

Abstract
There are a substantial number of drugs acting either directly or indirectly on the heart, but surprisingly, little is known about the metabolic capacity of heart muscle cells.
We therefore investigated the gene expression and protein activity of cytochrome P450 isozymes in cultures of adult cardiomyocytes of the rat.
Semi-quantitative CYP gene expression pattern suggests CYP1A1 and CYP2B1/2 to be key players in cardiomyocytes and upon treatment with Aroclor 1254 approximate 4 fold inductions could be observed for both gene families, when compared with appropriate controls.
The mRNA expression of most genes was sustained for prolonged periods of time, e.g. up to 120 h in culture and in the case of the CYP3A1 gene an approximate 10 fold induction was observed at the higher Aroclor 1254 dose level (10 μM) in 24 h old cultures.
The constitutively expressed genes, e.g. CYP2C11 and CYP2E1 are expressed throughout the entire culture period (5 days) and did not respond to Aroclor 1254 treatment.
CYP4A1 was mainly expressed in freshly isolated cardiomyocytes of control animals and its expression declined rapidly in culture.
There was good agreement between gene expression and translated protein activity using 7-ethoxyresorufin and testosterone as substrates.
The data reported herein should foster the routine use of freshly isolated and cultivated cardiomyocytes for drug profiling and toxicity studies.
Keywords: Cardiomyocyte culture, cytochrome P450 gene expression, metabolism of EROD and testosterone
Introduction
Cytochrome P450 genes encode a super-family of mixed function mono-oxygenases who are responsible for the oxidation of drugs and a wide range of structurally diverse chemicals (Smith et al., 1998). Cytochrome P450 genes are predominantly expressed in the liver, although specific isozymes are also expressed in extra hepatic tissue including lung, kidney and gastrointestinal tract (Kivisto et al., 1996a,1996b). The expression of individual P450 isozymes is often restricted to a particular cell type and can be responsible for tissue- and cell-specific toxicity as a consequence of P450-mediated metabolism of drugs and other chemicals (Smith et al., 1998).
There are a substantial number of drugs acting either directly or indirectly on the cardiovascular system, and the range of therapeutic agents which are available include medicines for the treatment of hypertension, acute and chronic heart failure, and other forms of chronic heart disease. The classes of drugs which are available for the treatment of heart disease include nitrates, beta-blockers, ACE inhibitors, ACE receptor blockers, antiarrhythmics and various cocktails of the latter, but there are also drugs with high cardiotoxicity such as anthracyclines (Pouna et al., 1996) or fenfluramines (Rajamani et al., 2000).
Many of the above named drugs are subject to cytochrome P450-mediated drug oxidation (Jurima-Romet et al., 1993, Stearns et al., 1995, Oldham et al., 1997, Tracy et al., 1999), but surprisingly, little is known about the metabolic capacity of the heart. Indeed, there is very limited information on the expression of P450 isozymes in heart tissue of laboratory animals (Stegeman et al., 1982, Geetha et al., 1991, Yamada et al., 1992, McCallum et al., 1993, Fulton et al., 1995, Xiao et al., 1998) despite their importance in the metabolism of drugs.
Cultures of primary cardiomyocytes are a valuable tool for studying the pharmacological and toxicological properties of drugs and chemicals. For instance, the proarrhythmogenic potential of new chemical entities can be studied in cultures of primary cardiomyocytes of adult rats as reported for the K-channel blocker amiodarone (Kessler-Icekson et al., 1996, Nuss et al., 1999). Next to evaluating the arrhythmogenic potential, investigation of drug–drug interactions or metabolic processes leading to adverse drug effects can also be studied with cultured cardiomyocytes but, once again, there is very limited information on the drug oxidation capacity of cardiomyocytes.
We investigated the gene expression and protein activity of cytochrome P450-dependent mono-oxygenases in cultures of adult cardiomyocytes of the rat, and for comparison in hepatocyte cultures to provide basic information on the metabolic capacity and gene regulation of drug metabolizing enzymes in heart tissue. We also treated cardiomyocyte cultures with Aroclor 1254, a complex mixture of individual polychlorinated biphenyl isomers and congeners, which are commonly used in genotoxicity studies and are well known for their ability to induce a wide range of drug metabolizing enzymes (Borlak et al., 1993; 1996).
Methods
Animals
All animal procedures described in this report were approved by the ethical committee of the local government of Hannover, Germany. Male Sprague Dawley rats weighing 199±17 g were obtained from Charles River (Sulzfeld, Germany). Food and water was given ad libitum.
Anaesthesia
Rats were anaesthetized with ketamin (anaesthetic) and xylazin-hydrochloride (muscle relaxant) with 0.1 ml of ketamin per 100 g body weight and 0.05 ml of xylazin-hydrochloride per 100 g body weight. In addition, 2000 international units of heparin were given intraperitoneally prior to surgery. All experimental results are obtained from n=3 individual animals.
