

Abstract
In the presence of L-NNA (100 μM), indomethacin (10 μM) and ODQ (10 μM), acetylcholine induced a concentration-dependent vasorelaxation of guinea-pig mesenteric and middle cerebral arteries precontracted with cirazoline or histamine, but not with high K+, indicating the contribution of an endothelium-derived hyperpolarizing factor (EDHF).
In cerebral arteries, charybdotoxin (ChTX; 0.1 μM) completely inhibited the indomethacin, L-NNA and ODQ-insensitive relaxation; iberiotoxin (IbTX, 0.1 μM), 4-aminopyridine (4-AP, 1 mM), or barium (30 μM) significantly reduced the response; in the mesenteric artery, ChTX and IbTX also reduced this relaxation. Glibenclamide (10 μM) had no affect in either the mesenteric or cerebral artery.
Neither clotrimazole (1 μM) nor 7-ethoxyresorufin (3 μM) affected EDHF-mediated relaxation in the mesenteric artery, but abolished or attenuated EDHF-mediated relaxations in the cerebral artery. AM404 (30 μM), a selective anandamide transport inhibitor, did not affect the vasorelaxation response to acetylcholine in the cerebral artery, but in the mesenteric artery potentiated the vasorelaxation response to acetylcholine in an IbTX, and apamin-sensitive, but SR 141816A-insensitive manner. Ouabain (100 μM) almost abolished EDHF-mediated relaxation in the mesenteric artery, but enhanced the relaxation in the cerebral artery whereas the addition of K+ (5–20 mM) to precontracted guinea-pig cerebral or mesenteric artery induced further vasoconstriction.
These data suggest that in the guinea-pig mesenteric and cerebral arteries different EDHFs mediate acetylcholine-induced relaxation, however, EDHF is unlikely to be mediated by K+.
Keywords: Acetylcholine, calcium-activated potassium channels, cytochrome P450, delayed rectifier potassium channels, endothelium-derived hyperpolarizing factor (EDHF), inwardly rectifying potassium channels, mesenteric artery, middle cerebral artery
Introduction
A substantial body of evidence supports the view that acetylcholine mediates endothelium-dependent relaxation of many vessels via the release of at least three endothelium-derived vasodilators: prostacyclin (PGI2), nitric oxide (NO) and endothelium-derived hyperpolarizing factor (EDHF) Gryglewski et al., 1986; Palmer et al., 1987; Taylor & Weston, 1988; Chen et al., 1988; Triggle et al., 1999). The regulation of the release, the cellular actions as well as the putative identity of EDHF have been the subject of a number of recent studies. It is assumed that EDHF-mediates vasorelaxation via the opening of the K+ channels, however, the identity and cellular mechanism(s) of action of EDHF remain poorly defined (Garland et al., 1995; Waldron et al., 1996; Guimaráes & Moura, 1999; Triggle et al., 1999). In porcine coronary artery (Hecker et al., 1994; Campbell et al., 1996; Popp et al., 1996) and rabbit carotid artery (Lischke et al., 1995; Dong et al., 1997a) evidence suggests that EDHF may be a cytochrome P450-derived epoxide. However, the evidence is inconsistent in the rat mesenteric artery (Fukao et al., 1997; Vanheel & de Voorde, 1997) and hepatic artery (Zygmunt et al., 1996). Evidence has also suggested than an endogenous cannabinoid, anandamide, may also be an EDHF (Randall et al., 1996; Randall & Kendall, 1997), however, this has been vigorously challenged (Plane et al., 1997; Zygmunt et al., 1997). Conversely, anandamide may be involved in mediating endothelium-dependent vasorelaxation via the stimulation of capacitative Ca2+ entry in endothelial cells and increasing the synthesis and release of an endothelium-derived vasorelaxant rather than a direct action on vascular smooth muscle (Mombouli, et al., 1999). A recent report (Edwards et al., 1998) suggests that endothelium-dependent hyperpolarization is mediated by a small increase (5–10 mM) in extracellular K+ that originates from endothelial cells subsequent to the opening of Ca2+-activated K+ channels.
The data presented in a number of recent studies have also emphasized the need to consider the role of direct endothelial-vascular smooth muscle cell communication in mediating endothelium-dependent relaxation in a variety of vascular preparations. Thus, Chaytor et al. (1998) reported that a peptide, Gap 27, which contains an amino acid sequence identical to that contained in the second extracellular loop of the connexin reported to be present in vascular tissue, attenuated the L-NAME insensitive endothelium-dependent relaxation to ACh in the rabbit mesenteric artery. Furthermore, Gap 27 also inhibited the ACh and ATP-induced relaxation in the rabbit aorta. Thus, endothelium-dependent hyperpolarization may reflect direct electrical communication from endothelial to VSMC and gap junctions may provide a preferred route for endothelial cell-derived factors to act on VSMCs. Of particular relevance to the current investigation are the recent studies by Yamamoto et al. (1998; 1999) that report the putative gap junction blocker 18 β-glycyrrhetinic acid blocked the ACh-mediated, ChTX-sensitive and endothelium-dependent hyperpolarization of guinea-pig mesenteric arterioles. These data suggest an essential role for myoendothelial gap junctions in mediating endothelium-dependent vasorelaxation in guinea-pig mesenteric arterioles.
Overall, the contribution of EDHF to endothelium-dependent relaxation appears to depend on tissue source, species and agonist employed to induce vasorelaxation (Nagao & Vanhoutte, 1993; Garland et al., 1995; Waldron et al., 1996). Unfortunately, there have been few systematic studies comparing the contribution of EDHF to endothelium-dependent relaxation in different vessels under identical experimental conditions. Furthermore, the contribution of EDHF to endothelium-dependent relaxation in small cerebral resistance arteries remains elusive (Petersson et al., 1997; Dong et al., 1998). Therefore, the present study was undertaken to systematically compare EDHF-mediated endothelium-dependent vasorelaxation in small cerebral and mesenteric resistance arteries from guinea-pigs under identical experimental conditions. Following such protocols we have found differences in the nature and cellular mechanism(s) of action of EDHF released from small cerebral and mesenteric resistance arteries. Our results suggest that there might be different EDHFs existing in cerebral versus peripheral vessels of guinea-pigs. Further studies, however, are required in order to assess the contribution of myoendothelial gap junctions to endothelium-dependent relaxation in guinea pig cerebral and mesenteric vessels. Some of the data presented here have been previously published in abstract form (Dong et al., 1997b).
