

Abstract
We evaluated the role of SH-groups in improvement of endothelial dysfunction with ACE-inhibitors in experimental heart failure. To this end, we compared the vasoprotective effect of chronic treatment with zofenopril (plus SH-group) versus lisinopril (no SH-group), or N-acetylcysteine (only SH-group) in myocardial infarcted (MI) heart failure rats.
After 11 weeks of treatment, aortas were obtained and studied as ring preparations for endothelium-dependent and -independent dilatation in continuous presence of indomethacin to avoid interference of vasoactive prostanoids, and the selective presence of the NOS-inhibitor L-NMMA to determine NO-contribution.
Total dilatation after receptor-dependent stimulation with acetylcholine (ACh) was attenuated (−49%, P<0.05) in untreated MI (n=11), compared to control rats with no-MI (n=8). This was in part due to impaired NO-contribution in MI (−50%, P<0.05 versus no-MI). At the same time the capacity for generation of biologically active NO after receptor-independent stimulation with A23187 remained intact.
Chronic treatment with n-acetylcysteine (n=8) selectively restored NO-contribution in total dilatation to ACh. In contrast, both ACE-inhibitors fully normalized total dilatation to ACh, including the part mediated by NO (no significant differences between zofenopril (n=10) and lisinopril (n=8)).
Zofenopril, but not lisinopril, additionally potentiated the effect of endogenous NO after A23187-induced release from the endothelium (+100%) as well as that of exogenous NO provided by nitroglycerin (+22%) and sodium nitrite (+36%) (for all P<0.05 versus no-MI).
We conclude that ACE-inhibition with a SH-group has a potential advantage in improvement of endothelial dysfunction through increased activity of NO after release from the endothelium into the vessel wall. Furthermore, this is the first study demonstrating the selective normalizing effect of N-actylcysteine on NO-contribution to ACh-induced dilatation in experimental heart failure.
Keywords: Myocardial-infarction, heart failure, N-acetylcysteine, ACE-inhibitor, SH-group, aorta, ACh, A23187, L-NMMA, NO
Introduction
Patients with chronic heart failure (CHF) display an attenuated response to the endothelium dependent vasodilator acetylcholine (ACh) in a setting of preserved responsiveness to the endothelium independent vasodilator nitroprusside (Kubo et al., 1991). Such manifestation of endothelial dysfunction has been implied to underlie the characteristically increased peripheral vascular resistance in CHF, thereby contributing to its progression (Drexler et al., 1993). Consequently, prevention or reversal of endothelial dysfunction is considered an important target for pharmacological intervention in CHF (Drexler et al., 1993). Angiotensin-converting enzyme inhibitors have been reported to favourably affect endothelial dysfunction both in clinical (Jeserich et al., 1995) and experimental CHF, including in the rat coronary occlusion-myocardial infarction (MI) model of heart failure (Thuillez et al., 1995).
Previous studies using this rat model of heart failure have reported progressive deterioration of endothelial function after MI, first demonstrable in the aorta at approximately 8 weeks (Ontkean et al., 1991; Teerlink et al., 1993; Thuillez et al., 1995; Buikema et al., 1997). These former studies also suggested little or no role for vasoactive prostanoids in neither the normal nor the abnormal aortic response to ACh in control and heart failure rats, respectively (Ontkean et al., 1991; Teerlink et al., 1993). More recent studies investigating endothelial dysfunction in this model suggest that attenuated dilatation to ACh in CHF may rather involve the impaired contribution of NO (Malmsjö et al., 1999). Recent findings also indicate that heart failure is associated with increased oxidative stress as well as an anti-oxidant deficit (Katz, 1997; Givertz & Colcucci, 1998; Keith et al., 1998), including in the rat MI-model (Hill & Singal, 1997). Both processes may contribute to increased NO-breakdown to underlie endothelial dysfunction in CHF. Reduction of oxidative stress has been implicated to explain the favourable effects of ACE-inhibition in other models of endothelial dysfunction (Rajagopalan et al., 1996; Fukui et al., 1997). Such a view also provides a rational for ACE-inhibitor therapy aimed at reduction of oxidative stress to improve endothelial dysfunction in CHF, thereby amply explaining previous observations. It additionally provides a rational for therapy with anti-oxidants and/or radical scavengers. It would thus appear that ACE-inhibitors containing a sulfhydryl (SH-) group with radical scavenging properties may have an additional advantage (van Gilst et al., 1991; Goldschmidt & Tallarida, 1991).
Both non-SH ACE-inhibitors and ACE-inhibitors containing SH-groups have been reported to exert beneficial effects on endothelial dysfunction in experimental CHF in separate studies (Ontkean et al., 1992; Thuillez et al., 1995; Buikema et al., 1997). So far, however, a direct comparison between ACE-inhibitors of both classes on endothelial dysfunction in CHF has been lacking. In the present study, MI-induced heart failure rats were studied for improvement of aortic endothelial dysfunction after chronic ACE-inhibition. The role of SH-groups in ACE-inhibition was evaluated by comparison of zofenopril (an ACE-inhibitor containing SH-groups) and lisinopril. To test for the role of SH-groups without ACE-inhibition we used n-acetylcysteine.
