphates. A 750 μl volume of the aqueous layer was loaded on columns packed with 0.5 ml of Dowex AG 1-X8 anion-exchanged resin (formate form, 100–200 mesh; BioRad) to separate radiolabelled components. The elution procedure consisted of three 3 ml washes with 5 mM inositol, then two elutions with 750 μl of 1.3 M ammonium formate with 0.1 M formic acid. The eluate was mixed with 10 ml Aquasol scintillation fluid (Packard Instrument Co.) and the total [3H]-inositol phosphates were determined by a liquid scintillation counter. Data were analysed by KaleidaGraph software to determine agonist potency.
Recombinant EP2, EP3, EP4, receptor; transient transfectants
COS-7 cells were transiently transfected using Lipofectin (Gibco–BRL Life Technologies, Gaithersburg, MD, U.S.A.) according to manufacturer's protocols. For binding studies, 2×106 cells were plated onto 150 mm dishes 24 h prior to transfection. Each plate was transfected with 50 μg plasmid DNA and 50 μl lipofectin. Cells were collected and membranes prepared at 48 h post-transfection, and frozen at −80°C until use.
Plasma membrane preparations were thawed at room temperature and used at a final 1 mg ml−1 concentration in a 500 μl volume. The binding of [3H]-17-phenyl PGF2α (specific activity 85 Ci mmol−1) and [3H]-PGE2 (specific activity 180 Ci mmol−1) were determined in duplicate and experiments were replicated three times. Incubations were for 60 min at 25°C and were terminated by the addition of 4 ml of ice-cold 50 μM TRIS-HCl, followed by rapid filtration through Whatman GF/B filters and three additional 4 ml washes in a cell harvester (Brandel). Competition studies were performed using a final concentration of 5 nM [3H]-17-phenyl PGF2α or 5 nM [3H]-PGE2 and non-specific binding determined with 10 μM of the respective unlabelled prostanoid.
Recombinant cat and human FP receptor; stable transfectants
HEK-293 cells were grown in DMEM with 10% foetal bovine serum (FBS), 250 μg ml−1 G418 (Life Technologies) and 200 μg ml−1 gentamicin or penicillin/streptomycin. Selection of stable transfectants was achieved with 200 μg ml−1 hygromycin, the optimal concentration being determined by hygromycin kill curve studies.
For transfections, the cells were grown to 50–60% confluency on 10 cm plates. The plasmid pCEP4 incorporating cDNA inserts for the human FP receptor (20 μg) was added to 500 μl of 250 mM CaCl2. HEPES buffered saline ×2 (2×HBS, 280 mM NaCl, 20 mM HEPES acid, 1.5 mM Na2 HPO4, pH 7.05–7.12) was then added dropwise to a total of 500 μl, with continuous vortexing at room temperature. After 30 min, 9 ml DMEM were added to the mixture. The DNA/DMEM/calcium phosphate was then added to the cells, which had been previously rinsed with 10 ml PBS. The cells were then incubated for 5 h at 37°C in humidified 95% air/5% CO2. The calcium phosphate solution was then removed and the cells were treated with 10% glycerol in DMEM for 2 min. The glycerol solution was then replaced by DMEM with 10% FBS. The cells were incubated overnight and the medium was replaced by DMEM/10% FBS containing 250 μg ml−1 G418 and penicillin/streptomycin. The following day hygromycin B was added to a final concentration of 200 μg ml−1.
Ten days after transfection, hygromycin B resistant clones were individually selected and transferred to a separate well on a 24-well plate. At confluence each clone was transferred to one well of a 6-well plate, and then expanded in a 10 cm dish. Cells were maintained under continuous hygromycin selection until use.
The stable cat FP receptor transfectants were similarly prepared using lipofectamine. Again cells were grown to 50–60% confluency on 10 cm plates and the plasmid pCEP4, incorporating cyclic DNA inserts for the feline FP receptor, was transfected using lipofectin. Hygromycin B selection was started 48 h post-transfection. Eight days after transfection, hygromycin B resistant clones were selected and transferred to 24-well plates.
Ca2+ signalling and radioligand binding competition studies were performed in an identical manner to that described for other cell studies.
EC50 values were determined for each individual tissue dose response curve and the mean EC50±s.e.mean was obtained for n experiments. The EC50 values were determined as the compound concentration (nM) which elicited a response one-half of the maximal response.
Materials
PGD2, PGE2, PGF2α, 17-phenyl PGF2α, U-46619 and fluprostenol were purchased from Cayman Chemical (Ann Arbor, MI, U.S.A.) 11β-12-epi-17-phenyl PGF2α, 1-decarboxyl 1-hydroxymethyl PGF2α (PGF2α 1-OH), and 2-decarboxyl-2-methoxymethyl PGF2α (PGF2α 1-OCH3) were synthesized at Allergan. [3H]-17-phenyl PGF2α was custom synthesized by Amersham. [3H]-PGE2 was purchased from Amersham (Arlington, NJ, U.S.A.).
Results
The contractile effects of PGF2α 1-OH, PGF2α 1-OCH3 and a variety of FP receptor agonists on the cat lung parenchyma are depicted in Figure 1. PGF2α 1-OH and PGF2α 1-OCH3 both contracted the cat lung parenchymal strip with a potency that closely resembled the activity of PGF2α, 17-phenyl PGF2α, and fluprostenol. The following rank order of potency was achieved with EC50 values (nM) in parentheses: fluprostenol (16)⩾PGF2α 1-OH (23)⩾U-46619 (27)⩾PGF2α 1-OCH3 (32)⩾17-phenyl PGF2α (44)⩾PGF2α (55)>PGE2 (458)>11β-12-epi-17-phenyl PGF2α (>10,000). 11β-12-epi-17-phenyl PGF2α was included to provide data on a relatively inactive PGF2α analogue to aid comparisons. PGE2 and U-46619 were examined and contracted the lung parenchymal strip but PGE2 had a potency more indicative of residual activity at FP and TP receptors rather than at EP receptors.