Methods
Preparation of arteries
Male guinea pigs (400–450 g) were killed by cervical dislocation after anaesthesia with halothane according to a research protocol consistent with the standards of the Canadian Council on Animal Care and approved by the local Animal Care Committee of the University of Calgary. Brains and small intestine were rapidly removed and placed in cold physiological salt solution (PSS) of the following composition (in mM): NaCl 118; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4 1.2; NaHCO3 12.5; dextrose 11.1. The pH of the PSS after saturation with 95% O2+5% CO2 gas mixture was 7.4. Solutions of high K+ PSS (40 mM) KCl) were made by equimolar replacement of NaCl with KCl. Segments of middle cerebral artery (150–200 μm i.d.) and the third order branches of mesenteric artery (200–250 μm i.d.) were gently dissected from the surface of the brain and the mesenteric vascular bed, respectively, and placed in fresh PSS. Where possible we used mesenteric vessels that approximated the size of the cerebral vessels studied (approximately 200 μm i.d.). Adherent connective tissue was removed and a 2 mm segment of artery was then prepared for recording isometric force development in a wire myograph as previously described (Waldron & Garland, 1994). Briefly, two tungsten wires (40 μm diameter) were inserted through the lumen of the vessel and the tissue placed in a 10 ml myograph chamber containing gassed PSS. One wire was then attached to a force transducer and the other connected to a micrometer. In some experiments, endothelial cells were removed from the vessels by repeatedly passing stainless steel cannulae of appropriate size through the vessel lumen and destruction of the endothelium was confirmed pharmacologically by loss of the relaxation response to acetylcholine (10 μM). Only vessels which after denudation had responses to contractile agents comparable to those from as endothelium-intact arteries were used for further studies. After a 30 min equilibration period, arteries were stretched in a stepwise fashion to a resting tension of 2 mN, which in preliminary studies was found to be the optimal preload for force development in these blood vessels. Tissues were routinely allowed to equilibrate for 2 h before the start of the experiments. Isometric tension was recorded at a sample interval of 0.2 s to a hard disc via an analog/digital converter (Axon Instruments, Foster City, U.S.A.) and analysed using computer software (Axotape 2.0, Axon Instruments).
Experimental protocols
The first series of experiments were performed to determine the contribution of EDHF to endothelium-dependent vasorelaxation in segments of the cerebral artery and the mesenteric artery. Arterial rings were precontracted with histamine (3 μM, cerebral artery) or cirazoline (1 μM, mesenteric artery) and subsequently relaxed by applying acetylcholine cumulatively in half-log increments from 3 nM to 3 μM. The second acetylcholine concentration-response curves were always performed after 30 min of washing with PSS and blockers (indomethacin (10 μM) plus NG-nitro-L-arginine (L-NNA, 100 μM); or a combination of both agents, plus high K+ PSS, were added to the tissues 30 min prior to determining the second concentration-response curve. Pretreatment with indomethacin and L-NNA did not affect the basal tone of the vessels, but increased the sensitivity of the vessels to histamine or cirazoline. To rule out the possibility of functional antagonism (in the presence of indomethacin plus L-NNA or a combination of both agents plus high K+ PSS) the concentration of histamine or cirazoline was adjusted to ensure a similar level of tone to control conditions. In a second series of experiments designed to pharmacologically characterize the EDHF response from either the cerebral artery or the mesenteric artery, initial concentration-response curves for acetylcholine were obtained in the presence of indomethacin and L-NNA, and then compared with a second exposure to acetylcholine performed under similar conditions plus the presence of another blocker. In these experiments, the second concentration-response curves were performed with: (a) either U73122 (1 μM) or AACOCF3 (1 μM) to inhibit phospholipase C (PLC) or phospholipase A2 (PLA2), respectively; (b) clotrimazole (1 μM) or 7-ethoxyresorufin (3 μM) to inhibit cytochrome P450 and AM404 (30 μM) to inhibit the carrier-mediated transport of anandamide. In a third series of experiments designed to investigate the cellular mechanisms of the vasodilation mediated by EDHF, initial concentration-response curves to acetylcholine were obtained in the presence of indomethacin and L-NNA, and then compared to the extent of relaxation during a second exposure to acetylcholine performed under similar conditions plus the presence of another potassium channel blocker: (a) ChTX (0.1 μM), to block intermediate conductance and large conductance Ca2+ activated K+ channels (IKCa and BKCa respectively); (b) IbTX (0.1 μM), to block large conductance Ca2+-activated K+ (BKCa) channels; (c) Apamin (3 μM), to block small conductance Ca2+-activated K+ (SKCa) channels; (d) Glibenclamide (10 μM), to block ATP-sensitive K+ (KATP) channels; (e) 4-aminopyridine (4-AP, 1 mM), to block voltage-gated K+ (KV) channels; (f) BaCl2 (30 μM), to block inwardly rectifying K+ (Kir) channels. Because pretreatment with ChTX and 4-AP sometimes increased the basal tone of the vessels, the concentration of histamine or cirazoline was appropriately decreased after pretreatment with ChTX or 4-AP in order to maintain tone comparable to that prior pretreatment with the K+ channel blockers. The other K+ channel blockers used in the present study did not affect the basal level of tone in the cerebral or the mesenteric artery. In most experiments, only two concentration-response curves were obtained for an individual tissue. In preliminary experiments, control curves to acetylcholine were performed in the absence of inhibitors to determine the effects of time and repeated exposure to acetylcholine on the magnitude of the vasorelaxation, however, under these conditions no significant difference was identified during three sequential concentration-response curves to acetylcholine.
Drugs
Indomethacin, glibenclamide, apamin, charybdotoxin, 4-AP, and clotrimazole were purchased from Sigma Chemical Company (St. Louis, U.S.A.). Iberiotoxin, NG-nitro-L-arginine were purchased from Research Biochemicals, Inc. (Natick, U.S.A.). 1H[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one (ODQ) was purchased from Tocris Cookson Inc. (St. Louis, U.S.A.), AM404 from Cayman Chemical (MI, U.S.A.), and SR 141716A from SRI International (Menlo Park, CA, U.S.A.). Stock solutions of clotrimazole (1 mM) and indomethacin (1 mM) were prepared in ethanol and 4% (w/v) NaHCO3, respectively, and SR 141716A and glibenclamide (10 mM) in dimethylsulphoxide. All other compounds dissolved freely in distilled water. At the final concentrations employed (0.1%), none of the solvents used had any effect on the vessels during preliminary experiments. A saturated solution of nitric oxide was prepared by bubbling gaseous nitric oxide (Liquid Carbonic INC.) for 20 min through a gas tight vial containing ice-cold distilled water which had been bubbled with research grade argon (Union Carbide) for 20 min, producing a saturated solution of exogenous nitric oxide of ∼1 mM. Nitric oxide in solution was serially diluted immediately before use in gas-tight vials using cold deoxygenated distilled water and then injected into the organ bath in volumes of 10–30 μl with a gas-tight syringe.