Methods
Coronary ligation-myocardial infarction
All procedures were reviewed and approved by the Animal Research Committee at the University of Groningen. Normotensive male Wistar rats (250–300 g) were obtained from Harlan (Zeist, The Netherlands) and housed at the Central Animal Laboratory, University of Groningen (The Netherlands). Rats were housed group-wise and had free access to food and water. At the time of operation anaesthesia was induced with isoflurane (2.0–2.5% in oxygen), after which rats were intubated and mechanically ventilated with this gas-mixture (Amsterdam Infant Ventilator, Hoek/Loos, Schiedam, The Netherlands). MI (n=87) was induced by direct coronary ligation (Pfeffer et al., 1979), as described previously (Pinto et al., 1993). Briefly, a left-sided thoracotomy was performed and the anterior descending coronary artery occluded with a 6–0 silk suture 1–2 mm after the bifurcation. Care was given to obtain a blanching of the myocardium distal to the ligature to confirm MI; if necessary the procedure was repeated by placement of a second or third ligature.
Stratification
One week after surgical procedures, survival amongst the MI-rats was 67% (58/87); the majority of the rats that died did so within 24 h after the operation. Surviving rats were anaesthetized (isoflurane 2.0% in oxygen) and subjected to ECG-measurements, based on which individual rats with ECG-evidence (averaged QT-interval prolongation in three precordial leads) for MI (n=50) were assigned to one of three active treatment regimens (n=12 each) or a no treatment (n=14) regimen. MI-operated rats with minor or no ECG-evidence for MI were additionally allocated to the no treatment regimen (i.e. no-MI, n=8) to function as a control group, thus generating the five experimental groups in total. As described previously (de Vries et al., 1997), ECG-stratification was used for a balanced distribution of MI-rats with different MI-size over the experimental groups only, and in all cases group stratification was checked at postmortem by quantitative left ventricular histopathology.
Treatment
Rats allocated to one of the three active treatment regimens received (mixed with the chow, Hope Farms, Woerden, The Netherlands) either zofenopril (ZOF, n=12), lisinopril (LIS, n=12) or N-acetylcysteine (NAC, n=12); the remaining rats (i.e. n=8 for no-MI, n=14 for MI) served as non treated controls. Based on drug analysis of the food (Dr S. Villa, Menarini Ricerche, Pomezia (Rome), Italy) together with the assessment of food-intake and body weight (BW), the average daily dose of ZOF, LIS and NAC was approximately 6 mg kg−1, 2 mg kg−1 and 40 mg kg−1, respectively. The dose of ZOF was chosen based on previous experiments in our laboratory (Pinto et al., 1991; van Wijngaarden et al., 1991). The dose of LIS was chosen relative to ZOF based on the clinical defined daily dose for LIS (10 mg) and ZOF (30 mg) (ATC INDEX, 1998). The dose of NAC was comparable to that used by Galen & Siebeneich (1998) and who reported its favourable effects on diabetes-induced endothelial dysfunction in rats. To avoid a potential effect of early pharmacological intervention on infarct-size and wound healing (van Gilst et al., 1994; Buikema et al., 1997), treatment was started at 2 weeks after induction of MI (i.e. established MI). Tail cuff systolic blood pressure was recorded immediately before surgical procedures and ECG-measurements at 1 week, and was repeated after 10 weeks of active treatment. Treatment lasted for 11–12 weeks and animals were studied at 13–14 weeks after initial surgical procedures. Three rats, all untreated MI, additionally died during the active treatment period. All groups were studied concurrently and at the same age.
Sacrifice
At sacrifice, heparin (5000 IU kg−1) was administered to anaesthetized rats (isoflurane in 2% oxygen) via the tail vein, before arterial blood was sampled from the abdominal aorta for assessment of plasma NOx concentrations. Plasma nitrite (NO2−) and nitrate (NO3−) concentrations (together referred to as NOx) were used as a measure for the production of NO radical. The assay used is based on the determination of nitrite, but not nitrate, in the Gries reaction, as previously described by Moshage et al. (1995).
The heart was rapidly excised and arrested in icy-cold 0.9% NaCl, and transferred to an organ perfusion set-up in which it was mounted. Retrograde perfusion of the aorta (at 38°C), essentially by the Langendorff method, was achieved immediately. The hearts started to beat spontaneously and after equilibration for 10 min baseline measurements were performed on left ventricular (LV-) pressure, systolic and diastolic dP dT−1, heart rate, and coronary flow, as described in detail elsewhere (van Gilst et al., 1991; Pinto et al., 1993). Thereafter, hearts were arrested in diastole in 2 mol l−1 KCl and weighed, before the atria and great vessels were dissected from the ventricles. The right ventricular free wall (RV) was separated from the left ventricle (LV), before ventricular weights were obtained. A transversal slice through the midst of the LV containing the infarcted area was fixed in Bouin's fluid, embedded in paraffin and 5 μm sections were cut for histological analysis. Infarct size was determined by planimetry and was expressed as the percentage of scare to total LV circumference, as described in detail elsewhere (van Gilst et al., 1994). Rats that were assigned to MI-groups but which appeared to have an infarct-size <20% (2/12 for ZOF, 4/12 for LIS and 4/12 for NAC) were excluded from analysis.