Figure 1.
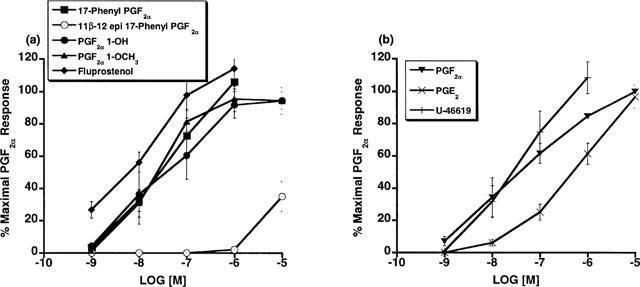
Cat lung parenchymal tissue: contractile effects of 17-phenyl PGF2α, Fluprostenol, PGF2α 1-OH, PGF2α 1-OCH3, 11β-12-epi-17-phenyl PGF2α (a), and PGF2α PGE2 and U-46619 (b) expressed as per cent maximal response to 10−5 M PGF2α. Points represent mean values±s.e.mean; n=4–6.
The unexpected high potency of PGF2α 1-OH and PGF2α 1-OCH3 was subsequently examined in a variety of FP receptor preparations, notably the recombinant feline FP receptor which was cloned from immortalized cat iris sphincter smooth muscle cells (Ocklind et al., 1995) and overexpressed as a stable transfectant. A very different activity profile was obtained at the recombinant cat FP receptor. On studying Ca2+ signalling in HEK 293 cells stably transfected with the cat FP receptor, PGF2α 1-OH and PGF2α 1-OCH3 exhibited only minimal activity compared to 17-phenyl PGF2α and PGF2α (Figure 2a). Thus, EC50 values for 17-phenyl PGF2α and PGF2α were 3 and 9.7 nM, respectively. PGF2α 1-OH and PGF2α 1-OCH3 exhibited no meaningful activity until concentrations exceeding 1 μM were attained. Similar results were obtained with the recombinant human FP receptor (Figure 2b). EC50 values for 17-phenyl PGF2α and PGF2α were 6.8 and 11.2 nM, respectively. Radioligand binding studies on the overexpressed, recombinant cat FP receptor revealed a similar activity profile. PGF2α 1-OH and PGF2α 1-OCH3 did not compete with [3H]-17-phenyl PGF2α for the recombinant cat FP receptor (Figure 3a) or the recombinant human FP receptor until relatively high concentrations were achieved (Figure 3b). Radioligand binding studies closely correlated with results obtained on Ca2+ signalling. Studies on total inositol phosphate formation were also performed for overexpressed, recombinant cat and human FP receptors. 17-phenyl PGF2α and PGF2α 1-OCH3 exhibited the same enormous separation in potency in stimulating total inositol phosphate production in recombinant cat (Figure 4a) and human (Figure 4b) FP receptor preparations that had been observed for both Ca2+ signalling and radioligand binding competition studies. PGF2α 1-OH, however, appeared more potent in stimulating total inositol formation than would have been predicted from the Ca2+ signalling and binding studies. Thus, measurable EC50 values of 639 and 567 were obtained for PGF2α 1-OH at recombinant human and feline FP receptors, respectively. 17-phenyl PGF2α remained substantially more potent (EC50=9.7 nM cat and 6.8 nM human) but the PGF2α 1-OH total inositol phosphate responses seemed sufficiently different from those obtained for Ca2+ signalling and radioligand binding competition studies to merit further investigation. One plausible explanation for the discrepancies involving PGF2α 1-OH, albeit not very likely, was the possibility that part of the molecule was oxidized to the free acid PGF2α. This was not investigated in the cell systems but a bioassay was performed for the cat lung parenchymal tissue assay. Following 15 min of incubation with graded doses of PGF2α 1-OH, the physiological solution was assayed using radioligand binding competition studies at the recombinant human FP receptor. Using this radioligand binding bioassay, no competition for 17-phenyl PGF2α binding sites was apparent (Figure 5). Samples of PGF2α obtained from tissue bath fluid, as a positive control, inhibited 17-phenyl PGF2α binding. The results of this bioassay study indicate that the effects of PGF2α 1-OH on the cat lung parenchyma, which typically reach maximum in about 10 min, cannot be ascribed to the formation of biologically active levels of PGF2α.
Figure 2.
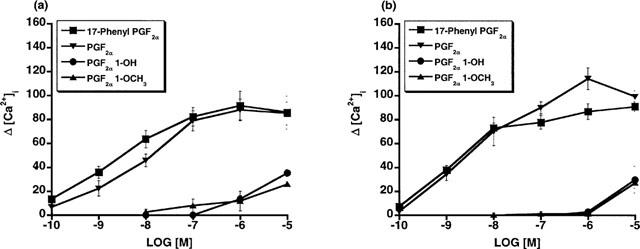
Recombinant feline (a) and human (b) FP receptors: Ca2+ transient signals in response to 17-phenyl PGF2α, PGF2α, PGF2α 1-OH, and PGF2α 1-OCH3 expressed as increased intracellular Ca2+ concentration. Points represent mean values±s.e.mean; n=3–4.
Figure 3.