Statistical analysis
Vasorelaxant responses were measured as percentage inhibition of the contraction induced by histamine or cirazoline. pEC50 values were determined as the negative log molar concentration of acetylcholine which caused 50% of the maximal vasorelaxant effect. All data were expressed as means±s.e.m. Where appropriate, Student's t-test for paired data and one way analysis of variance (ANOVA) were used and considered significant at P<0.05. In all experiments, n equals the number of animals from which vessels were removed.
Results
Contribution of EDHF to vasorelaxation
In the mesenteric artery precontracted with cirazoline, acetylcholine induced a concentration-dependent relaxation, that was completely abolished by removal of the endothelium (n=4, data not shown). A combination pretreatment with the cyclo-oxygenase inhibitor, indomethacin (10 μM) and the NO synthase inhibitor, L-NNA (100 μM), significantly shifted the concentration-response curves for acetylcholine to the right (Figure 1). The pEC50 value for the pretreatment was described (7.67±0.07 vs 7.06±0.03, P<0.01, n=6) with the maximal relaxation to acetylcholine unchanged (105±3% vs 100±1%, P>0.05, n=6). In the presence of indomethacin and L-NNA, the soluble guanylyl cyclase inhibitor ODQ (10 μM) produced no significant inhibition of the concentration-response curve to acetylcholine (Figure 1 and Table 1). However, after vessels were pretreated with indomethacin and L-NNA and then contracted with high K+ PSS (40 mM), the acetylcholine-induced vasorelaxation was completely blocked (Figure 1 and Table 1). In the cerebral artery, a combination of indomethacin and L-NNA significantly inhibited the concentration-response curves to acetylcholine (pEC50 value: 7.3±0.03 vs 6.80±0.08 and the maximal relaxation: 99±4% vs 76±5%, P<0.05, n=6 for both). ODQ (10 μM) produced no additional inhibition, however, after the cerebral artery was precontracted with high K+ PSS (40 mM), the acetylcholine-induced vasorelaxation was completely blocked (Figure 1 and Table 2).
Figure 1.
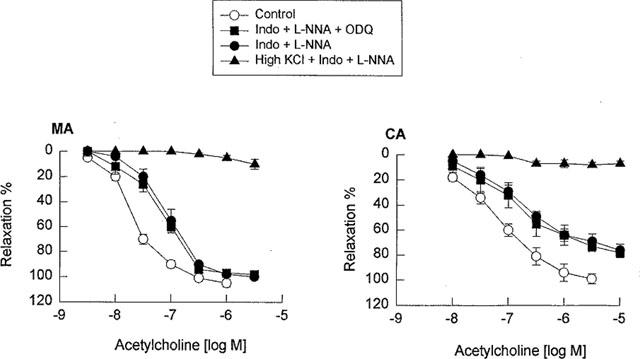
Concentration-response curves for acetylcholine in the guinea-pig mesenteric artery (MA) and middle cerebral artery (CA) in the absence and the presence of a combination of indomethacin (10 μM) and L-NNA (100 μM) or indomethacin and L-NNA plus ODQ (10 μM) or high K+ PPS (40 mM). Data are shown as the mean±s.e.mean from 5–7 separate experiments.
Table 1.
Effects of different treatments on EDHF-mediated vasorelaxation of the guinea-pig mesenteric arteries
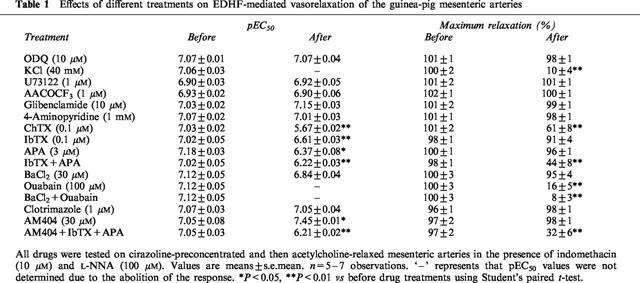
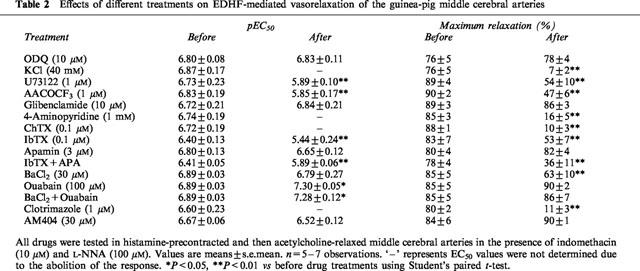
Table 2.
Effects of different treatments on EDHF-mediated vasorelaxation of the guinea-pig middle cerebral arteries
Role of K+ channels in EDHF-mediated vasorelaxation
To systematically compare the contribution of K+ channels to EDHF-mediated relaxation of the mesenteric and cerebral arteries, all vessels in this series of experiments were pretreated with indomethacin (10 μM) plus L-NNA (100 μM), and subsequently a variety of K+ channel blockers were evaluated. Glibenclamide (10 μM) did not affect the vasorelaxation response to acetylcholine in either the mesenteric or cerebral artery. 4-AP (1 mM) did not significantly inhibit the vasorelaxation response to acetylcholine in the mesenteric artery, but almost abolished the vasorelaxation response to acetylcholine in the cerebral artery. ChTX (0.1 μM) significantly inhibited the vasorelaxation response to acetylcholine in the mesenteric artery, but abolished the vasorelaxation response to acetylcholine in the cerebral arteries (Figure 2, Tables 1 and 2). Since ChTX may also inhibit K+ channels other than BKCa (Chandy & Gutman, 1995), the effects of IbTX, which is a more selective blocker of BKCa channels, and also apamin, a selective blocker of SKCa channels and the combination of these two were assessed. Pretreatment of the mesenteric artery with IbTX (0.1 μM), apamin (3 μM), or the combination of both compounds did not alter the basal tone of the mesenteric or cerebral artery, but significantly inhibited the vasorelaxation response to acetylcholine, with both the pEC50 values and maximal relaxations being decreased (Figure 2b and Table 1). In the cerebral artery, apamin alone had no significant effect, IbTX significantly decreased both the pEC50 values and the maximal relaxations in response to acetylcholine, and the combination of IbTX in addition to apamin produced no additional inhibitory action compared with IbTX alone (Figure 2b and Table 2). 30 μM Ba2+, which at this concentration has been reported to be selective for Kir, (Standen & Stanfield, 1978; Quayle et al., 1993), reduced the vasorelaxation response to acetylcholine in the cerebral artery, but not in the mesenteric artery (Figure 3 Tables 1 and 2).
Figure 2.