Organ baths studies with isolated aortic rings
Immediately after removal of the heart, the thoracic descending aorta was excised and placed in a Krebs bicarbonate solution of the following composition (mmol l−1): NaCl, 120.4; KCl, 5.9; CaCl2, 2.5; MgCl2, 1.2; NaH2PO4, 1.2; glucose, 11.5; NaHCO3, 25.0; continuously aerated with 95% O2 and 5% CO2. The vessel was cleaned of adhering fat tissue and rings of 2 mm in length were cut with a sharp razor blade, with care not to touch the luminal surface. Rings were mounted between two stirrups in organ baths filled 10 ml of Krebs solution containing 10 μmol l−1 indomethacin to avoid potential production and interference of vasoactive prostanoids. One stirrup was anchored inside the organ bath and the other connected to a displacement transducer to determine isotonic changes, as described previously (Buikema et al., 1996). Rings were subjected to 14 mN and allowed to stabilize for 60 min before they were primed and checked for viability by evoking a contraction with 1 μmol l−1 phenylephrine (PhE).
After washout and renewed stabilization, rings were pre-contracted with 1 μmol l−1 PhE for subsequent determination of the dilatory response to the endothelium-dependent vasodilators acetylcholine (ACh: 10 nmol l−1 to 10 μmol l−1) and calcium ionophore A23187 (1 nmol l−1 to 1 μmol l−1) – both in absence and presence of 100 μmol l−1 NG-mono-methyl-L-arginine (L-NMMA)) – and to the endothelium-independent vasodilators nitroglycerin (NTG: 1 nmol l−1 to 10 μmol l−1) and sodium nitrite (SN: 10 μmol l−1 to 10 mmol l−1) – in absence of L-NMMA only. Drugs were given in cumulative fashion and all concentration ranges represent the final bath concentrations.
Drugs
Krebs buffer and drug solutions were freshly prepared daily. Except nitroglycerin (University Hospital Pharmacy, Groningen, The Netherlands) and sodium nitrite (E. Merck, Darmstadt, Germany), all compounds were purchased from Sigma (St. Louis, MO, U.S.A.).
Calculations and statistical analysis
Vasodilator responses to ACh, A23187, NTG and SN were expressed as a percentage of PhE-induced pre-contraction before individual concentration-response curves were generated and the Area Under each individual Curve (AUC) determined (SigmaPlot scientific graphing software package, Jandell Scientific). The AUC was used to represent the individual response-size to a given vasodilator in a given condition, and where appropriate, to calculate the individual difference in response-size between different conditions (i.e. difference in response-size in presence of indomethacin versus indomethacin plus L-NMMA for ACh and A23187, respectively). The AUC was also used to calculate and present the average response-size per group, and for subsequent analysis of differences in the response-size among experimental groups. The preference for AUC to present and analyse response-size rather than using whole concentration-response curve was based on practical reasons and reasons of clarity only.
Unless stated otherwise, all data are expressed as mean±s.e.mean. Untreated MI-rats were compared to no-MI rats using Students' unpaired t-test. The four experimental MI-groups were compared among each other using oneway-analysis of variances (ANOVA) in combination with Dunnett post-hoc analysis for multiple comparison, using untreated MI as control category (SPSS for windows Standard Version 8.0). Differences were considered significant at a level of P<0.05 (two-tailed).
Results
Characteristics of myocardial infarction-induced heart failure-rats
We have previously shown that LV pressure and dP dT−1 are significantly decreased and become progressively attenuated in rats with MI-induced heart failure (Pinto et al., 1993). Concurrent haemodynamic abnormalities in this MI-model of heart failure are related to increased cardiac weight and infarct-size (Pfeffer et al., 1979; Tsunoda et al., 1986). In the present study, untreated MI-rats – displaying an infarct size of 34% on average – were characterized by LV-dysfunction (Table 1), reduced arterial blood pressure (Table 2), and increased right and left ventricular weights (Table 3), all of which are indicative of MI-induced heart failure.
Table 1.
Infarct size and baseline cardiac function for all groups
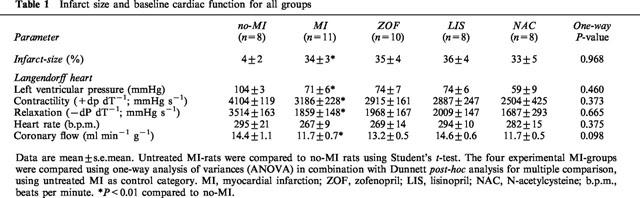
Table 2.
Time course of body weight and blood pressure for all groups
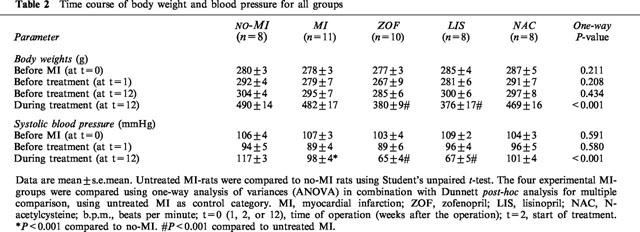
Table 3.
Final body and cardiac weights and cardiac:body weight ratios for all groups
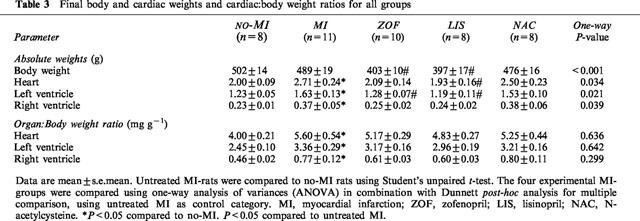
Mean infarct size was evenly balanced between the four experimental MI-groups (Table 2). Active treatment with zofenopril and lisinopril, but not n-acetylcysteine, significantly reduced body weight and systolic blood pressure to a similar extent (Table 2). Although this was accompanied by some tendency towards normalization of ventricular weight ratios, statistically significant reduction of heart weight ratios was not observed with any of the active treatment regimens (Table 3).