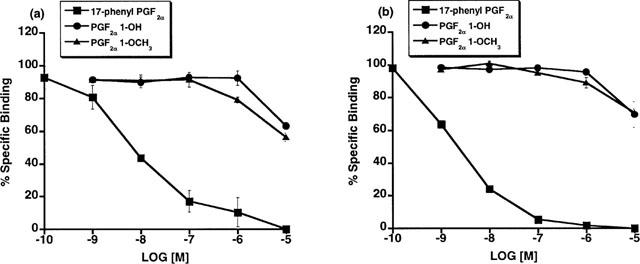
Recombinant feline (a) and human (b) FP receptors: radioligand binding competition studies vs [3H]-17-phenyl PGF2α employing unlabelled 17-phenyl PGF2α, PGF2α 1-OH, and PGF2α 1-OCH3. Points represent mean values±s.e.mean; n=3 experiments performed in duplicate.
Figure 4.
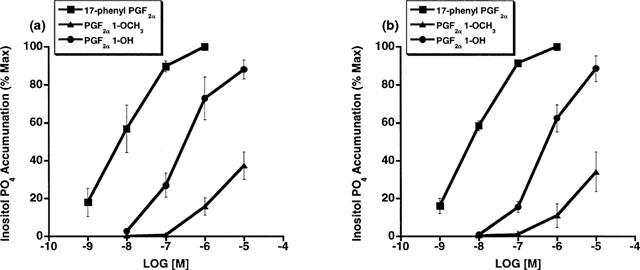
Recombinant feline (a) and human (b) FP receptors: formation of [3H]-inositol phosphates in response to 17-phenyl PGF2α, PGF2α 1-OH, and PGF2α 1-OCH3. Points represent mean values±s.e.mean. (a) feline: 17-phenyl PGF2α n=3, PGF2α 1-OH n=4, PGF2α 1-OCH3 n=3 experiment performed in duplicate. (b) human: 17-phenyl PGF2α n=6, PGF2α 1-OH n=3, PGF2α 1-OCH3 n=3 experiment performed in duplicate.
Figure 5.
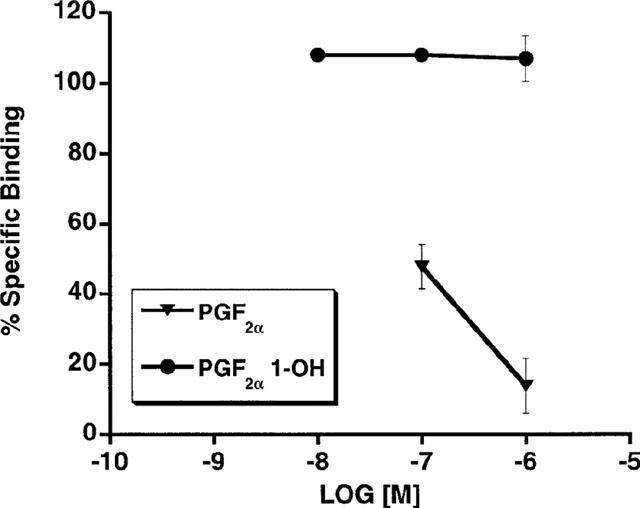
Bioassay for cat lung parenchymal tissue: following 15 min exposure to 10−7, 10−6 or 10−5 M PGF2α 1-OH: the physiological solutions were sampled and bioassayed using radioligand binding competition studies vs [3H]-17-phenyl PGF2α at recombinant human FP receptors. Competition afforded by PGF2α, the potential metabolite of PGF2α 1-OH, was evaluated for comparison. Points represent mean values±s.e.mean; n=3 experiments performed in duplicate.
Since several of the studies reported herein employed overexpressed recombinant receptors, further studies involving FP receptors expressed at constitutive levels were performed. Firstly, the effects of PGF2α 1-OH and PGF2α 1-OCH3 on human dermal fibroblasts, which constitutively express the FP receptor, were examined. PGF2α 1-OH and PGF2α 1-OCH3 elicited no measurable Ca2+ signal in human dermal fibroblasts over a 10 nM to 10 μM concentration range (Figure 6a). In marked contrast, PGF2α, and 17-phenyl PGF2α potently increased intracellular [Ca2+] with EC50 values of 49.3, and 15 nM respectively. Similar studies were performed in Swiss 3T3 cells with identical results (Figure 6b). Moreover, functional antagonist studies in Swiss 3T3 cells revealed that pretreatment with 1 μM PGF2α 1-OH or PGF2α 1-OCH3 did not attenuate Ca2+ signal responses to PGF2α or fluprostenol (Figure 7). Secondly, the effects of 17-phenyl PGF2α were compared to those of PGF2α 1-OH and PGF2α 1-OCH3 in the rat uterus. 17-phenyl PGF2α potently produced a dose-dependent increase in rat uterine contraction (EC50=7.8 nM), whereas PGF2α 1-OH and PGF2α 1-OCH3 were much less active with EC50 values of 957 and >10,000 nM, respectively (Figure 8).
Figure 6.
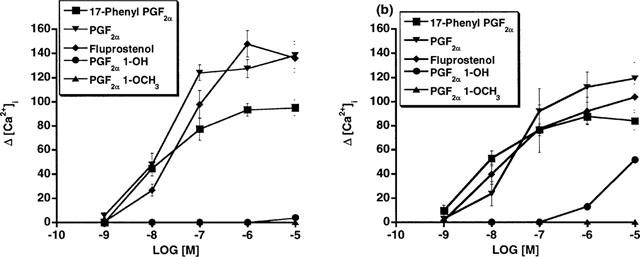
Human dermal and Swiss 3T3 fibroblasts constitutively expressing FP receptors: Ca2+ transient signals in response to 17-phenyl PGF2α, PGF2α, fluprostenol, PGF2α 1-OH and PGF2α 1-OCH3 expressed as increased intracellular Ca2+ concentration in (a) human dermal fibroblasts and (b) Swiss 3T3 cells. Points represent mean value±s.e.mean; n=4–5.
Figure 7.