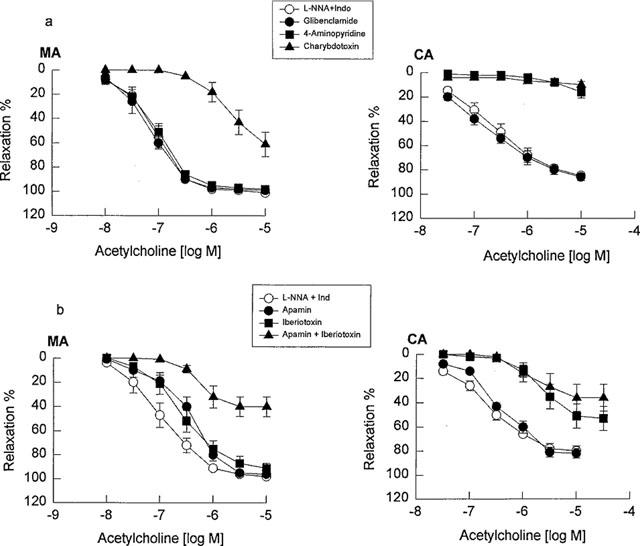
Concentration-response curves for acetylcholine in the guinea-pig mesenteric artery (MA) and middle cerebral artery (CA) in the presence of a combination of L-NNA (100 μM) plus indomethacin (10 μM) as the control compared with the vessels pretreated with L-NNA plus indomethacin and K+ channel blockers. (a) Glibenclamide (10 μM), 4-aminopyridine (1 mM) or charybdotoxin (0.1 μM); (b) apamin (3 μM), iberiotoxin (0.1 μM) or a combination of apamin and iberiotoxin. Data are shown as the mean±s.e.mean from 5–7 separate experiments.
Figure 3.
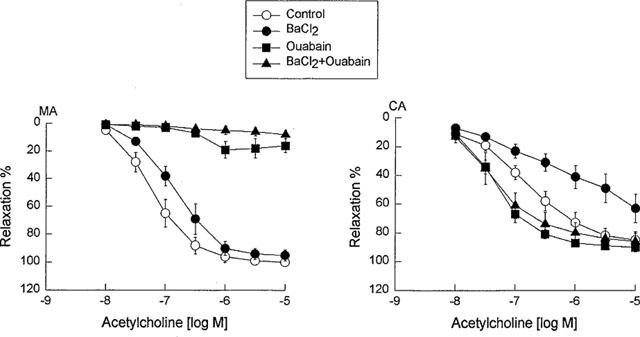
Concentration-response curves for acetylcholine in the guinea-pig mesenteric artery (MA) and middle cerebral artery (CA) in the presence of a combination of NG-nitro-L-arginine (100 μ) plus indomethacin (10 μM) as a control and vessels pretreated with BaCl2 (30 μM), ouabain (100 μM) or their combination. Data are shown as the mean±s.e.mean from 8–10 separate experiments.
Role of cytochrome P450 products in EDHF-mediated relaxation
To investigate the possibility that EDHF released from the mesenteric and cerebral arteries is a cytochrome P450 metabolite, all vessels in this series of experiments were pretreated with indomethacin (10 μM) plus L-NNA (100 μM), and these inhibitors of cytochrome P450 were evaluated. Pretreated with clotrimazole (1 μM), an imidazole that binds to the heme moiety of the enzyme (Matsuura et al., 1991), did not significantly inhibit the vasorelaxation response to acetylcholine in the mesenteric artery, but abolished the vasorelaxation response to acetylcholine in the cerebral artery (Figure 4, Tables 1 and 2). Similarly, 7-ethoxyresorufin (3 μM), a competitive substrate of cytochrome P450 enzymes (Tassaneeyakul et al., 1993), did not affect the vasorelaxation response to acetylcholine in the mesenteric artery, but significantly inhibited the vasorelaxation response to acetylcholine in the cerebral artery. The maximal relaxation attributed to EDHF in the mesenteric artery was not reduced in the presence of 3 μM 7-ethoxyresorufin (101±2% vs 99±3%, n=3, P>0.05), whereas pretreatment with 7-ethoxyresorufin reduced the maximal relaxation of the cerebral artery (80±4% vs 48±5%, n=3, P<0.01). Consistently, pretreatment with U73122 (1 μM), a PLC inhibitor and AACOCF3 (1 μM), a PLA2 inhibitor, significantly inhibited EDHF-mediated vasorelaxation in the cerebral artery without effects in the mesenteric artery (Figure 5, Tables 1 and 2). To address the potential non-specific effects of clotrimazole and AACOCF3 on smooth muscle cell calcium and KCa channels, their actions were investigated on histamine-induced contraction and NS1619-induced relaxation of endothelium-denuded cerebral artery. Both clotrimazole and AACOCF3 at the concentrations applied in the present study (n=4, data not shown) neither affected histamine (1 μM)-induced contraction nor inhibited the relaxation induced by NS1619 (30 μM), a putative KCa channel-opener (Olesen et al., 1994).
Figure 4.
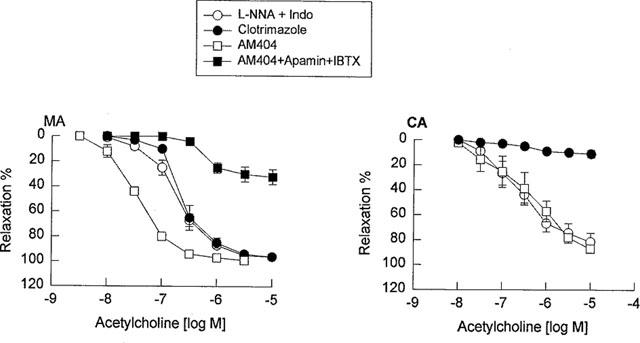
Concentration-response curves for acetylcholine in the guinea-pig mesenteric artery (MA) and middle cerebral artery (CA) in the presence of a combination of L-NNA (100 μM) plus indomethacin (10 μM) as the control and vessels pretreated with: clotrimazole (1 μM), AM404 (30 μM) or combination of AM404, apamin (3 μM) and iberiotoxin (0.1 μM). Data shown as the mean±s.e.mean from 5–6 separate experiments.
Figure 5.
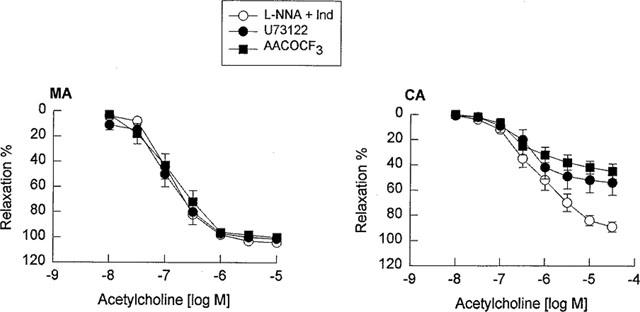
Concentration-response curves for acetylcholine in the guinea-pig mesenteric artery (MA) and middle cerebral artery (CA) in the presence of a combination of L-NNA (100 μM) plus indomethacin (10 μM) as the control and vessels pretreated with U73122 (1 μM) or AACOCF3 (1 μM). Data are shown as the mean±s.e.mean from six separate experiments.