Isolation and cultivation of adult cardiomyocytes
The thorax was opened by surgical procedures and the aorta ascendens was anatomically prepared. The heart was perfused in situ with the washing solution for 1 min (Joklik medium (mg l−1): NaCl 6500, KCL 400, MgCl2 200, NaH2PO4 1327, NaHCO3 2000, glucose 2000, arginine 105, cystine 24, glutamine 294, histidine 31, isoleucine 52, leucine 52, lysine 58, methionine 15, phenylalanine 32, threonine 48, tryptophane 10, tyrosine 36, valine 48, cholinchloride 1, folic acid 1, myo-inositol 2, nicotinamide 1, calcium pantothenate 1, pyridoxal 1, riboflavine 0.1, thiamine 1, streptomycin 50 and penicillin 75,000 u ml−1; Biochrom, Berlin, Germany), 500 μM EDTA and 15 mM butanedione monoxime (BDM) (Fluka, Deisenhofen, Germany). After the initial wash, the heart was swiftly removed and mounted onto the perfusion apparatus. Then, the washing solution was pumped for 5 min through the heart and immediately thereafter the enzyme solution for the isolation of cardiomyocytes (Joklik medium with 15 mM BDM, 1% bovine serum albumin, 20 μM calcium chloride, 1 mg ml−1 collagenase II; Worthington, Freehold, U.S.A.) was pumped through the heart (Ismatec MCP pump, flow rate 4 ml min−1) for 25 min at 37°C. The heart tissue became tender at the end of the perfusion period and was transferred into a petri dish containing 20 ml of the mincing solution (Joklik medium with 5% foetal calf serum). The heart was dissected with surgical scissors to obtain small pieces (<1 mm3). Then, a cell suspension was prepared and filtered through a nylon mesh (Becton Dickinson, Heidelberg, Germany) of the size of 100 μm. For 3 min the filtrate was centrifuged at 56×g at room temperature. The resultant supernatant was discarded and a further 30 ml mincing solution was added to the cell pellet. Then, the cell suspension was transferred into a culture flask (75 cm2, Corning Costar, Bodenheim, Germany) and put into the CO2 incubator. After 15 min, a 2 mM calcium solution was cautiously added to the medium. The calcium concentration was then increased over 2 h in a total of eight steps to obtain a final 1 mM concentration. Cells were also examined and photographed under phase contrast microscopy (Zeiss, Jena, Germany) to assess qualitatively and quantitatively the number of spherical and rod-shaped cells.
The cells were counted with the Neubauer's counting chamber (Hecht, Germany). Cells were cultured at 37°C at 5% CO2 for 5 days. Purity of culture was >95% and further improved by preplating cell suspension on plastic flasks for 2 h as previously reported (Chajek et al., 1977). Myocytes were cultured in the presence of Aroclor 1254 (0.1, 1.0 and 10.0 μM; dissolved in dimethylsulphoxide) and controls received the vehicle only, e.g. dimethylsulphoxide only.
Isolation and cultivation of hepatocytes
For isolation of hepatocytes a modified method described by Seglen et al. (1979) was used. In brief, after midline incision the portal vein was cannulated and the liver was first perfused in situ with 100 ml calcium-free Krebs Ringer buffer (KRB) for 10 min, then with 100 ml KRB and EDTA (1 mmol l−1). After that the liver was perfused for 8–10 min with KRB supplemented with Collagenase type IV (Worthington, Freehold, U.S.A.) and 0.5 mM calcium chloride (Sigma, Deisenhofen, Germany). Following perfusion, the liver capsule was gently removed and the dissolved liver tissue was filtrated through a nylon mesh (pore size, 100 μm) and washed twice with the washing buffer (1000 ml Hanks balanced salt solution (PAA, Germany) supplemented with 2.4 g HEPES (Sigma) and 2 g bovine serum albumin (Sigma). The cell pellet was resuspended in William's E medium (Biochrom, Berlin, Germany) supplemented with 5% FCS (Biochrom, Germany), 9.6 μg ml−1 prednisolone, 0.014 μg ml−1 glucagon (Novo, Germany), 0.16 u ml−1 insulin (Hoechst, Germany), 200 u ml−1 penicillin and 200 u ml−1 streptomycin (Karlsruhe, Germany).
Hepatocytes were counted in a haemocytometer in the presence of 0.04% trypan-blue solution. Two million hepatocytes per dish were cultured between two layers of collagen in a modification of the method of Dunn et al. (1989) and were used for further experiments.
RNA and cDNA
RNA was isolated from cells shortly after isolation and also after 1, 2, 3 and 5 days of culture using the SV total RNA Isolation System (Promega, Mannheim, Germany) according to the manufacturer's recommendation. Quality of isolated RNA was checked using a 1.0% agarose gel. Four μg total RNA from each sample were used for reverse transcription. RNA and random-primer (Roche, Mannheim, Germany) were preheated for 10 min at 70°C. Then 1×RT–AMV-buffer, dNTPs (1.0 nM, Roche, Germany), 40 u RNAse inhibitor (Stratagene, Amsterdam, Netherlands), 20 u AMV–RT (Promega), were added and diethyl pyrocarbonate (Sigma) treated water was added to a final volume of 20 μl. Then, reverse transcription was carried out for 60 min at 42°C and was stopped by heating to 95°C for 5 min. The resulting cDNA was frozen at −20°C until further experimentation.
Thermocycler RT–PCR
For the PCR amplification of cDNA, a 25 μl reaction mixture was prepared containing 10× polymerase reaction buffer, 1.5 mM MgCl2, 0.4 nM dNTPs (Roche), 400 nM concentration of the 3′ and 5′-specific primers as shown in Table 1 (synthesized by Gibco), 1 u Taq-polymerase (Roche), and 1 μl of cDNA.
Table 1.
Used oligonucleotide primers in the RT–PCR

PCR reactions were carried out in a thermal cycler (T3, Biometra, Germany) using the following melting, annealing and extension cycling conditions: denaturation for 30 s at 94°C, annealing for 60 s at 57°C and extension for 60 s at 72°C (29 cycles) for CYP isoforms and GAPDH. DNA contamination was checked for by direct amplification of RNA extracts prior to conversion of RNA to cDNA and any possible contamination could be excluded. PCR-reactions were done within the linear range of amplification and were separated using a 1.8% agarose gel and were stained with ethidium bromide and photographed on a transilluminator (see Figure 1). A semiquantitative measurement was done using the program NIH Image V.1.62.