Studies with isolated aortic rings
In the present study potential interference of vasoactive prostanoids in dilatory responses was avoided with indomethacin to block cyclo-oxygenase in all vascular experiments – hence, a condition hereafter referred to as ‘total dilatation'.
Endothelial dysfunction in MI-induced heart failure rats
Consistent with the studies mentioned in the introduction section, total aortic dilatation after receptor-dependent stimulation with ACh (calculated and presented as AUC in Figure 1; see also Methods) was significantly reduced at 13–14 weeks after induction of MI (−50% on average, see also Table 4) in our rats. Inhibition of NOS using L-NMMA significantly reduced total dilatation to ACh (P<0.001, not specified in Figure 1) in the present study, indicating the important involvement of NO. However, this L-NMMA-sensitive part of the dilator response was significantly smaller in untreated MI-rats (−49% on average), indicating a decreased NO-contribution to ACh-induced dilatation as compared to control rats with no-MI. Moreover, the part of the total dilatation to ACh resistant to L-NMMA (hence, and indomethacin) was also significantly decreased in untreated MI-rats (−49% on average), pointing at a decreased contribution of a non-prostanoid/non-NO vasodilator substance as well. Consequently, despite significant reduction in total dilatation to ACh in untreated MI the relative contribution of L-NNMA-sensitive (i.e. NO) and -resistant (i.e. non-prostanoid/non-NO) vasodilator pathways in response to ACh remained unchanged (see also Figure 4).
Figure 1.
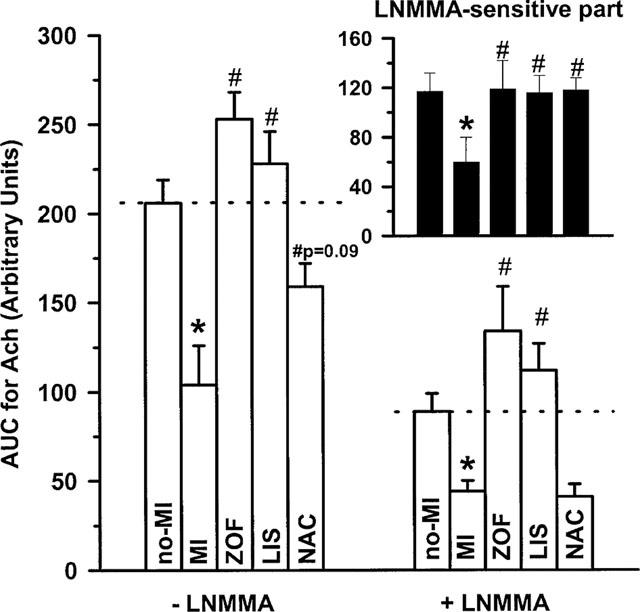
Response-size (displayed as the area under the curve; AUC) of acetylcholine (ACh)-induced dilatation in isolated aortic rings from treated (zofenopril, ZOF, n=10; lisinopril, LIS, n=8; N-acetylcysteine, NAC, n=8) and untreated rats with chronic myocardial infarction (MI, n=11) or no-MI (n=8). Dilatations were studied in presence of 10 μmol l−1 indomethacin, either with or without 100 μmol l−1 NG-mono-methy-L-arginine (L-NMMA) as indicated. The response-difference between both conditions representing the L-NMMA-sensitive part of the dilatation is shown in the small graph on the right-side top; identification of the bars from left to right is in the same order as below. Data are mean±s.e.mean; * indicates P<0.05 versus no-MI and # indicates P<0.05 versus MI.
Table 4.
Summary of directional changes (relative to no-MI) in aortic vasodilator responses to endothelium-dependent and -independent stimuli in treated and untreated rats with experimental myocardial infarction
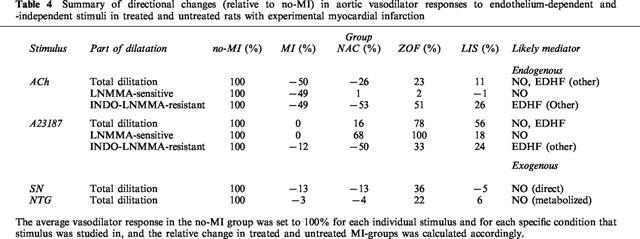
Figure 4.
Relative contribution of L-NMMA-sensitive (i.e. NO-mediated) and L-NMMA-resistant (i.e. EDHF/other) vasodilator pathways to acetylcholine (ACh) and A23187-induced dilatation in isolated aortic rings from treated (zofenopril, ZOF; lisinopril, LIS; N-acetylcysteine, NAC) and untreated rats with chronic myocardial infarction (MI, n=11) or no-MI (n=8).
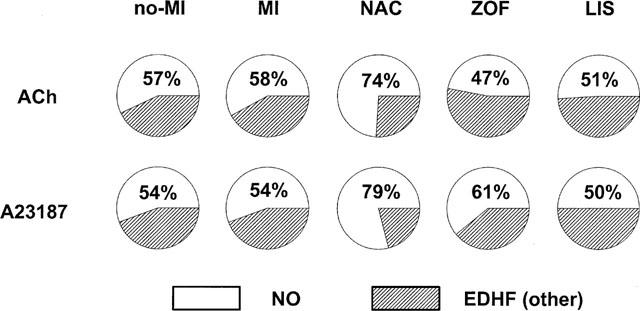
In contrast to ACh, total dilatation after receptor-independent stimulation with A23187 was unaffected in untreated MI, compared to controls with no-MI (Figure 2). Also the L-NMMA-sensitive NO-mediated part of the response to A23187 was not attenuated in untreated MI, indicating that the capacity of the endothelium for generation of biologically active NO was intact in our heart failure rats. Since the L-NMMA-resistant part remained similarly intact, the relative contribution of both dilator pathways (i.e. NO and non-prostanoid/non-NO) in response to A23187 remained unchanged in untreated MI (see also Figure 4).