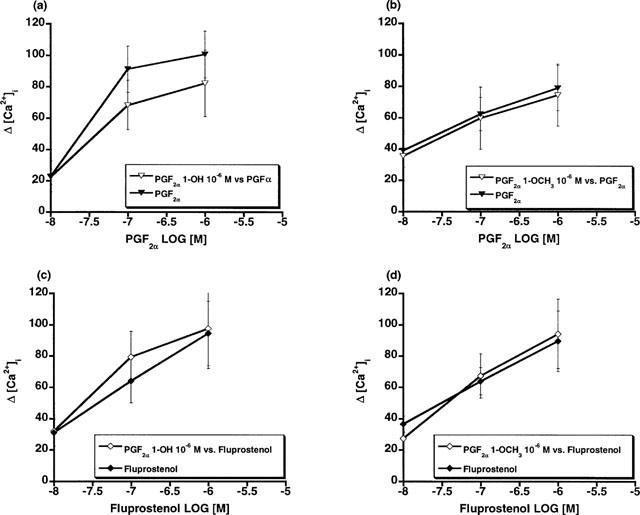
Swiss 3T3 cells: effects of PGF2α on intracellular Ca2+ concentration after pretreatment with (a) 10−6 M PGF2α 1-OH or (b) 10−6 M PGF2α 1-OCH3 and the effect of fluprostenol on intracellular Ca2+ concentration after pretreatment with (c) 10−6 M PGF2α 1-OH or (d) 10−6 M PGF2α 1-OCH3. Points are mean values±s.e.mean; n=4–5.
Figure 8.
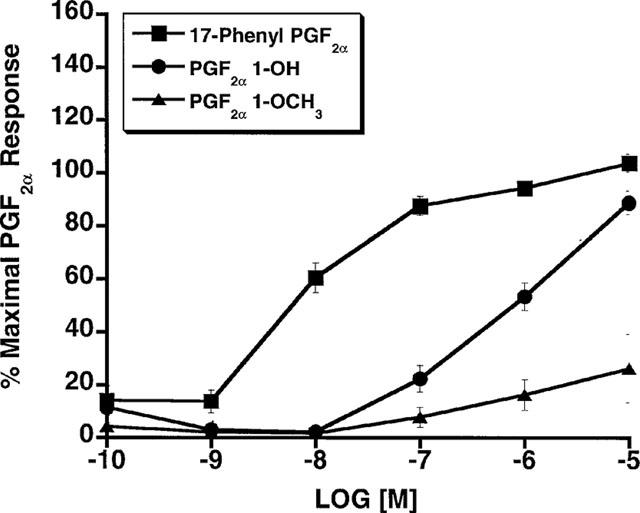
Rat uterus: contractile effects of 17-phenyl PGF2α, PGF2α 1-OH and PGF2α 1-OCH3 expressed as per cent maximal response to 10−5 M PGF2α. Points represent mean values±s.e.mean; n=6.
Radioligand binding competition studies using [3H]-17-phenyl PGF2α in plasma membrane fractions prepared from the cat lung parenchyma (Figure 9) indicated the presence of an FP receptor according to the rank order of competition: PGF2α (29.5)>17-phenyl PGF2α (45-5)>PGD2 (83.4)>PGE2 (800)>U-46619 (3065); I.C.50 (nM) values are in parentheses. PGF2α 1-OH offered relatively modest competition and an IC50 value of 932 nM was obtained. The results for PGF2α 1-OCH3 are not reported since the minimal level of specific binding was not achieved for these experiments and this data set was viewed as unsatisfactory. 11β-12-epi-17-phenyl PGF2α did not compete with [3H]-17-phenyl PGF2α for its binding sites. Radioligand binding competition studies vs [3H]-17-phenyl PGF2α and [3H]-PGF2α in Swiss 3T3 cells are depicted in Figure 10. PGF2α 1-OH did not effectively compete with radiolabelled 17-phenyl PGF2α or PGF2α for FP receptor binding sites in Swiss 3T3 cells. PGF2α 1-OCH3 very weakly competed with [3H]-17-phenyl PGF2α for the Swiss 3T3 cell FP receptor and an IC50 value of 4974 nM was obtained. Specific binding for the PGF2α 1-OCH3 binding experiment was 25% less than typical (Woodward et al., 1995).
Figure 9.
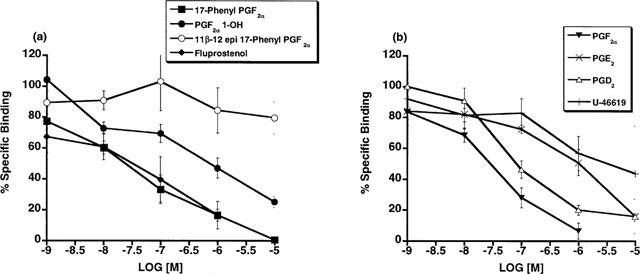
Cat lung parenchymal tissue: radioligand binding competition studies vs [3H]-17-phenyl PGF2α employing unlabelled 17-phenyl PGF2α, fluprostenol, PGF2α 1-OH, 11β-12-epi-17-phenyl PGF2α in (a) and PGF2α PGE2, PGD2, and U-46619 in (b). Points represent mean values±s.e.mean; n=3 experiments performed in duplicate.
Figure 10.
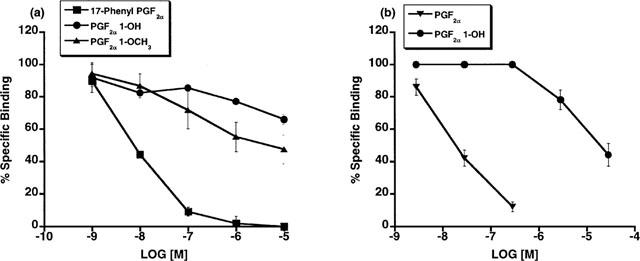
Swiss 3T3 cells: radioligand binding competition studies vs [3H]-17-phenyl PGF2α (a) and [3H]-PGF2α (b) employing unlabelled PGF2α, PGF2α 1-OH, PGF2α 1-OCH3, and 17-phenyl PGF2α. Points represent mean values±s.e.mean; n=3 experiments performed in duplicate.