Figure 1.
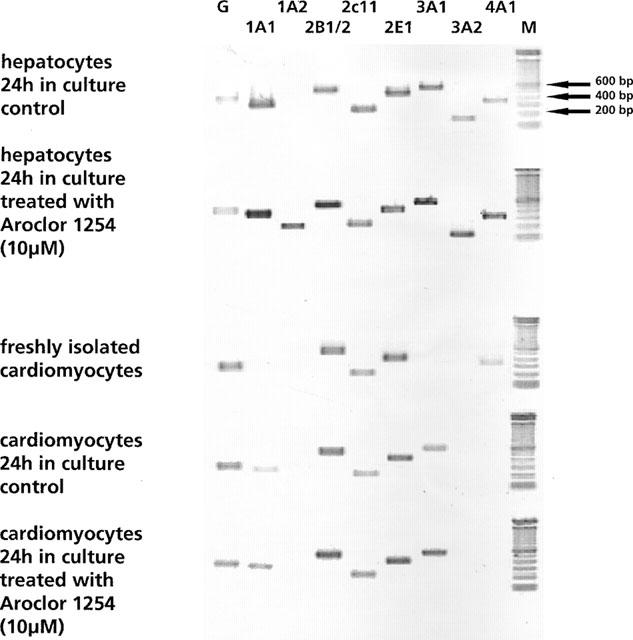
mRNA expression of cytochrome P450 isozymes and GAPDH. (M=molecular weight marker, G=GAPDH).
EROD assay
This assay was done essentially as described by Grant et al. (1987). Control cells and cells treated with Aroclor 1254 were incubated with 10 μM of 7-ethoxyresorufin and 10 μM dicumarol (Sigma) and aliquots of media were removed from the cultures at time points 0, 1, 2 and 4 h. Five hundred μl of glycine buffer (pH 10.3) was added to 500 μl of the samples and subsequent fluorometric analysis was carried out on a spectrofluoro-photometer (RF-1501, Schimadzu). Additionally, aliquots of culture media were incubated at 37°C with 100 u ml−1 of β-glucuronidase (Sigma) overnight to assess the product release of β-glucuronide conjugates. The fluorometric analysis of the resultant product resorufin was done at an excitation wave length of 530 nm and an emission wave length at 585 nm. Calibration of the system was done with appropriate standards at a concentration range of up to 100 nM.
Testosterone assay
To estimate the activity of individual CYP isozymes cardiomyocyte cultures are exposed to culture medium containing 170 μM testosterone. Finasteride (25 μM) is added as a 5α-reductase inhibitor because of its contribution to the formation of certain testosterone metabolites. Testosterone and its metabolites are analysed by HPLC according to Arlotto et al. (1991), with slight modifications as described below. 11α-Hydroxyprogesterone was used as an internal standard for the quantitative determination of testosterone and its metabolites using the following procedures: 1 μg 11-α-hydroxyprogesterone is added to 1 ml of cell culture supernatant. Following addition of 100 μl isopropanol the samples are extracted with 5 ml ethyl acetate by gentle shaking for 20 min. Extracts are evaporated to dryness and the residues are reconstituted in 100 μl of the mobile phase (water/methanol/acetonitrile/, 60/25/15, v/v/v) and 80 μl of the sample are injected onto the HPLC system (Hewlett Packard HP1100). The mobile phase is delivered at a flow rate of 1 ml min−1 using the HP 1100 Quaternary Pump. Chromatographic separation of cytochrome P450 produced testosterone metabolites is achieved on a C18 Nucleosil column, 250×4 mM and a particle size of 5 μm (Macherey-Nagel, Germany). Chromatographic separation is done at a temperature of 30°C and testosterone and metabolites are detected by UV-absorption at 238 nm using synthetic reference standards.
The mobile phase consisted of water (solvent A), methanol (solvent B) and acetonitrile (solvent C) and analysis is initiated with an isocratic elution of 60% A, 25% B and 15% C for 12 min, followed by 45% A, 40% B and 15% C for 3 min and finally 45% A, 45% B and 10% C thereafter. The total run time is 30 min per sample.
Control cells and cells treated with Aroclor 1254 (10.0 μM, 24 h) were incubated with 170 μM testosterone and 25 μM finasteride and were analysed at time points 0, 1, 2, 4 and 24 h.
Statistical analysis
Data represents mean±standard deviation. The Wilcoxon signed rank test was used and differences were considered significant at P<0.05 compared with the appropriate controls.
Results
Morphological examination
The purity of cardiomyocyte cultures was on average >95% using standard procedures including phase contrast light microscopy. Cardiomyocytes were rod shaped and their morphological appearance did not indicate cytoskeletal damage. With the trypan-blue exclusion test an average cell vitality was determined to be 92±4.9%.
Gene expression data (qualitative analysis)
Figure 1 depicts ethidium bromide stained amplification products of 8 CYP mono-oxygenase genes and the house keeping gene GAPDH from freshly isolated cardiomyocytes, cultures of control and Aroclor 1254 treated cardiomyocytes and hepatocytes, 24 h post isolation.
With freshly isolated cardiomyocytes, amplification of CYP2B1/2 and CYP2E1 resulted in clearly visible bands (see Figure 1) and in the case of CYP2C11 transcripts were also detected, albeit, at much lower levels. mRNA transcripts of CYP4A1 were additionally detected in freshly isolated cardiomyocytes.