Figure 2.
Response-size (displayed as the area under the curve; AUC) of A23187-induced dilatation in isolated aortic rings from treated (zofenopril, ZOF, n=10; lisinopril, LIS, n=8; N-acetylcysteine, NAC, n=8) and untreated rats with chronic myocardial infarction (MI, n=11) or no-MI (n=8). Dilatations were studied in the presence of 10 μmol l−1 indomethacin, either with or without 100 μmol l−1 NG-mono-methy-L-arginine (L-NMMA) as indicated. The response-difference between both conditions representing the L-NMMA-sensitive part of the dilatation is shown in the small graph on the right-side top; identification of the bars from left to right is in the same order as below. Data are mean±s.e.mean; # indicates P<0.05 versus MI.
The intact response to the SN (i.e. a direct donor of exogenous NO; Feelisch, 1991) rules out that a potential impairment at the level of vascular smooth muscle reactivity to NO might have accounted for the attenuated response to ACh in untreated MI-rats in the present study (Figure 3B). The intact response to NTG – a donor of exogenous NO that requires additional metabolic activation to release NO (Feelisch, 1991)–further indicates that the availability of cofactors (i.e. thiols) for bio-transformation of NTG was not limited in our heart failure rats (Figure 3A).
Figure 3.
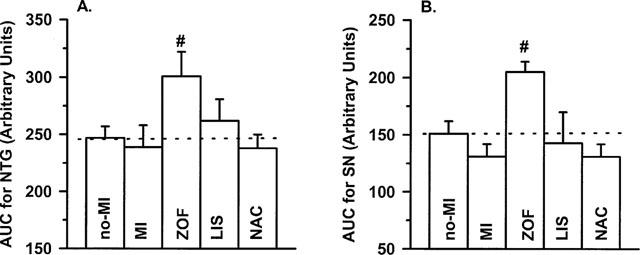
Response-size (displayed as area under the curve; AUC) of A: nitroglycerin (NTG) and B: sodium nitrite (SN)-induced dilatation in isolated aortic rings from treated (zofenopril, ZOF, n=10; lisinopril, LIS, n=8; N-acetylcysteine, NAC, n=8) and untreated rats with chronic myocardial infarction (MI, n=11) or no-MI (n=8). Dilatations were studied in presence of 10 μmol l−1 indomethacin. Data are mean±s.e.mean; # indicates P<0.05 versus MI.
Effects of chronic treatment with n-acetylcysteine, zofenopril and lisinopril
Impaired total dilatation to ACh in untreated MI (i.e. compared to no-MI) was moderately increased (P=0.09) after treatment with NAC, and significantly improved after treatment with zofenopril and lisinopril (i.e. all compared untreated MI; Figure 1). Apparently, the increase after NAC was due to a selective normalizing effect on NO-contribution since the L-NMMA-sensitive part of the dilatation to ACh was significantly increased after NAC, whereas the L-NMMA-resistant part was not significantly altered. Consequently, the relative contribution of the L-NMMA-sensitive pathway (i.e. NO) to ACh-induced dilation increased from 50% on average in untreated MI to approximately 74% on average after treatment with NAC (Figure 4). In contrast, ACE-inhibition significantly increased the contribution of both L-NMMA-sensitive and L-NMMA-resistant endothelial vasodilators after ACh without dramatically changing relative contribution of both vasodilator pathways in response to ACh; if anything relative NO-contribution after ACh tended to be lower after ACE-inhibition (Figure 4).
Intact total dilatation to A23187 in untreated MI (i.e. compared to no-MI) remained unaffected after treatment with NAC and lisinopril, but was significantly increased after zofenopril (Figure 2). Apparently, the increase after zofenopril involved an increased contribution of NO since the L-NMMA-sensitive part of the dilation to ACh was significantly increased after zofenopril (+100% on average). Of note – in spite of the fact that statistical significance was not achieved –a similar trend was observed after NAC, which also tended to increase the L-NMMA-sensitive part (+68% on average), as it seemed, at the expense of the L-NMMA-resistant part (−50% on average, again P=NS). Interestingly, the relative contribution of the L-NMMA-sensitive dilator pathway (i.e. NO) to total dilatation after A23187 increased from approximately 54% on average in untreated rats (i.e. no-MI and MI) to approximately 61 and 79% on average after zofenopril and NAC, respectively (i.e. SH-treated rats), irrespective of absolute differences in total dilatation (i.e. increased after zofenopril and unchanged after NAC), and this shift was not observed after lisinopril (Figure 4).
Finally, neither NAC nor lisinopril treatment significantly affected endothelium-independent dilatation to SN or NTG (Figure 3). By contrast, endothelium-independent dilator responses to both drugs were significantly increased after zofenopril.