The activities of PGF2α 1-OH, PGF2α 1-OCH3, PGF2α, and 17-phenyl PGF2α in PGF2α sensitive preparations are summarized in Table 1. Finally, the activity of a panel of non-human and human prostanoid receptors (natural and recombinant) was examined. PGF2α 1-OH and PGF2α 1-OCH3 had no meaningful activity at DP, EP1, EP2, EP3, EP4, FP, IP or TP receptors. These data are summarized in Table 2
Table 1.
Effects of PGF2α 1-OH, PGF2α 1-OCH3, 17-phenyl PGF2α, and PGF2α on various PGF2α responsive preparations
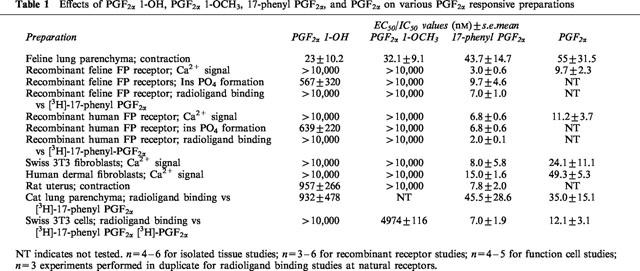
Table 2.
Effects of PGF2α 1-OH and PGF2α 1-OCH3 at DP, EP1, EP2, EP3, EP4, IP and TP receptors

Discussion
There are distinct structural requirements for optimal interaction with PGF2α-sensitive receptors, notably the presence of a carboxylic acid group at the C1 position (Schaaf, 1976; Schaaf & Hess, 1979). Thus, replacement of the −COOH group of PGF2α by an amide or hydroxyl substituent resulted in a marked decrease in agonist activity (Maddox et al., 1978; Schaaf & Hess, 1979). In view of this, we found that replacement of the carboxylic acid moiety of PGF2α by non-acidic hydroxyl (OH) and methoxy (OCH3) groups resulted in an unprecedented activity profile. PGF2α 1-OH (AGN 190910) and PGF2α 1-OCH3 (AGN 191129) exhibited only very weak activity at recombinant and natural FP receptors from cat, rat, mouse and human. In the cat lung parenchymal strip, PGF2α 1-OH and PGF2α 1-OCH3 exhibited much greater activity and were essentially equipotent to PGF2α and the potent, selective FP receptor agonist 17-phenyl PGF2α (Magerlein et al., 1975; Miller et al., 1975; Woodward et al., 1995). The pharmacology of PGF2α 1-OH and PGF2α 1-OCH3 was further investigated by performing a number of functional and radioligand binding studies.
The sum of the data presented herein indicates that PGF2α 1-OH and PGF2α 1-OCH3 interact very weakly with the FP receptor as described in the prostanoid receptor classification (Coleman et al., 1994). Compared to 17-phenyl PGF2α, PGF2α 1-OH and PGF2α 1-OCH3 were at least three orders of magnitude less potent in producing a Ca2+ signal in (i) human dermal fibroblasts and Swiss mouse 3T3 cells constitutively expressing the FP receptor and (ii) in stable transfectants in HEK cells overexpressing either the human or cat FP receptor. The results of radioligand binding competition studies were consistent with the functional studies. PGF2α 1-OH and PGF2α 1-OCH3 did not compete with [3H]-17-phenyl PGF2α for the cat and human FP receptor until they achieved a 1 μM concentration. Moreover, functional competition studies in Swiss 3T3 cells demonstrated that pretreatment with 1 μM PGF2α 1-OH or PGF2α 1-OCH3 did not attenuate the Ca2+ signal elicited by either PGF2α or the potent and selective FP receptor agonist fluprostenol. The results obtained for PGF2α 1-OH in these FP receptor studies essentially correlate with a previous report on the gerbil colon where PGF2α 1-OH was more than two orders of magnitude less potent than PGF2α (Maddox et al., 1978). In addition, these investigators reported that PGF2α 1-OH did not antagonize the response to PGF2α (Maddox et al., 1978).
The finding that replacement of the carboxylic acid group with a non-acidic group resulted in dramatically reduced affinity for the FP receptor was further examined by studying total inositol phosphate formation. The use of LiCl to inhibit inositol phosphate degradation by phosphatases allows the functional response to be amplified and the utilization of signal amplification has been suggested to be of value in more precisely rank ordering FP agonist potencies (Griffin et al., 1997; 1998). Moreover, the studies reported herein were performed using overexpressed recombinant receptors. This amplification assay offered an opportunity to potentially reveal responses at the 10 and 100 nM concentrations of PGF2α 1-OH and PGF2α 1-OCH3 that might have been overlooked in the more physiological setting provided by the Ca2+ signalling assay. PGF2α 1-OH and PGF2α 1-OCH3 did not, however, produce activity at concentrations where PGF2α and other FP agonists exerted marked activity. As in the case of the Ca2+ signalling and radioligand binding studies, the potency of PGF2α 1-OCH3 in stimulating inositol phosphate formation was about two to three orders of magnitude less than that of 17-phenyl PGF2α. PGF2α 1-OH was less than two orders of magnitude the potency of 17-phenyl-PGF2α in the inositol phosphate assay. Nevertheless, the potent effects of 10 and 100 nM PGF2α 1-OH and PGF2α 1-OCH3 observed in the cat lung parenchymal strip preparation could not be replicated at the FP receptor, even using an amplification assay in cells overexpressing FP receptors.