The CYP gene expression pattern in 24 h-old cultures shows additional signals for CYP1A1 and CYP3A1 and treatment with Aroclor 1254 resulted in transcriptional activation of the CYP1A1, CYP2B1/2 and CYP3A1 gene with expression levels being similar to that of GAPDH. For comparison, results with rat hepatocyte cultures from control and treated (10 μM Aroclor 1254) cells are additionally shown in Figure 1 and 4. Based on our RT–PCR assay an array of CYP genes were detected and this included CYP1A1, CYP1A2, CYP2B1/2, CYP2C11, CYP2E1, CYP3A1, CYP3A2 and CYP4A1. Furthermore, treatment of rat hepatocyte cultures with Aroclor 1254 (10 μM) resulted in transcriptional activation on the following genes: CYP1A1, CYP1A2, CYP2B1/2, CYP3A1, CYP3A2 and CYP4A1. Noticeably, the mRNA transcripts for CYP4A1 were undetectable in 24 h-old cardiomyocyte cultures, which may indicate differences in the half lives of individual CYP gene mRNA transcripts in cultures of cardiomyocytes.
Figure 4.
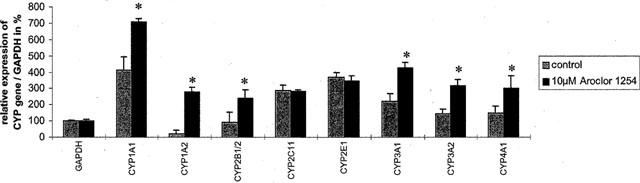
mRNA expression of cytochrome P450 isoforms relative to GAPDH in hepatocyte cultures. The GAPDH signal was set to 100%. Data represent mean±standard deviation for n=3 individual cell culture experiments with approximately 2 million cells per culture dish (*, P=<0.05 compared with controls).
CYP gene expression relative to GAPDH (semi-quantitative analysis)
The gene expression of individual CYP isozymes in freshly isolated and in cultured cardiomyocytes is shown in Figures 2 and 3. CYP1A1 mRNA transcripts could not be detected in freshly isolated cardiomyocytes, but its expression was evident in 24, 48 and 72 h-old cultures. Treatment with Aroclor 1254 resulted at the higher dose level in a 4 fold mRNA induction when compared to the 24 h control. This induction of CYP1A1 mRNA transcripts declined rapidly after 48 and 72 h and the gene expression was close to zero at day 5 in culture.
Figure 2.
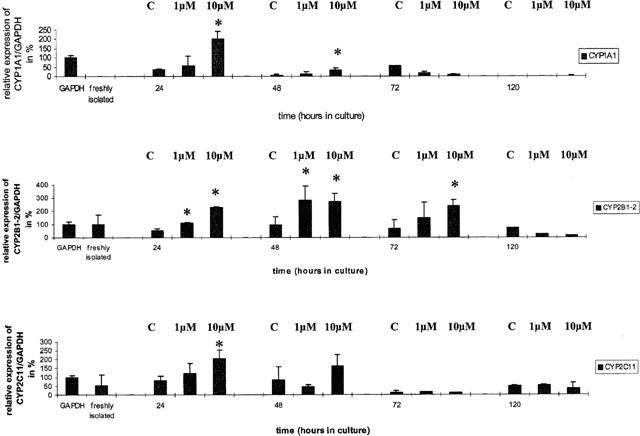
mRNA expression of cytochrome P450 1A1, 2B1/2 and 2C11 relative to GAPDH in cardiomyocyte cultures. The GAPDH signal was set to 100%. Data represent mean±standard deviation for n=3 individual cell culture experiments with approximately 2 million cells per culture dish (**, P=<0.05 compared with controls). (C=control, 1 μM and 10 μM=concentration of Aroclor 1254).
Figure 3.
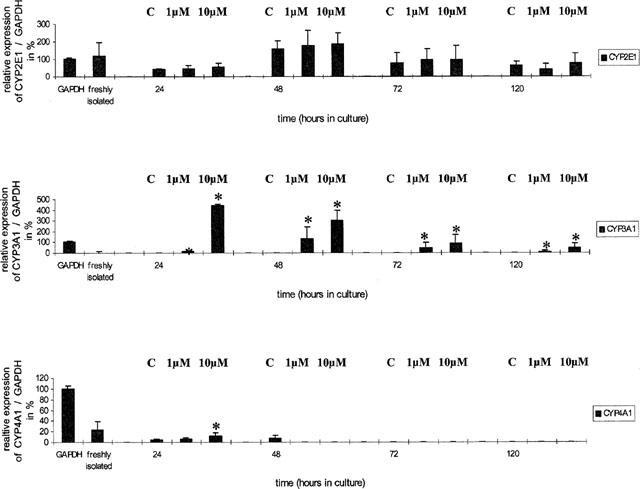
mRNA expression of cytochrome P450 2E1, 3A1 and 4A1 relative to GAPDH in cardiomyocyte cultures. The GAPDH signal was set to 100%. Data represent mean±standard deviation for n=3 individual cell culture experiments with approximately 2 million cells per culture dish (*, P=<0.05 compared with controls). (C=control, 1 μM and 10 μM=concentration of Aroclor 1254).
With CYP2B1/2 a dose-dependent induction of mRNA transcripts could be observed in 24 and 72 h old cultures, but this was not seen with 48 h-old cultures. Overall, the induction was marginal and on average less than 3 fold.
The gene expression of CYP2C11 in freshly isolated and in cultured cardiomyocytes showed some variation which on average was close to 50% of the densitometric signal obtained for GAPDH.
CYP2E1 mRNA expression was detected throughout the culture. With freshly isolated cardiomyocytes, CYP2E1 mRNA expression was similar to that of GAPDH but declined in 24 h-old cultures to about 50%. Treatment with Aroclor 1254 at both dose levels had no effect on the gene expression of CYP2E1, and in 72 h and 120 h-old cultures expression levels were approximately 50% of freshly isolated cardiomyocytes.