A summary of directional changes in the above vasodilator responses in treated and untreated MI-rats relative to control rats with no-MI is given in Table 4. Furthermore, a summary of relative contributions of L-NMMA-sensitive and -resistant vasodilator pathways in response to ACh and A23187 in individual groups is given in Figure 4.
Plasma NOx
Plasma NOx concentrations, which were not significantly altered after MI, were profoundly increased after zofenopril and lisinopril, but not after NAC (Figure 5). This increase was more pronounced after zofenopril as compared to lisinopril, although statistical significance between both ACE-inhibitors was not reached.
Figure 5.
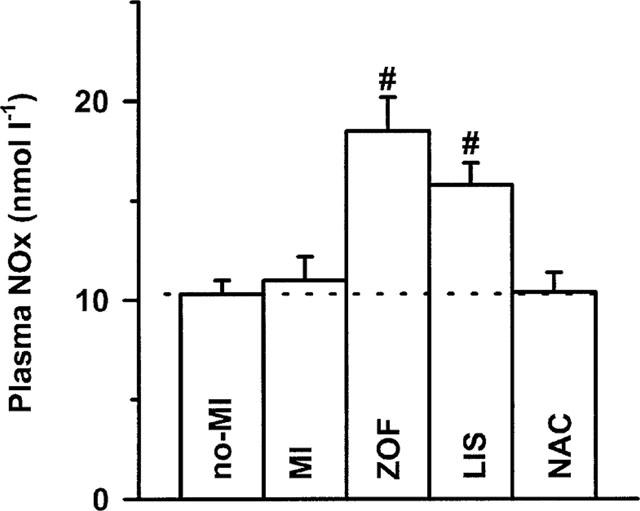
Plasma NOx levels in treated (zofenopril, ZOF, n=10; lisinopril, LIS, n=8; N-acetylcysteine, NAC, n=8) and untreated rats with chronic myocardial infarction (MI, n=11) or no-MI (n=8). Data are mean±s.e.mean; # indicates P<0.05 versus MI.
Discussion
The role of the SH-group in improvement of aortic endothelial dysfunction with ACE-inhibitors in experimental CHF was studied in MI-induced heart failure rats, chronically treated with zofenopril (ACE-inhibitor with SH-group) or lisinopril (ACE-inhibitor only), or with n-acetylcysteine (SH-group only). Results of our vascular studies first of all confirm that in rats with heart failure secondary to experimental MI, endothelium-dependent dilatation is attenuated after receptor-dependent stimulation with ACh but not after receptor-independent stimulation with A23187. Using L-NMMA, we demonstrated that impaired NO-contribution in part underlies the attenuated dilator response to ACh, despite an intact capacity for generation of biologically active NO after A23187. Our results also confirm that chronic ACE-inhibition (irrespective of SH-groups) prevents deterioration of the dilator response to ACh in heart failure, as it appeared through preservation of both the NO- as well as a non-prostanoid/non-NO vasodilator pathway. We here additionally report that chronic ACE-inhibition with a SH-group (i.e. zofenopril) – but not without a SH-group (i.e. lisinopril) – also potentiated the effect of endogenous NO after A23187-induced release from the endothelium as well as that of exogenous NO provided by NTG and SN. Moreover, this is the first study to demonstrate the effect of n-acetylcysteine (i.e. SH-groups per se) in selectively normalizing NO-contribution to ACh-induced dilatation in experimental heart failure.
Aortic endothelial dysfunction in rats with heart failure secondary to experimental myocardial infarction
Our present results with ACh are in agreement with recent studies reporting marked down-regulation of NO-mediated dilatation after ACh and P2Y-receptor stimulation in isolated mesenteric arteries in this model (Malmsjö et al., 1999). On the other hand, our results with A23187 also confirm previous findings by Ontkean et al. (1991), indicating that the capacity for generation of biologically active NO is still intact in CHF. The discrepancy between ACh and A23187 probably reflects the different mechanisms for endothelial cell activation (i.e. involvement of receptors after ACh but not A23187) and/or transfer of a relaxing factor(s) from the endothelium to vascular smooth muscle (i.e. involvement of gap-junctions after ACh but not A23187; Hutcheson et al., 1999). Underlying mechanisms that may explain the above decrease in NO-contribution after ACh include alterations/defects at the receptor level and/or signal transduction pathways (subsequently) leading to impaired NOS-activation (i.e. decreased NO-formation). It may also include the concomitant or increased stimulation of NO-inactivating pathways (i.e. increased NO-breakdown) after receptor-dependent stimulation, leading to decreased availability of biologically active NO after ACh. Recently, Bauersachs et al. (1999) reported impaired ACh-induced dilatation in CHF rats to be associated with elevated aortic NADPH-stimulated O2− production. They also showed that this could be partially restored by pretreatment with exogenous superoxide dismutase, and concluded that inactivation of NO may be an essential mechanism for endothelial dysfunction in heart failure. Finally, a potential role of impairment at the level of gap junctions involved in the transfer of a factor after ACh – but not A23187 – in CHF has yet to be studied.
The part of the total dilatation to ACh resistant to L-NMMA (and, hence, indomethacin) was also decreased in untreated MI-rats in the present study, pointing at decreased contribution of a non-prostanoid/non-NO vasodilator substance. Although EDHF would be the most likely candidate according to the criteria denoted by the literature, true identification of EDHF also requires evidence for hyperpolarization and relaxation of vascular smooth muscle cells that can be blocked by certain potassium channel blockers. Since we did not include such experiments, our studies do not allow to comment on the precise nature of the L-NMMA-resistant part of total dilatation after ACh other than that it was decreased in untreated MI and likely to have been mediated by EDHF.