Studies on the effects of PGF2α 1-OH and PGF2α 1-OCH3 on inositol phosphate formation in the cells stably transfected with recombinant feline and human FP receptors revealed a difference in the two compounds that essentially translated into functional activity in the rat isolated uterus. In both preparations PGF2α 1-OH produced a maximal response that closely approached the 17-phenyl PGF2α maximum, although PGF2α 1-OH was decidedly less potent that 17-phenyl PGF2α in both instances. PGF2α 1-OCH3 was less potent PGF2α 1-OH in the recombinant FP receptor inositol phosphate and rat uterus assays and PGF2α 1-OCH3 did not achieve 50% of the maximal 17-phenyl PGF2α response. This activity profile differed markedly from that in the feline lung parenchyma where both PGF2α 1-OH and PGF2α 1-OCH3 produced a similar maximum to that obtained for PGF2α, 17-phenyl PGF2α, and fluprostenol. This sets the feline lung parenchyma apart from other PGF2α-sensitive preparations by virtue of its exquisite responsiveness to these non-ionizable C1 modified PGF2α analogues.
The effects of PGF2α 1-OH and PGF2α 1-OCH3 were examined at all other prostanoid receptors described in the current classification to investigate the possibility that effects on the cat lung parenchyma may result from stimulation of a DP, EP, IP or TP receptor subtype. This does not seem to be the case. These compounds exhibited no meaningful functional activity at human DP or IP receptors (inhibition of ADP induced platelet aggregation), rabbit DP, EP2 and EP4 receptors (relaxation of the rabbit jugular vein), human EP1 receptors (Ca2+ signal in HEL cells), guinea-pig EP1 receptors (contraction of the ileum), EP3 receptors (inhibition of field stimulation vas deferens contraction), and human TP receptors (platelet aggregation and Ca2+ signal in HEL cells). In addition, radioligand binding competition studies were performed on COS-7 cells transiently transfected with human EP2, EP3D, and EP4 receptors (Regan et al., 1994), PGF2α 1-OH and PGF2α 1-OCH3 exhibited no meaningful affinity for any of these recombinant human receptors.
The effects of PGF2α 1-OH and PGF2α 1-OCH3 on the cat lung parenchyma are not consistent with activity at any known prostanoid receptor. These C1 modified PGF2α analogues are potent myotropic agents in the cat lung parenchyma and exhibit similar potency to PGF2α and other classical FP receptor agonists. Since it was impossible to reasonably ascribe the activity of PGF2α 1-OH and PGF2α 1-OCH3 to interaction with the FP or any other known prostanoid receptor, one further avenue of investigation was pursued. The possibility of conversion to the free acid metabolite (PGF2α) was considered. The physiological solution from tissue baths containing the cat lung parenchymal strip and graded doses of PGF2α 1-OH were sampled and analysed for PGF2α using a bioassay involving radioligand binding competition studies at recombinant human FP receptors. No evidence for conversion to the free acid metabolite was detected. Moreover, the rat uterus study indicates that low potency of C1 modified PGF2α analogues is not a phenomenon confined to cell preparations and provides a direct isolated tissue comparison with the cat lung parenchymal preparation.
The pharmacology of cat lung parenchymal responses was further investigated using radioligand binding competition studies. PGF2α 1-OH appeared to exert more affinity (∼1000 nM) for 17-phenyl PGF2α binding sites in the cat lung parenchyma than for any of the recombinant receptors (>10,000 nM). Nevertheless, its IC50 (932 nM) was substantially less than that obtained for 17-phenyl PGF2α (45.5 nM) or PGF2α (35 nM). It appears, therefore, that the cat lung does not contain a homogenous population of receptors that accept PGF2α 1-OH and PGF2α with equal affinity. One interpretation is that the cat lung parenchyma comprises two distinct subpopulations of receptor, one of which can recognize PGF2α 1-OH, the other population being the classical FP receptor (Coleman et al., 1984; Abramovitz et al., 1994). Evidence for the presence of the classical FP receptor in cat lung parenchymal tissue was obtained from radioligand binding competition studies. Here the typical rank order of potency for an FP receptor was essentially obtained i.e. PGF2α⩾17-phenyl PGF2α>PGD2>PGE2=U-46619 (Coleman et al., 1984; Woodward & Lawrence, 1994; Abramovitz et al., 1994; Woodward et al., 1995). The identity of the receptor that recognizes PGF2α 1-OH and PGF2α 1-OCH3 in the cat lung parenchyma remains uncertain but does not appear to be the classical FP receptor, given the results of radioligand binding studies on plasma membrane preparations from lung parenchymal tissue and studies on the recombinant cat FP receptor.
Considering the data presented herein, it is very difficult to ascribe the potent activities of PGF2α 1-OH and PGF2α 1-OCH3 to interaction with the FP receptor. One interpretation that would be consistent with the data is the presence of two distinct PFG2α-sensitive receptors in the cat lung, the identity of one of these receptors being the classical FP receptor. The identity of the target for PGF2α 1-OH and PGF2α 1-OCH3 cannot be so readily determined. It may represent a second, previously unidentified FP receptor. If this were the case, however, greater potency for PGF2α 1-OH in the radioligand binding competition assay would be predicted, unless the PGF2α 1-OH sensitive receptor were present as a small fraction of the total population of PGF2α-sensitive receptors present in the cat lung parenchyma. Alternatively, the natural ligand for the PGF2α 1-OH and PGF2α 1-OCH3 sensitive receptors may not be PGF2α but rather a yet to be discovered neutral lipid. Postulates implicating the classical FP receptor would require mechanisms unprecedented for the G protein coupled superfamily of receptors. Reconciliation with the results obtained in radioligand and functional antagonism studies would require drug-FP receptor association and dissociation rates so rapid that they do not interfere with PGF2α binding and cannot be reliably measured. Reconciliation of the different functional activities requires speculation to explain activity in the cat lung parenchyma, for example a plasma membrane Ca2+ channel that is operant regardless of drug–FP receptor interaction kinetics. Historical precedent would favour the concept that the protein target for PGF2α 1-OH and PGF2α 1-OCH3 is unique and is not a known prostanoid receptor.