In the case of CYP3A1, highly significant effects are observed upon treatment with Aroclor 1254 with mRNA transcripts being induced after 24, 48, 72 and 120 h of culture. The densitometric quantification of CYP3A1 mRNA is difficult, because of its low expression in untreated animals (controls) but its expression relative to GAPDH was in the order of 4 fold and 3 fold within 24 h and 48 h of cardiomyocyte cultivation (see Figure 3).
Finally, CYP4A1 mRNA transcripts were only detected in freshly isolated and up to 48 h cultivated cardiomyocytes, the level being 20 and 5% of the densitometric signals obtained for GAPDH. When compared within the 24 h group, treatment with Aroclor 1254 (10 μM) resulted in a small, but significant induction (see Figure 3).
The gene expression in hepatocyte cultures was clearly more abundant when compared with cardiomyocyte cultures and overall treatment with Aroclor 1254 (10 μM) resulted in higher expression levels as evidenced by the ratio of CYP gene/GAPDH shown in Figure 4.
Enzyme activities with CYP marker substrates
The protein activity of individual CYP isoforms were studied in control and Aroclor 1254 treated cardiomyocyte and hepatocyte cultures. Ethoxyresorufin is a substrate for CYP1A1 and a dose- and time-dependent increase in CYP1A1 activity was observed as reflected by the cumulative release of resorufin (see Figure 5a). The approximately 5 fold increase in CYP1A1 activity at the 10.0 μM dose of Aroclor 1254 agrees well with the induction estimated for CYP1A1 mRNA. When aliquots of cardiomyocyte culture media were digested with β-glucuronidase no difference in the amount of resorufin (product release) was seen, thus indicating that cardiomyocytes have neglectable glucuronidase activity towards this substrate (see Figure 5b).
Figure 5.
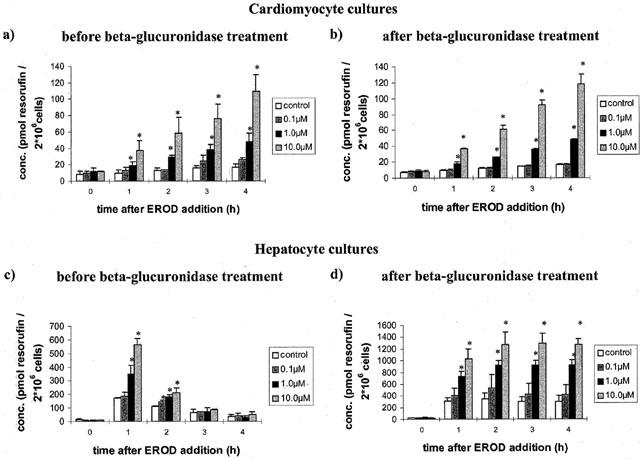
Metabolism of 7-ethoxyresorufin in control and Aroclor 1254 (10 μM) treated cardiomyocytes before (a) and after (b) treatment with 100 u ml−1 β-glucuronidase 24 h post isolation and cultivation. Metabolism of 7-ethoxyresorufin in control and Aroclor 1254 (10 μM) treated hepatocytes before (c) and after (d) treatment with 100 u ml−1 β-glucuronidase 24 h post isolation and cultivation. Data represent mean±standard deviation for n=3 individual cell culture experiments with approximately 2 million cells per culture dish. *, P=<0.05 compared with controls.
In strong contrast, with hepatocyte cultures the amount of resorufin produced declined steadily over time and was close to control values after 4 h of incubation (see Figure 5c). When, however, these samples were treated with the enzyme β-glucuronidase a linear and dose-dependent increase in EROD-activity could be observed with an approximate 12 fold increase in activity at the highest dose level (see Figure 5d). When compared with cardiomyocytes the EROD activity of hepatocyte cultures was up to 40 fold and 25 fold higher in controls and treated cells, respectively.
No difference in the metabolism of testosterone among the various treatment groups was obvious and thus, the metabolism of testosterone for control cardiomyocytes exposed to Aroclor 1254 have been combined (Figure 6a). There is a time-dependent increase in the release of 6-β-Hydroxy-testosterone (HT) and androstendione, but no other metabolites could be identified and the levels of product release were similar, e.g. 30 and 50 pmol per 2×106 cells ml−1 h−1 culture medium for 6β- HT and androstendione, respectively (see Figure 6a). In hepatocyte cultures the product release was much higher, the level being 550 and 5000 pmol per 2×106 cells ml−1 h−1 culture medium for 6-β-HT and androstendione, respectively. Additional metabolites were observed in hepatocyte cultures and contain 16-α-testosterone, 2-α-testosterone, 16-β-testosterone and 7-α-testosterone.
Figure 6.
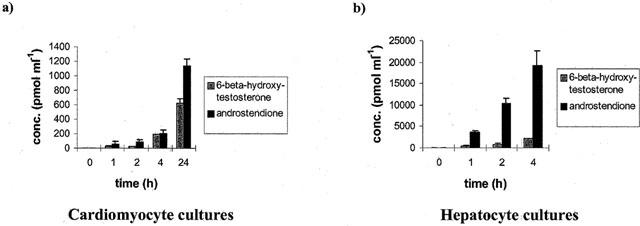
Testosterone metabolism in cardiomyocytes (a) and hepatocytes (b) 24 h post isolation and cultivation. The release of 6-β-hydroxy-testosterone and androstendione is shown. Data represent mean±standard deviation for n=3 individual cell culture experiments with approximately 2 million cells per culture dish.