Effects of chronic treatment with N-acetylcysteine and ACE-inhibitors
Recently, chronic treatment with N-acetylcysteine was reported to abrogate the attenuated aortic dilatation to ACh in streptozotocin-induced diabetic rats (Galen & Siebeneich, 1998). To explain their findings, the authors of the latter study discuss the direct (i.e. antioxidant) radical scavenging properties of n-acetylcysteine in relation to the enhanced .OH formation in experimental diabetic rats. More generally, they point at increased oxidative stress (i.e. increased production of reactive oxygen) that may underlie endothelial dysfunction (hence, NO-breakdown) in various cardiovascular disorders. This corroborates with our above discussion on endothelial dysfunction in heart failure rats, and is consistent with our present finding that N-acetylcysteine selectively restored the NO-mediated – but not the non-prostanoid/non-NO-mediated – dilatation to ACh in MI-rats.
Reduction of oxidative stress has also been implicated to explain the favourable effects of ACE-inhibition in (various) models of endothelial dysfunction, especially those related to increased angiotensin II-activity (Rajagopalan et al., 1996; Fukui et al., 1997). This seems to include heart failure in which the activated (tissue) renin-angiotensin system (RAS) has been implied to underlie and to contribute to the deterioration of cardiovascular function (Hirsch et al., 1990; Pinto et al., 1995). The important implication with respect to the present study would then be that both ACE-inhibitors may have exerted their beneficial effect on ACh mostly by interference at ‘trigger-level' (i.e. prevention of RAS-related increase in oxidative stress and subsequent impairment of biologically active NO) while N-acetylcysteine may have dealt relatively more with the ‘consequence-level' (i.e. scavenging of RAS-related increase in oxidative stress that underlies diminished NO activity).
Normalization or even potentiation of ACh-induced dilatation after chronic ACE-inhibition may also involve the accumulation of endogenous bradykinin (and possibly angiotensin 1–7) to increase NO-availability (Hornig & Drexler, 1997). The latter may be substantiated by the significantly increased plasma levels of NO-metabolites (NOX) after ACE-inhibition, but not after N-acetylcysteine, in the present study. In addition to NO, bradykinin may also increase vasodilator prostanoids and EDHF (Hornig & Drexler, 1997). The latter might have accounted for the intact (or even increased) part of response to ACh after ACE-inhibition that was mediated by a non-prostanoid/non-NO vasodilator.
Similarities and discrepancies between lisinopril, zofenopril and N-acetylcysteine
In contrast to ACh, both ACE-inhibitors clearly differed in their effect on dilator responses to NTG, SN, as well as A23187. Zofenopril, but not lisinopril, potentiated the response to the NTG and SN, as well as the NO-contribution to the dilator response to A23187. These results demonstrate the enhanced effect of endogenous and exogenous NO after chronic treatment with an ACE-inhibitor with SH-group compared to those without.
Such data are very suggestive for a favourable interaction between NO and the SH-group, but seem to be contradicted by our results obtained after N-acetylcysteine treatment, which lacked the above potentiating effects of zofenopril. One plausible explanation for this discrepancy may be that N-actylcysteine and zofenopril deliver/provide their SH-groups to different compartments in the vascular wall. N-acetylcysteine is a hydrophilic SH-donor and anti-oxidant which easily penetrates the cell membrane (Mazor et al., 1996). There, it could function as an antioxidant to protect NO from acute breakdown during synthesis in endothelial cells after ACh-induced stimulation.
Zofenopril on the other hand – being an ACE-inhibitor – may be anticipated to be importantly localized on circulating and bound ACE, and that (in addition to the intracellular compartment) its SH-groups are available to the extra-cellular environment in the vascular wall. Here, it could function to stabilize NO (either after release from the endothelium, or when administered exogenously) through formation of R-SNO and/or prevent its breakdown by scavenging free radicals in the extra-cellular environment. Hence, this closely relates to discussions regarding the discrepancies between NO and EDRF and whether or not EDRF rather may be a closely related adduct of NO, such as RS-NO (Meyers et al., 1990).
Limitations to the study
Important confounding factors in the present study are the differential effects on blood pressure (altered haemodynamics after both ACE-inhibitors but not after N-acetylcysteine) and plasma levels of NOX, clearly reflecting the different mechanisms of action between ACE-inhibitors and N-acetylcysteine. This difference is further exemplified by the property of n-acetylcysteine to favourably affect the contribution of NO in endothelium-dependent dilation in heart failure rats, irrespectively whether total dilatation is impaired (i.e. after ACh) or intact (i.e. after A23187). Another complicating factor is the proposed difference in the site of action between zofenopril (intra- and extra-cellular) and n-acetylcysteine (relatively more intra-cellular). All these factors may in part have contributed to the differential effects on ACh-induced relaxation, and complicate evaluation of the role of the SH-group in improvement of endothelial function with ACE-inhibitors in heart failure.
Furthermore, to address this issue we employed aortic endothelial dysfunction as a read-out model, mainly because of practical reasons. It is well recognized, however, that the small resistance arteries are more important in increased vascular resistance in heart failure. Studies performed on mesenteric arteries of heart failure rats have reported preservation (Buus et al., 1999) as well as attenuation of ACh-induced relaxation (Mulder et al. 1996), but these are not further discussed in the present paper for reasons of space-limitation. It should be noted, however, that important differences exist between vessels of different size and of different vascular beds regarding the relative contribution of different endothelial mediators to ACh-induced relaxation, so that our present results with the aorta not automatically apply for other vessels. Rather they may serve to develop future studies evaluating resistance vessels for the above issue.