Acknowledgments
The authors would like to express their appreciation to Linda Johnson for preparing the figures and manuscript.
Abbreviations
- ACD
acid citrate-dextrose
- ADP
adenosine diphosphate
- DMEM
Dulbecco's modified Eagles medium
- DMSO
dimethylsulphoxide
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol-bis (β-aminoethyl ether) N,N,N′,N′-tetraacetic acid
- FBS
foetal bovine serum
- HEPES
N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulphonic acid]
- PGD2
prostaglandin D2
- PGE2
prostaglandin E2
- PGF2α
prostaglandin F2α
- PGI2
prostaglandin I2
- PRP
platelet rich plasma
- TRIS
tromethamine
- TxA2
thromboxane A2
References
- ABRAMOVITZ M., BOIE Y., NGUYEN T., RUSHMORE T.H., BAYNE M.A., METTERS K.M., SLIPETZ D.M., GRYGORCZYK R. Cloning and expression of a cDNA for the human prostanoid FP receptor. J. Biol. Chem. 1994;269:2632–2636. [PubMed] [Google Scholar]
- CHEN J., CHAMPA-RODRIGUES M.L., WOODWARD D.F. Identification of a prostanoid FP receptor population producing endothelium-dependent vasorelaxation in the rabbit jugular vein. Br. J. Pharmacol. 1995;116:3035–3041. doi: 10.1111/j.1476-5381.1995.tb15960.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLEMAN R.A., HUMPHREY P.P.A., KENNEDY I., LUMLEY P. Prostanoid receptors: the development of a working classification. Trends Pharmacol. Sci. 1984;5:303–306. [Google Scholar]
- COLEMAN R.A., SMITH W.L., NARUMIYA S. VIII. International Union of Pharmacology classification of prostanoid receptors: properties, distribution and structure of the receptors and their subtypes. Pharmacol. Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- FERNANDES B., CRANKSHAW D. Functional characterization of the prostanoid DP receptors in human myometrium. Eur. J. Pharmacol. 1995;283:73–81. doi: 10.1016/0014-2999(95)00288-v. [DOI] [PubMed] [Google Scholar]
- FUNK C.D., FURCI L., FITZGERALD G.A., GRYGORCZYK R., ROCHETTE C., BAYNE M.A., ABRAMOVITA M., ADAM M., METTERS K.M. Cloning and expression of a cDNA for the human prostaglandin E receptor EP1 subtype. J. Biol. Chem. 1993;268:26767–26772. [PubMed] [Google Scholar]
- GILES H., LEFF P., BOLOFO M.L., ROBERTSON A.D. The classification of prostaglandin DP-receptors in platelets and vasculature using BW A 868 C, a novel, selective and potent competitive antagonist. Br. J. Pharmacol. 1989;96:291–300. doi: 10.1111/j.1476-5381.1989.tb11816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIFFIN B.W., MAGNINO P.E., PANG I-H., SHARIF N.A. Pharmacological characterization of an FP prostaglandin receptor on rat vascular smooth muscle cells (A7r5) coupled to phosphoinositide turnover and intracellular calcium mobilization. J. Pharmacol. Exp. Ther. 1998;286:411–418. [PubMed] [Google Scholar]
- GRIFFIN B.W., WILLIAMS G.W., CRIDER J.Y., SHARIF N.A. FP prostaglandin receptors mediating inositol phosphates generation and calcium mobilization in Swiss 3T3 cells: a pharmacological study. J. Pharmacol. Exp. Ther. 1997;281:845–854. [PubMed] [Google Scholar]
- HALUSHKA P.V., ALLAN C.J., DAVIS-BRUNO K.L. Thromboxane A2 receptors. J. Lipid Mediat. Cell Signal. 1995;12:361–378. doi: 10.1016/0929-7855(95)00023-j. [DOI] [PubMed] [Google Scholar]
- HARRIS D.N,. , MICHEL I.M., GOLDENBERG H.J., HARTL K.S., ALLEN G.T., STEINBACHER T.E., SCHUMACHER W.A., HAN W.C., HALL S.E., FLOYD D.M., OGLETREE M.L. Pharmacological characterization of potent, long-acting thromboxane receptor antagonists, SQ 33,261 and SQ 33,552. J. Pharmacol. Exp. Ther. 1992;261:131–137. [PubMed] [Google Scholar]
- KENNEDY I., COLEMAN R.A., HUMPHREY P.P.A., LEVY G.P., LUMLEY P. Studies on the characterization of prostanoid receptors; a proposed classification. Prostaglandins. 1982;24:667–689. doi: 10.1016/0090-6980(82)90036-3. [DOI] [PubMed] [Google Scholar]
- LAWRENCE R.A., JONES R.L. Investigation of the prostaglandin E (EP-) receptor subtype mediating relaxation of the rabbit jugular vein. Br. J. Pharmacol. 1992;63:1339–1446. doi: 10.1111/j.1476-5381.1992.tb09063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWRENCE R.A., JONES R.L., WILSON N.H. Characterization of receptors involved in the direct and indirect actions of prostaglandins E and I on the guinea pig ileum. Br. J. Pharmacol. 1992;105:271–278. doi: 10.1111/j.1476-5381.1992.tb14245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Y.J., JACKSON D.M., BLACKHAM A. Effects of BW A 868C, a selective prostaglandin DP receptor antagonist, in dog isolated vascular preparations. Eur. J. Pharmacol. 1996;303:187–192. doi: 10.1016/0014-2999(96)00037-4. [DOI] [PubMed] [Google Scholar]