When the metabolism of testosterone was assessed (after 4 h incubation) in up to 5-day-old cultures a decline in the production of 6-β-hydroxy-testosterone and androstendione was observed in cardiomyocyte cultures, the level being 40% and 10%, respectively 120 h post-isolation (Figure 7). Again, this decline in metabolic capacity correlates well with the gene expression data for the various CYP isoforms described above.
Figure 7.
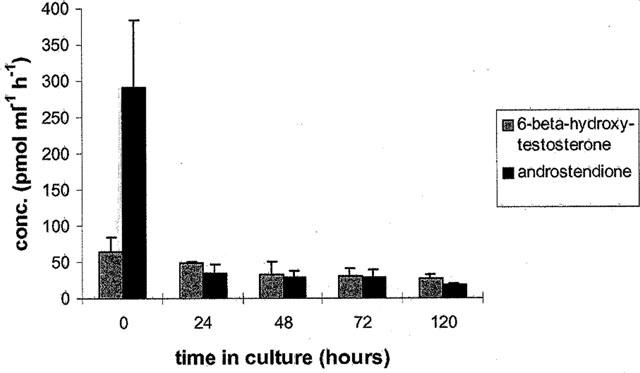
Testosterone metabolism in freshly isolated and cultivated (24, 48, 72 and 120 h) cardiomyocytes. The release of 6-β-hydroxy-testosterone and androstendione is shown. Data represent mean±standard deviation for n=3 individual cell culture experiments with approximately 2 million cells per culture dish.
Discussion
We investigated the gene expression and enzyme activity of cytochrome P450 isozymes in cultures of adult cardiomyocytes and hepatocytes of the rat. Unequivocal evidence is presented to show the expression of a number of important drug oxidizing enzymes in cultures of adult rat cardiomyocytes at the gene and enzyme activity level. With the RT–PCR assay, transcripts for CYP1A1, CYP2B1/2, CYP2C11, CYP2E1, CYP3A1 and CYP4A1 could be amplified in cultures of cardiomyocytes, albeit at different expression levels. CYP1A1 and CYP2B1/2 were key players with regard to expression and mRNA induction and the mRNA expression of most genes was sustained for prolonged periods of time, e.g. up to 120 h in culture. In the case of the CYP3A1 gene an approximate 10 fold induction was noticeable in 24 h-old cultures at the higher Aroclor 1254 dose (10 μM) when compared with freshly isolated cells. The expression of constitutive genes CYP2C11 and CYP2E1 remained stable along the entire culture period, e.g. 5 days, and the latter genes did not respond to Aroclor 1254 treatment with the exception of CYP2C11 in 24 h-old cultures. Interestingly CYP4A1 was mainly expressed in freshly isolated cardiomyocytes of control animals but its expression declined thereafter to levels below detection. In view of its role in fatty acid metabolism (Simpson, 1997) future studies should focus on CYP4A1 gene regulation with regard to arachidonic acid metabolism. Arachidonic acid and some of its metabolic derivatives are important messengers in signal transduction cascades and may impact gene regulation in cardiomyocytes.
There was also good agreement between the gene expression and enzyme activity data, as shown by the EROD and testosterone metabolism reported in Figures 5, 6a,b and 7, and our findings with rat hepatocytes confirm previous results of several investigators, which show cytochrome P450 protein expression to be predominant in the liver (Smith et al., 1998; Clarke, 1998; Hansen et al., 2000).
To the best of our knowledge the concomitant gene expression and enzyme activity of cytochrome P450 isozymes in cultures of cardiomyocytes of adult rats has not been reported, as yet, and thus no comparison to reported literature findings can be made. Nevertheless, Stegemann et al. (1982) have reported high levels of P450 in fish cardiac microsomes of Stenotomus chrysops. These authors could show that treatment of Stenotomus chrysops (scup) with β-naphthophlavone produced a strong, approximately 10 fold, induction of spectrally measured P450 in cardiac microsomes with specific P450 content reaching levels similar to those induced in scup liver. In our study a mere 4 fold induction was observed in cultures of cardiomyocytes, but a similar 12 fold increase in EROD activity was recorded in assays with Aroclor 1254 treated hepatocytes.
One of the first reports on cytochrome P450 mRNA expression in human heart tissue was published by Price and coworkers (Price et al. 1992). These authors reported the relative expression of aromatase cytochrome P450 in human foetal tissue as determined by competitive polymerase chain reaction and cytochrome P450 aromatase mRNA was identified in all foetal tissues including liver, lung, brain, skin and testine, kidney, spleen and the heart. Nonetheless, foetal liver contained far more P450 aromatase mRNA when compared to any of the other tissues studied and again, this is in accordance with the findings of this study where high levels of P450 mRNA were detected in hepatocytes.