Other candidates for EDHF in the arteries
To investigate the chemical nature of EDHF, all vessels in this series of experiments were pretreated with indomethacin (10 μM) plus L-NNA (100 μM) and then a variety of inhibitors were evaluated. Co-treatment with AM404 (30 μM), an anandamide transport inhibitor (Beltramo et al., 1997), had no effect on the vasorelaxation response to acetylcholine in the cerebral artery, but potentiated the vasorelaxation response to acetylcholine in the mesenteric artery. A combination of IbTX (0.1 μM) and apamin (3 μM) reversed the AM404-induced enhancement of acetylcholine-mediated relaxation in the mesenteric artery, and the response was similar to that observed in IbTX and apamin alone (Figure 4). AM404 (30 μM), in a manner similar to ODQ (10 μM), also significantly inhibited the relaxation induced by NO at 0.1 to 1 μM in the mesenteric artery (Figure 6), and furthermore, the enhancement effect of AM404 on the acetylcholine-induced relaxation was not inhibited by the cannabinoid CB1 receptor antagonist SR 141716A (30 μM, n=6, data not shown).
Figure 6.
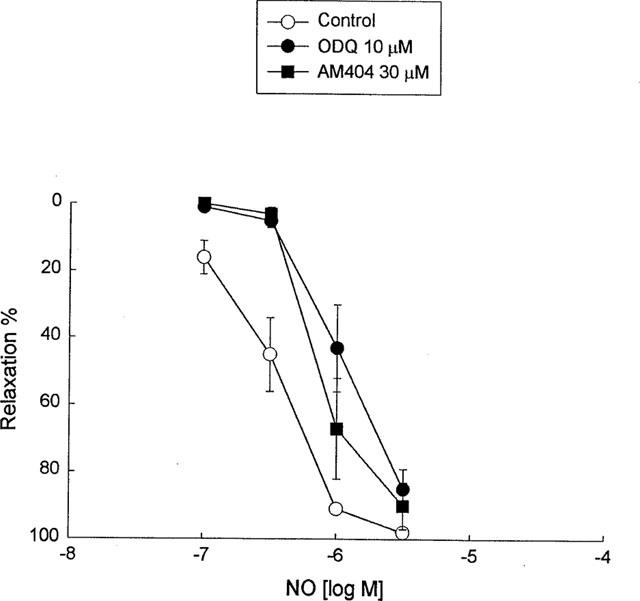
The inhibitory effects of AM404 (30 μM) and ODQ (10 μM) on NO-mediated relaxation of endothelial-denuded guinea-pig mesenteric artery. Data shown are as the mean±s.e.mean from six separate experiments.
To investigate the contribution of an increase in extracellular K+ as a potential mediator of EDHF, we investigated the effects of Ba2+ (30 μM), ouabain (100 μM), either alone or in combination, on the L-NNA and indomethacin resistant component of acetylcholine-mediated relaxation in the cerebral artery and the mesenteric artery. Either pretreatment with ouabain alone for 30 min, or in combination with Ba2+ for the same duration, almost abolished EDHF-mediated relaxation in the mesenteric artery, but enhanced the relaxation in the cerebral artery (Figure 3). Moreover, addition of K+ in the concentration range of 5–20 mM did not induce vasorelaxation, but rather induced contraction of both mesenteric artery precontracted with cirazoline and cerebral artery precontracted with histamine. (n=5, data not shown).
Discussion
The present comparative study of guinea-pig cerebral vs mesenteric resistance vessels, conducted under identical experimental conditions, indicates the heterogeneous nature of EDHF-mediated vasorelaxation. These data suggest that EDHF or the cellular process that mediated EDHF, cannot be readily attributed to a single factor or pathway.
Different contribution of EDHF
In the present study, acetylcholine-induced relaxations of the mesenteric and cerebral arteries were completely abolished by removal of the endothelium, indicating that acetylcholine mediates endothelium-dependent relaxation. In the presence of the NO synthase inhibitor L-NNA plus the cyclo-oxygenase inhibitor indomethacin, the acetylcholine-induced vasorelaxation remained largely unaffected. Cohen et al. (1997) has recently reported that a combination of NG-nitro-L-arginine methyl ester (L-NAME, 30 μM) and L-NNA (100 μM) did not completely inhibit NO release and vasorelaxation in the rabbit carotid artery and, thus, in the present studies we combined ODQ (10 μM), a soluble guanylyl cyclase inhibitor with L-NNA (100 μM) and indomethacin (10 μM). As apparent from the data presented in Figure 1, ODQ did not further inhibit the L-NNA/indomethacin-insensitive relaxation response to acetylcholine in either the guinea-pig mesenteric or cerebral artery. Furthermore, LY83583 (10 μM), a non-selective soluble guanylyl cyclase inhibitor, which also inhibits NO synthase and inactivates NO (Kontos & Wei, 1993), produced no additional inhibition of the L-NNA/indomethacin-insensitive relaxation to acetylcholine in the guinea-pig cerebral artery (unpublished observations). These results suggest that the L-NNA/indomethacin-insensitive acetylcholine-induced relaxations in guinea-pig mesenteric and cerebral arteries are neither mediated by NO nor soluble guanylyl cyclase and independent of relaxant prostanoids. Our data are thus in contrast to those of Cohen et al. (1997) that were obtained in the rabbit carotid, and, perhaps, reflecting differences between conduit and resistance-sized vessels. The L-NAME/indomethacin-insensitive relaxation responses in both cerebral and mesenteric arteries were, however, abolished when arterial segments were preconstricted with high K+PSS, which causes membrane depolarization and increases the membrane conductance for K+ as the membrane potential shifts toward the new EK. High K+ blockade of the L-NAME/indomethacin-insensitive relaxation establishes that the endothelium-dependent mediators in both cerebral and mesenteric arteries have the properties expected of an EDHF and that membrane hyperpolarization is fundamental to EDHF-induced relaxation. Our results are consistent with those reported in the rat mesenteric artery (Adeagbo & Triggle, 1993; Waldron & Garland, 1994) and the guinea-pig basilar artery (Petersson et al., 1997), in which EDHF appears to be a major contributor to endothelium-dependent vasorelaxation. The results also suggest that the LNNA/indomethacin-insensitive relaxation response to acetylcholine is of greater importance in the mesenteric artery than in the cerebral artery (pEC50 values 7.04 in mesenteric artery vs 6.70 in the cerebral artery, maximal relaxation 99% in the mesenteric artery vs 83% in the cerebral artery, Figure 1).