- LYDFORD S.J., MCKECHNIE K. Characterization of the prostaglandin E2 sensitive (EP)-receptor in the rat isolated trachea. Br. J. Pharmacol. 1994;112:133–136. doi: 10.1111/j.1476-5381.1994.tb13042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MADDOX Y.T., RAMWELL P.W., SHINER C.S., COREY E.J. Amide and 1-amino derivatives of F prostaglandins as prostaglandin antagonists. Nature. 1978;273:549–552. doi: 10.1038/273549a0. [DOI] [PubMed] [Google Scholar]
- MAGERLEIN B.J., BUNDY G.L., LINCOLN F.H., YOUNGDALE G.A. Synthesis of 17-phenyl-18, 19, 20-trinorprostaglandins. Prostaglandins. 1975;9:5–8. doi: 10.1016/s0090-6980(75)80112-2. [DOI] [PubMed] [Google Scholar]
- MILLER W.L., WEEKS J.R., LAUDERDALE J.W., KIRTON K.T. Biological activities of 17-phenyl-18, 19, 20-trinorprostaglandins. Prostaglandins. 1975;9:9–17. doi: 10.1016/s0090-6980(75)80113-4. [DOI] [PubMed] [Google Scholar]
- MILNE S.A., ARMSTRONG R.A., WOODWARD D.F. Comparison of the EP receptor subtypes mediating relaxation of the rabbit jugular vein and pig saphenous veins. Prostaglandins. 1995;49:225–237. doi: 10.1016/0090-6980(95)00018-6. [DOI] [PubMed] [Google Scholar]
- OCKLIND A., JOUSUFZAI S.K., GHOSH S., COCA-PRADOS M., STJERNSCHANTZ J., ABDEL-LATIF A.A. Immortalization of cat iris sphincter smooth muscle cells by SV40 virus: growth, morphological, biochemical and pharmacological characteristics. Exp. Eye Res. 1995;61:535–546. doi: 10.1016/s0014-4835(05)80047-8. [DOI] [PubMed] [Google Scholar]
- RANGACHARI P.K., BETT P-A., PRIOR E.T., ROBERTS L.J. Effects of a selective DP receptor agonist (BW 245C) and antagonist (BW A868C) on the canine colonic epithelium: an argument for a different DP receptor. J. Pharmacol. Exp. Ther. 1995;275:611–617. [PubMed] [Google Scholar]
- REGAN J.W., BAILEY T.J., PIERCE K.L., ZHENG D., DONELLO J., KEDZIE K.M., WOODWARD D.F., FAIRBAIRN C.E., GIL D.W. Molecular cloning and expression of human EP3 receptors: evidence of 3 variants with differing carboxyl termini. Br. J. Pharmacol. 1994;112:377–385. doi: 10.1111/j.1476-5381.1994.tb13082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAAF T.K. Prostaglandin structure-activity relationships Annual Reports in Medicinal Chemistry 1976New York, San Francisco, London: Academic Press; 80–88.ed. Gordon, M., Francis, J., Whitfield, G., Hess, H.-J., Shen, T.Y., and Counsell, R. pp [Google Scholar]
- SCHAAF T.K., HESS H-J. Synthesis and biological activity of carboxyl-terminus modified prostaglandin analogs. J. Med. Chem. 1979;22:1340–1346. doi: 10.1021/jm00197a012. [DOI] [PubMed] [Google Scholar]
- SCHAEFER M., HOFMANN T., SCHULTZ G., GUDERMANN T. A new prostaglandin E receptor mediates calcium influx and acrosome reaction in human spermatozoa. Proc. Natl. Acad. Sci. 1998;95:3008–3013. doi: 10.1073/pnas.95.6.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKECHI H., MATSUMURA K., WATANABE Y., KATO K., NOYORI R., SUZUKI M. A novel subtype of the prostacyclin receptor expressed in the central nervous system. J. Biol. Chem. 1996;271:5901–5906. doi: 10.1074/jbc.271.10.5901. [DOI] [PubMed] [Google Scholar]
- WISE H., QIAN Y-M., JONES R.L. A study of prostacyclin mimetics distinguishes neuronal from neutrophil IP receptors. Eur. J. Pharmacol. 1995;278:265–269. doi: 10.1016/0014-2999(95)00173-i. [DOI] [PubMed] [Google Scholar]
- WOODWARD D.F., FAIRBAIRN C.E,. , KRAUSS A.H-P., LAWRENCE R.A., PROTZMAN C.E. Radioligand binding analysis of receptor subtypes in two FP receptor preparations that exhibit different functional rank orders of potency in response to prostaglandins. J. Pharmacol. Exp. Ther. 1995;273:285–291. [PubMed] [Google Scholar]
- WOODWARD D.F., HAWLEY S.B., WILLIAMS L.S., RALSTON T.L., PROTZMAN C.E., SPADA C.S., NIEVES A.L. Studies on the ocular pharmacology of prostaglandin D2. Invest. Ophthalmol. Vis. Sci. 1990;31:138–146. [PubMed] [Google Scholar]
- WOODWARD D.F., LAWRENCE R.A. Identification of a single (FP) receptor associated with prostanoid-induced Ca2+ signals in Swiss 3T3 cells. Biochem. Pharmacol. 1994;47:1567–1574. doi: 10.1016/0006-2952(94)90533-9. [DOI] [PubMed] [Google Scholar]
- YAMAGUCHI D., HAHN T.J., BEEKER T.G., KLEEMAN C.R., MUALLEM S. Relationship of cAMP and calcium messenger systems in prostaglandin stimulated UMR-106 cells. J. Biol. Chem. 1988;263:10745–10753. [PubMed] [Google Scholar]