Also, in a study of Fulton et al. (1995), the cytochrome P450-dependent effects of bradykinin in the rat heart was investigated. The authors report that vasodilator responses were reduced when the CYP P450 inhibitory drug clotrimazole at a 1 μM concentration was used, and although NO and prostaglandins do not mediate vasodilator response to bradykinin in the rat heart, the investigations of Fulton et al. clearly suggest a major role for a P450-dependent mechanism via arachidonic acids metabolism. The molecular cloning, expression and functional significance of a specific cytochrome P450 in rat heart cardiomyocytes was reported by Wu and coworkers (Wu et al. 1997). In the latter study, metabolism of arachidonic acid and 11,12-epoxyeicosatrienoic was governed by P450 2J2, and this particular enzyme may play an important role in heart ischaemia. We show P450 4A1 mRNA expression in freshly isolated and 24 h-old cardiomyocytes and in view of its role in fatty acid metabolism future studies should focus on the relative contribution of this enzyme in the metabolism of arachidonic acid. In view of the very low CYP activities in heart we suggest that cardiomyocytes are unlikely to be an important quantitative contributer to CYP-mediated arachidonic acid metabolism. Nevertheless, a small amount of locally formed arachidonic acid products may have critical roles as biomediators. There is also evidence that cytochrome P450 modulates contractility of rat cardiomyocytes through metabolism of arachidonic acid (Xiao et al., 1998). The discovery of P450 in cardiomyocytes is intriguing and it is surprising that so little is known about the metabolic capacity of the heart. Several cardioselective drugs, such as β-blockers, calcium antagonists, inhibitors of angiotensin converting enzymes and angiotensin converting enzyme receptor antagonists are all metabolized by cytochrome P450-dependent pathways (Jurima-Romet et al., 1993; Stearns et al., 1995; Tracy et al., 1999; Oldham & Clarke, 1997). The discovery of cytochrome P450 in cardiomyocytes have implications for pharmacotherapy from both a safety and efficacy perspective, and it is tempting to speculate that the level of expression of cytochrome P450 enzymes might provide an explanation for drug therapy failure or cardiac toxicity upon drug treatment. The finding of steroid (testosterone) metabolism in cardiomyocytes provides further evidence for the metabolic competence of this tissue and its importance in the metabolism of endogenous compounds.
Abbreviations
- ACE
angiotensin converting enzyme
- AMV-RT
avian myeloblastosis virus-reverse transcriptase
- BDM
butanedione monoxime
- cDNA
copy DNA
- CYP
cytochrome P
- dNTPs
deoxy-nucleotide triphosphate
- EDTA
ethylenediaminetetraacetic acid
- EROD
7-ethoxyresorufin O-deethylase
- GAPDH
glyceraldehyde-3-phosphate-dehydrogenase
- HEPES
Hydroxyethyl-piperazino-ethansulphonic acid
- PCR
polymerase chain reaction
- NO
nitric oxide
- SV
Simian Virus
References
- ARLOTTO M.P., TRANT J.M., ESTABROOK R.W. Measurement of steroid hydroxylation reactions by high-performance liquid chromatography as indicator of P450 identity and function. Methods Enzymol. 1991;206:454–462. doi: 10.1016/0076-6879(91)06114-i. [DOI] [PubMed] [Google Scholar]
- BORLAK J., SCOTT A., HENDERSON C.J., JENKE H.J., WOLF C.R. Transfer of PCBs via lactation simultaneously induces the expression of P450 isoenzymes and the protooncogenes c-Ha-ras and c-raf in neonates. Biochem. Pharmacol. 1993;51:517–529. doi: 10.1016/0006-2952(95)02228-7. [DOI] [PubMed] [Google Scholar]
- BORLAK J., SCOTT A., HENDERSON C.J., JENKE H.J., WOLF C.R. Transplacental transfer of polychlorinated biphenyls induces simultaneously the expression of P450 isoenzymes and the protooncogenes c-Ha-ras and c-raf. Biochem. Pharmacol. 1996;45:1373–1386. doi: 10.1016/0006-2952(93)90035-u. [DOI] [PubMed] [Google Scholar]
- CHAJEK T., STEIN O., STEIN Y. Rat heart in culture as a tool to elucidate the cellular origin of lipoprotein lipase. Biochim. Biophys. Acta. 1977;488:104–144. doi: 10.1016/0005-2760(77)90131-x. [DOI] [PubMed] [Google Scholar]
- CLARKE S.E. In vitro assessment of human cytochrome P450. Xenobiotica. 1998;28:1167–1202. doi: 10.1080/004982598238877. [DOI] [PubMed] [Google Scholar]
- DUNN J.C., YARMUSH M.L., KOEBE H.G., TOMPKINS R.G. Hepatocyte function and extracellular matrix geometry: long-term culture in a sandwich configuration. FASEB J. 1989;3:174–177. doi: 10.1096/fasebj.3.2.2914628. [DOI] [PubMed] [Google Scholar]
- FULTON D., MAHBOUBI K., MCGIFF J.C., QUILLEY J. Cytochrome P450-dependent effects of bradykinin in the rat heart. Br. J. Pharmacol. 1995;114:99–102. doi: 10.1111/j.1476-5381.1995.tb14911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEETHA A., MARAR T., DEVI C.S. Effect of alpha-tocopherol on doxorubicin-induced changes in rat liver and heart microsomes. Indian. J. Exp. Biol. 1991;2:782–785. [PubMed] [Google Scholar]
- GRANT M.H., BURKE M.D., HAWKSWORTH G.M., DUTHIE S.J., ENGESET J., PETRIE J.C. Human adult hepatocytes in primary monolayer culture. Maintenance of mixed function oxidase and conjugation pathways of drug metabolism. Biochem. Pharmacol. 1987;36:2311–2316. doi: 10.1016/0006-2952(87)90596-x. [DOI] [PubMed] [Google Scholar]