Cellular targets for EDHF
Extensive investigation indicates that the cellular modes of action of EDHF show tissue and species variability (Garland et al., 1995; Waldron et al., 1996; Triggle et al., 1999). Four kinds of K+ channels have been reported to mediate the vascular actions of EDHF: (a) KCa channels (Popp et al., 1996; Li et al., 1997); (b) Kdr channels (Zakharenko, 1996; Petersson et al., 1997); (c) Kir channels (Edwards et al., 1998); (d) KATP channels (Liu & Flavahan, 1997). In most studies, however, the effects of EDHF are inhibited by KCa channel blockers (Popp et al., 1996; Dong et al., 1997a, 1997b; Petersson et al., 1997). Three kinds of KCa channels have been described: large conductance (BKCa), intermediate conductance (IKCa) and small conductance (SKCa; Cook & Quast, 1990). In the present study, three K+ channel blockers with different selectivity for subtypes of KCa channels were used: (1) ChTX was employed as a nonselective blocker of KCa as it is known to also inhibit other K+ channels, for example voltage-gated K+ channels well as an intermediate conductance, IKCa, K+ channels (Cook & Quast, 1990; Nelson & Quayle, 1995). (2) IbTX was used as a highly selective and potent blocker of BKCa channels (Galvez et al., 1990). (3) Apamin was utilized in our studies as a selective blocker of SKCa channels (Cook & Quast, 1990; Nelson & Quayle, 1995). Of importance was our observation that 4-AP, which has been shown to block KV, including Kdr in many tissues, abolished EDHF-mediated relaxation in the cerebral artery, but had no effect in the mesenteric artery. Finally, Ba2+, which at concentration of 30 μM is putatively a selective Kir channel blocker, inhibited EDHF-mediated relaxation in the cerebral artery, but was without effect in the mesenteric artery. In contrast, ouabain inhibited EDHF-mediated relaxation in the mesenteric artery, but potentiated the EDHF-response in the cerebral artery. The different profiles of inhibition that we have reported for ChTX, IbTX, apamin, 4-AP, Ba2+ and ouabain against the vasorelaxation response attributed to EDHF suggests that the cellular targets of EDHF differ in guinea-pig mesenteric and cerebral arteries. Taking into account the limited selectivity of some of the K+ channel blockers that we have used in this study, our data suggests that KCa (SKCa, IKCa, BKCa) channels are involved in EDHF-mediated relaxation in the mesenteric artery, and most likely, Kir, and KV, channels are involved in the cerebral artery.
Nature of EDHF
As discussed above, our data indicate that the pharmacological properties of EDHF, or the cellular events mediating the response to EDHF, differ in the cerebral and mesenteric arteries thus suggesting the heterogeneity of EDHF. Although the chemical identity of EDHF remains elusive, three candidate molecules have received recent attention: (1) A cytochrome P450 metabolite of arachidonic acid, possibly an EET (Hecker et al., 1994; Campbell et al., 1996; Popp et al., 1996); (2) Endothelial cell-derived increase in extracellular K+ (Edwards et al., 1998); (3) An endogenous cannabinoid, anandamide (Randall et al., 1996; Randall & Kendall, 1997). These hypotheses have, however, been vigorously challenged notably with respect to the specificity of the pharmacological agents applied and, in addition, the data reported also illustrates laboratory-dependent differences that may reflect variabilities in the experimental conditions. (Zygmunt et al., 1996; 1997; Fukao et al., 1997; Plane et al., 1997; Vanheel & de Voorde, 1997). In the present study, identical experimental conditions were applied to our comparison of the contribution and pharmacological properties of EDHF in the guinea pig mesenteric and cerebral arteries.
Clotrimazole, at a concentration which did not affect the contraction evoked by cirazoline or histamine, or the relaxation response to the KCa channel opener NS1619, abolished the EDHF-mediated relaxation in the cerebral artery. 7-ethoxyresorufin also significantly inhibited the vasorelaxation response to acetylcholine in the cerebral artery. Consistently, pretreatment with U73122, a PLC inhibitor, and AACOCF3, a PLA2 inhibitor, significantly inhibited EDHF-mediated vasorelaxation in the cerebral artery. Taken together these findings suggest that a cytochrome P450-reaction may be involved in the production or vascular action of EDHF in the cerebral artery, however, with the experimental protocol employed we cannot conclude as to whether the effects of cytochrome P450 inhibition are at the level of the endothelial or the vascular smooth muscle cell or both. In contrast, EDHF-mediated relaxation in the mesenteric artery was not affected by inhibiting either cytochrome P450 enzymes, PLC or PLA2, but was significantly enhanced by AM404, which would be expected to potentiate the activity of anandamide by blocking the cellular re-uptake this endogenous cannabinoid (Beltramo et al., 1997). The effects of AM404 were attenuated by a combination of IbTX and apamin, however, the specificity of AM404 is questionable since it also inhibited NO-mediated vasorelaxation of the mesenteric artery and the AM404-induced enhancement of the acetylcholine-induced relaxation was not inhibited by SR 141716A, an agent that targets anandamide at its receptor. Thus, our data suggest that an endogenous cannabinoid is unlikely to be involved in the EDHF-mediated relaxation in the mesenteric artery. Of interest is the report from Mombouli et al. (1999) that annandamide increased cytosolic Ca2+ in endothelial cells and thus may enhance the release of endothelial cell-derived vasorelaxant factors.
Edwards et al. (1998) have recently suggested that EDHF is endothelial cell-derived K+, which, in the 5–20 mM concentration range hyperpolarizes and relaxes adjacent rat artery smooth muscle cells via the activation of Ba2+-sensitive Kir channels and Na+, K+-ATPase. We also tested this hypothesis in guinea-pig arteries. Exposure to ouabain for 30 min, alone or in combination with Ba2+ almost abolished EDHF-mediated relaxation in mesenteric artery, but K+, applied at low concentrations, rather than inducing relaxation further contracted the mesenteric and cerebral arteries. Furthermore, in the cerebral artery ouabain potentiated the EDHF-mediated relaxation. These data suggest that K+ is not an EDHF in guinea-pig arteries although Ba2+-sensitive K+ channels may be involved in the EDHF-mediated relaxation of cerebral artery and Na+, K+-ATPase may be involved in the EDHF-mediated relaxation of mesenteric artery. Our results are consistent with those reported by Quignard et al. (1999), in which they found that K+ and EDHF are unlikely the same entity in guinea-pig carotid and porcine coronary arteries. Furthermore, a report by Ding et al. (2000) has also indicated that K+ does not fulfil the expected criteria for an EDHF in mouse saphenous arteries.