- HANSEN T., BORLAK J., BADER A. Cytochrome P450 enzyme activity and protein expression in primary porcine enterocyte and hepatocyte cultures. Xenobiotica. 2000;30:27–46. doi: 10.1080/004982500237802. [DOI] [PubMed] [Google Scholar]
- JURIMA-ROMET M., HUANG H.S. Comparative cytotoxicity of angiotensin-converting enzyme inhibitors in cultured rat hepatocytes. Biochem. Pharmacol. 1993;46:2163–2170. doi: 10.1016/0006-2952(93)90605-v. [DOI] [PubMed] [Google Scholar]
- KESSLER-ICEKSON G., SCHLESINGER H., DJALDETTI M., BRODIE C., SAMPSON S.R., BERGMAN M. Cardiomyocytes in culture – a model to study the cellular actions of amiodarone. Isr. J. Med. Sci. 1996;32:1212–1216. [PubMed] [Google Scholar]
- KIVISTO K.T., BOOKJANS G., FROMM M.F., GRIESE E.U., MUNZEL P., KROEMER H.K. Expression of CYP3A4, CYP3A5 and CYP3A7 in human duodenal tissue. Br. J. Clin. Pharmacol. 1996b;42:387–389. doi: 10.1046/j.1365-2125.1996.42615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIVISTO K.T., GRIESE E.U., FRITZ P., LINDER A., HAKKOLA J., RAUNIO H., BEAUNE P., KROEMER H.K. Expression of cytochrome P450 3A enzymes in human lung: a combined RT-PCR and immunohistochemical analysis of normal tissue and lung tumours. Naunyn Schmiedebergs Arch. Pharmacol. 1996a;353:207–212. doi: 10.1007/BF00168759. [DOI] [PubMed] [Google Scholar]
- MCCALLUM G.P., HORTON J.E., FALKNER K.C., BEND J.R. Microsomal cytochrome P450 1A1 dependent monooxygenase activity in guinea pig heart: induction, inhibition, and increased activity by addition of exogenous NADPH-cytochrome P450 reductase. Can. J. Physiol. Pharmacol. 1993;71:151–156. doi: 10.1139/y93-021. [DOI] [PubMed] [Google Scholar]
- NUSS H.B., KAAB S., KASS D.A., TOMASELLI G.F., MARBAN E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am. J. Physiol. 1999;277:H80–H91. doi: 10.1152/ajpheart.1999.277.1.H80. [DOI] [PubMed] [Google Scholar]
- OLDHAM H.G., CLARKE S.E. In vitro identification of the human cytochrome P450 enzymes involved in the metabolism of R(+)- and S(−)-carvedilol. Drug Metab. Dispos. 1997;25:970–977. [PubMed] [Google Scholar]
- POUNA P., BONORON-ADELE S., GOUVERNEUR G., TARIOSSE L., BESSE P., ROBERT J. Development of the model of rat isolated perfused heart for the evaluation of anthracycline cardiotoxicity and its circumvention. Br. J. Pharmacol. 1996;117:1593–1599. doi: 10.1111/j.1476-5381.1996.tb15326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRICE T., AITKEN J., SIMPSON E.R. Relative expression of aromatase cytochrome P450 in human fetal tissues as determined by competitive polymerase chain reaction amplification. J. Clin. Endocrinol. Metab. 1992;74:879–883. doi: 10.1210/jcem.74.4.1548354. [DOI] [PubMed] [Google Scholar]
- RAJAMANI S., STUDENIK C., LEMMENS-GRUBER R., HEISTRACHER P. Cardiotoxic effects of fenfluramine hydrochloride on isolated cardiac preparations and ventricular myocytes of guinea-pigs. Br. J. Pharmacol. 2000;129:843–852. doi: 10.1038/sj.bjp.0703118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEGLEN P.O. Hepatocyte suspensions and cultures as tools in experimental carcinogenesis. J. Toxicol. Environ. Health. 1979;5:551–560. doi: 10.1080/15287397909529766. [DOI] [PubMed] [Google Scholar]
- SIMPSON A.E. The cytochrome P450 4 (CYP4) family. Gen. Pharmacol. 1997;28:351–359. doi: 10.1016/s0306-3623(96)00246-7. [DOI] [PubMed] [Google Scholar]
- SMITH G., STUBBINS M.J., HARRIES L.W., WOLF C.R. Molecular genetics of the human cytochrome P450 monooxygenase superfamily. Xenobiotica. 1998;28:1129–1165. doi: 10.1080/004982598238868. [DOI] [PubMed] [Google Scholar]
- STEARNS R.A., CHAKRAVARTY P.K., CHEN R., CHIU S.H. Biotransformation of losartan to its active carboxylic acid metabolite in human liver microsomes. Role of cytochrome P4502C and 3A subfamily members. Drug Metab. Dispos. 1995;23:207–215. [PubMed] [Google Scholar]
- STEGEMAN J.J., WOODIN B.R., KLOTZ A.V., WOLKE R.E., ORME-JOHNSON N.R. Cytochrome P-450 and monooxygenase activity in cardiac microsomes from the fish Stenotomus chrysops. Mol. Pharmacol. 1982;21:517–526. [PubMed] [Google Scholar]
- TRACY T.S., KORZEKWA K.R., GONZALEZ F.J., WAINER I.W. Cytochrome P450 isoforms involved in metabolism of the enantiomers of verapamil and norverapamil. Br. J. Clin. Pharmacol. 1999;47:545–552. doi: 10.1046/j.1365-2125.1999.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU S., CHEN W., MURPHY E., GABEL S., TOMER K.B., FOLEY J., STEENBERGEN C., FALCK J.R., MOOMAW C.R., ZELDIN C. Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. J. Biol. Chem. 1997;272:12551–12559. doi: 10.1074/jbc.272.19.12551. [DOI] [PubMed] [Google Scholar]
- XIAO Y.F., HUANG L., MORGAN J.P. Cytochrome P450: a novel system modulating Ca2+ channels and concentration in mammalian heart cells. J. Physiol. 1998;508:377–392. doi: 10.1111/j.1469-7793.1998.777bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMADA H., KANEKO H., TAKEUCHI K., OGURI K., YOSHIMURA H. Tissue-specific expression, induction, and inhibition through metabolic intermediate-complex formation of guinea pig cytochrome P450 belonging to the CYP2B subfamily. Arch. Biochem. Biophys. 1992;299:248–254. doi: 10.1016/0003-9861(92)90271-w. [DOI] [PubMed] [Google Scholar]