The role of myoendothelial cell gap junctions in contributing to endothelium-dependent relaxation in guinea-pig mesenteric and middle cerebral arteries was not assessed in the present study. This is an important area for future investigation. Chaytor et al. (1998) have reported that the Gap 27 junction peptide, when applied at a high concentration of 300 μM, attenuated the L-NAME-insensitive dependent relaxation to ACh in rabbit mesenteric artery and aorta. The specificity of action of the Gap 27 peptide was supported by the failure of Gap 27 to affect relaxation to the endothelium-independent vasodilator, sodium nitroprusside. Nonetheless, the specificity of action of Gap 27 has been questioned by others (van Breemen and Laher, personal communication) and thus further studies are warranted. Other studies have reported that the lipophilic aglycone, glycyrrhetinic acid, can also reversibly prevent gap junction communication. This has been demonstrated in rabbit iliac artery (Taylor et al., 1998) and also guinea-pig submucosal arterioles (Yamamoto et al., 1998, 1999). The studies are of particular interest to the current study since they provide data from arterioles downstream from those that we have used (200–250 μm). It remains to be determined whether myoendothelial gap junctions are also involved in contributing to the non-NO/PGI2 endothelium-dependent relaxation that we have described in guinea-pig mesenteric arterioles.
In conclusion, our data suggest that EDHF is not a single substance and that the nature of EDHF shows considerable tissue variability. In the guinea-pig cerebral artery, a cytochrome P450 product may be involved in the production and/or action of EDHF and EDHF-mediated vasorelaxation involves the activation of IKCa, BKCa, KV, and Kir channels. In contrast, in the mesenteric artery, our data indicates the lack of a role for a cytochrome P450 product, however, EDHF mediates vasorelaxation via the activation of KCa (SKCa, IKCa, BKCa) channels. Our data demonstrating that pretreatment with the cannabinoid transport inhibitor AM404 potentiates EDHF-mediated relaxation in the mesenteric artery indicates that further studies concerning the role for an endogenous cannabinoid in the regulation of EDHF production/release/action are required. Our study does not exclude possible role for gap junction proteins in mediating, or contributing to endothelium-dependent hyperpolarization in the vessels studied. Finally, we can provide no convincing data in support of the hypothesis that an increase in extracellular K+ is an EDHF in guinea-pig mesenteric or cerebral artery.
Acknowledgments
This work was supported by an Alberta Heritage Foundation for Medical Research award (to H. Dong), a HSFA operating grant to W.C. Cole, and an MRC operating grant support to C.R. Triggle.
Abbreviations
- 4-AP
4-aminopyridine
- BKCa
large conductance Ca2+-activated K+ channels
- ChTX
charybdotoxin
- EDHF
endothelium-derived hyperpolarizing factor
- EET
epoxyeicosatrienoic acid
- IbTX
iberiotoxin
- IKCa
intermediate conductance K+ channels
- KATP
ATP-sensitive K+ channels
- Kdr
voltage-gated delayed rectifier K+ channels
- Kir
inwardly rectifying K+ channels
- KV
voltage-gated K+ channels
- L-NNA
NG-nitro-L-arginine
- ODQ
1H[1,2,4]oxadiozolo[4,3,-a]quinoxalin-1-one
- PLA2
phospholipase A2
- PLC
phospholipase C
- SKCa
small conductance Ca2+-activated K+ channels
References
- ADEAGBO A.S.O., TRIGGLE C.R. Varying extracellular [K+]: A functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- BELTRAMO M., STELLA N., CALIGNANO A., LIN S.Y., MAKRIYANNIS A., PIOMELLI D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1096. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., GEBREMEDHIN D., PRATT P.F., HARDER D.R. Identification of epoxyeicosatrienoic acid as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHANDY K.G., GUTMAN G.A. Voltage-gated potassium channel genes Ligand- and Voltage-gated Ion Channels 1995Boca Raton: CRC Press; 2–71.ed. North, R.A. pp [Google Scholar]
- CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN R.A., PLANE F., NAJIBI S., HUK I., MALINSKI T., GARLAND C.J. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc. Natl. Acad. Sci. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOK N.S., QUAST U. Potassium channel pharmacology Potassium channels: structure, classification, function and therapeutic potential 1990New York: Halstead Press; 181–255.ed. Cook, N.S. pp [Google Scholar]
- DING H., KUBES P., TRIGGLE C.R. Potassium- and acetylcholine-induced vasorelaxation in mice lacking endothelial nitric synthase. Br. J. Pharmacol. 2000;129:1194–2000. doi: 10.1038/sj.bjp.0703144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG H., COLE W.C., TRIGGLE C.R. Differences of endothelium-dependent vasorelaxation mediated by EDHF in small resistance cerebral versus peripheral vessels of guinea pig. Biophys. J. 1997b;72:A383. [Google Scholar]
- DONG H., WALDRON G.J., COLE W.C., TRIGGLE C.R. Roles of calcium-activated and voltage-gated delayed rectifier potassium channels in endothelium-dependent vasorelaxation of the rabbit middle cerebral artery. Br. J. Pharmacol. 1998;123:821–832. doi: 10.1038/sj.bjp.0701680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG H., WALDRON G.J., GALIPEAU D., COLE W.C., TRIGGLE C.R. NO/PGI2-independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. Br. J. Pharmacol. 1997a;120:695–701. doi: 10.1038/sj.bjp.0700945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FUKAO M., HATTORI Y., KANNO M., SAKUMA, KITABATAKE A. Evidence against a role of cytochrome P450-derived arachidonic acid metabolites in endothelium-dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery. Br. J. Pharmacol. 1997;120:439–446. doi: 10.1038/sj.bjp.0700932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALVEZ A., GIMINEZ-GALLEGO G., REUBEN J.P., ROY-CONTANCIN L., FEIGENBAUM P., KACZOROWSKI G.J., GARCIA M.L. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J. Biol. Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: A role in the control of vascular tone. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- GRYGLEWSKI R.J., MONCADA S., PALMER R.M.J. Bioassay of prostacyclin and endothelium-derived relaxing factor (EDRF) from porcine aortic endothelial cells. Br. J. Pharmacol. 1986;87:685–694. doi: 10.1111/j.1476-5381.1986.tb14586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUIMARÁES S., MOURA D. Advances in vascular neuroeffector mechanisms. Trends Pharmacol. Sci. 1999;20:90–93. doi: 10.1016/s0165-6147(99)01299-7. [DOI] [PubMed] [Google Scholar]
- HECKER M., BARA A.T., BAUERSACHS J., BUSSE R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONTOS H.A., WEI E.P. Hydroxyl radical-dependent inactivation of guanylate cyclase in cerebral arteries by methylene blue and by LY83583. Stroke. 1993;24:427–434. doi: 10.1161/01.str.24.3.427. [DOI] [PubMed] [Google Scholar]
- LI P.L., ZAO A.P., CAMPBELL W.B. Regulation of potassium channels in coronary arterial smooth muscle by endothelium-derived vasodilators. Hypertension. 1997;29:262–267. doi: 10.1161/01.hyp.29.1.262. [DOI] [PubMed] [Google Scholar]
- LISCHKE V., BUSSE R., HECKER M. Selective inhibition by barbiturates of the synthesis of endothelium-derived hyperpolarizing factor in the rabbit carotid artery. Br. J. Pharmacol. 1995;115:969–974. doi: 10.1111/j.1476-5381.1995.tb15905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Q., FLAVAHAN N.A. Hypoxic dilation of porcine small coronary arteries: role of endothelium and KATP-channels. Br. J. Pharmacol. 1997;120:728–734. doi: 10.1038/sj.bjp.0700939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUURA Y., KOTANI E., IIO T., FUKUDA T., TOBINAGA S., YOSHIDA T., KUROIWA Y. Structure-activity relationships in the induction of hepatic mircosomal cytochrome P450 by clotrimazole and its structurally related compounds in rats. Biochem. Pharmacol. 1991;41:1949–1956. doi: 10.1016/0006-2952(91)90135-r. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.-V., SCHAEFFER G., HOLZMANN S., KOSTNER G.M., GRAIER W.F. Anandamide-induced mobilization of cytosolic Ca2+ in endothelial cells. Br. J. Pharmacol. 1999;126:1593–1600. doi: 10.1038/sj.bjp.0702483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGAO T., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor and endothelium-dependent relaxations. Am. J. Respir. Cell. Mol. Biol. 1993;8:1–6. doi: 10.1165/ajrcmb/8.1.1. [DOI] [PubMed] [Google Scholar]
- NELSON M.T., QUAYLE J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- OLESEN S.P., MUNCH E., MOLDT P., DREJER J. Selective activation of Ca2+-dependent K+ channels by novel benzimidazolone. Eur. J. Pharmacol. 1994;251:53–59. doi: 10.1016/0014-2999(94)90442-1. [DOI] [PubMed] [Google Scholar]
- PALMER R.M.J., FERRIGE A.G., MONCADA S. Nitric oxide release accounts for biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- PETERSSON J., ZYGMUNT P.D., HÖGESTÄTT E.D. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLANE F., HOLLAND M., WALDRON G.J., GARLAND C.J., BOYLE J.P. Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. Br. J. Pharmacol. 1997;121:1509–1511. doi: 10.1038/sj.bjp.0701361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I., BUSSE R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAYLE J.M., MCCARRON J.G., BRAYDEN J.E., NELSON M.T. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am. J. Physiol. 1993;265:C1363–C1370. doi: 10.1152/ajpcell.1993.265.5.C1363. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J.F., FÉLÉTOU M., THOLLON C., VILAINE J.-P., DUHAULT J., VANHOUTTE M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANDALL M.D., ALEXANDER S.P.H., BENNETT T., BOYD E.A., FRY J.R., GARDINER S.M., KEMP P.A., MCCULLOCH A.I., KENDALL D.A. An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem. Biophys. Res. Commun. 1996;229:114–120. doi: 10.1006/bbrc.1996.1766. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., KENDALL D.A. Involvement of a cannabinoid in endothelium-derived hyperpolarizing factor-mediated coronary vasorelaxation. Eur. J. Pharmacol. 1997;335:205–209. doi: 10.1016/s0014-2999(97)01237-5. [DOI] [PubMed] [Google Scholar]
- STANDEN N.B., STANFIELD P.R. A potential- and time-dependent blockade of inward rectification in frog skeletal muscle fibres by barium and strontium ions. J. Physiol. 1978;280:169–191. doi: 10.1113/jphysiol.1978.sp012379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TASSANEEYAKUL W., BIRKETT D.J., VERONESE M.E., MCMANUS M.E., TUKEY R.H., QUATTROCHI L.C., GELBOIN H.V., MINERS J.O. Specificity of substrate and inhibitor probes for human cytochromes P450 1A1 and 1A2. J. Pharmacol. Exp. Ther. 1993;265:401–407. [PubMed] [Google Scholar]
- TAYLOR H.J., CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Inhibition of the gap junctional component of endothelium-dependent relaxations in rabbit iliac artery by 18-alpha glycyrrhetinic acid. Br. J. Pharmacol. 1998;125:1–3. doi: 10.1038/sj.bjp.0702078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAYLOR S.G., WESTON A.H. Endothelium-derived hyperpolarizing factor: a new endogenous inhibitor from the vascular endothelium. Trends Pharmacol. Sci. 1988;9:272–274. doi: 10.1016/0165-6147(88)90003-x. [DOI] [PubMed] [Google Scholar]
- TRIGGLE C.R., DONG H., WALDRON G.J., COLE W.C. Endothelium-derived hyperpolarizing factor(s): Species and tissue heterogeneity. Clin. Exp. Pharmacol. Physiol. 1999;26:176–179. doi: 10.1046/j.1440-1681.1999.03007.x. [DOI] [PubMed] [Google Scholar]
- VANHEEL B., DE VOORDE J.V. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat main mesenteric artery. J. Physiol. 1997;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALDRON G.J., DONG H., COLE W.C., TRIGGLE C.R. Endothelium-dependent hyperpolarization of vascular smooth muscle: role for a non-nitric oxide synthase product. Acta Pharmacol. Sin. 1996;17:3–7. [PubMed] [Google Scholar]
- WALDRON G.J., GARLAND C.J. Contribution of both nitric oxide and a change in membrane potential to acetylcholine-induced relaxation in the rat small mesenteric artery. Br. J. Pharmacol. 1994;112:831–836. doi: 10.1111/j.1476-5381.1994.tb13154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO Y., FUKUTA H., NAKAHIRA Y., SUZUKI H. Blockade by 18beta-glycyrrhetinic acid of intercellular electrical coupling in guinea-pig arterioles. J. Physiol. 1998;511:501–508. doi: 10.1111/j.1469-7793.1998.501bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO Y., IMAEDA K., SUZUKI H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAKHARENKO S.S. A role of delayed-rectifier potassium currents in EDHF-dependent hyperpolarization and relaxation of rat mesenteric small arteries Hypertension 199628709(abstract) [Google Scholar]
- ZYGMUNT P.M., EDWARDS G., WESTON A.H., DAVIS S.C., HÖGESTÄTT E.D. Effects of cytochrome P450 inhibitors on EDHF-mediated relaxation in the rat hepatic artery. Br. J. Pharmacol. 1996;118:1147–1152. doi: 10.1111/j.1476-5381.1996.tb15517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGESTÄTT E.D., WALDECK K., EDWARDS G., KIRKUP A.J., WESTON A.H. Studies on the effects of anandamide in rat hepatic artery. Br. J. Pharmacol. 1997;122:1679–1686. doi: 10.1038/sj.bjp.0701601. [DOI] [PMC free article] [PubMed] [Google Scholar]