Conclusions
ACE-inhibitors with a SH-group have a potential advantage in improvement of endothelial dysfunction in CHF through increased protection/stability of NO after release from the endothelium from into the vessel wall. In addition, this is the first study demonstrating the selective normalizing effect of N-acetylcysteine on NO-contribution to ACh-induced dilatation in experimental heart failure.
Acknowledgments
The authors wish to express their gratitude to Egbert Scholtens, Bianca Meijeringh and Azuwerus van Buiten for biotechnical assistance in experimental animal procedures and vascular studies, and to Dr Stefano Evangelista from Menarini Ricerche S.P.A. (Firenze, Italy) for supplying zofenopril and financial support of this study. R.A. Tio was supported by the Netherlands Heart Foundation (grant D95-019).
Abbreviations
- ACh
acetylcholine
- AUC
area under curve
- BW
body weight
- CHF
chronic heart failure
- INDO
indomethacin
- LIS
lisinopril
- L-NMMA
NG-mono-methyl-L-arginine
- MI
myocardial infarction
- NAC
n-acetylcysteine
- NO
nitric oxide
- NT
not treated
- NTG
nitroglycerin
- PhE
phenylephrine
- RAS
renin-angiotensin system
- SH
sulfhydryl
- SN
sodium nitrite
- RV and LV
right and left ventricle
- ZOF
zofenopril
References
- BAUERSACHS J., BOULOUMIE A., FRACCEROLLO D., HU K., BUSSE R., ERTL G. Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression: role of enhanced vascular superoxide production. Circulation. 1999;100:292–298. doi: 10.1161/01.cir.100.3.292. [DOI] [PubMed] [Google Scholar]
- BUIKEMA H., PINTO Y.M., ROOKS G., GRANDJEAN J.G., SCHUNKERT H., VAN GILST W.H. The deletion polymorphism of the angiotensin-converting enzyme gene is related to phenotypic differences in human arteries. Eur. Heart. J. 1996;17:787–794. doi: 10.1093/oxfordjournals.eurheartj.a014947. [DOI] [PubMed] [Google Scholar]
- BUIKEMA H., VAN VELDHUISEN D.J., HEGEMAN H., VAN GILST W.H. Early pharmacologic intervention may prevent the deterioration in endothelial function after experimental myocardial infarction in rats: effects of ibopamine and captopril. J. Cardiac. Failure. 1997;3:125–132. doi: 10.1016/s1071-9164(97)90046-4. [DOI] [PubMed] [Google Scholar]
- BUUS N.H., KAHR O., MULVANY M.J. Effect of short- and long-term heart failure on small artery morphology and endothelial function in the rat. J. Cardiovasc. Pharmacol. 1999;34:34–40. doi: 10.1097/00005344-199907000-00006. [DOI] [PubMed] [Google Scholar]
- DE VRIES R.J.M., ANTHONIO R., VAN VELDHUISEN D.J., SCHOLTENS E., BUIKEMA H., VAN GILST W.H. Effects of amlodipine on endothelial function in rats with chronic heart failure after experimental myocardial infarction. J. Cardiovasc. Pharmacol. 1997;30:683–689. doi: 10.1097/00005344-199711000-00020. [DOI] [PubMed] [Google Scholar]
- DREXLER H., HAYOZ D., MÜNZEL T., JUST H., ZELIS R., BRUNNER H.R. Endothelial function in congestive heart failure. Am. Heart. J. 1993;126:761–764. doi: 10.1016/0002-8703(93)90926-z. [DOI] [PubMed] [Google Scholar]
- FEELISCH M. The biochemical pathways of nitric oxide formation from nitrovasodilators: appropiate choice of exogenous NO donors and aspects of preparation and handling of aqueous NO solutions. J. Cardiovasc. Pharmacol. 1991;17:27–36. [Google Scholar]
- FUKUI T., ISHIZAKA N., RAJAGOPALAN S., LAURSEN J.B., CAPERS Q., IV, TAYLOR W.R., HARRISON D.G., DE LEON H., WILCOX J.N., GRIENDLING K.K. p22Phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 1997;80:45–51. doi: 10.1161/01.res.80.1.45. [DOI] [PubMed] [Google Scholar]
- GALEN M.P., SIEBENEICH W. Oral administration of the antioxidant, N-acetylcysteine, abrogates diabetes-induced endothelial dysfunction. J. Cardiovasc. Pharmacol. 1998;32:101–105. doi: 10.1097/00005344-199807000-00016. [DOI] [PubMed] [Google Scholar]
- GIVERTZ M.M., COLCUCCI W.S. New targets for heart failure therapy; endothelin, inflammatory cytokines, and oxidative stress. Lancet. 1998;352 Suppl.1:S135–S138. doi: 10.1016/s0140-6736(98)90017-4. [DOI] [PubMed] [Google Scholar]
- GOLDSCHMIDT J.E., TALLARIDA R.J. Pharmacological evidence that captopril possesses an endothelium-mediated component of vasodilation: effect of sulfhydryl groups on endothelium-derived relaxing factor. J. Pharmacol. Exp. Ther. 1991;257:1136–1145. [PubMed] [Google Scholar]
- HILL M.F., SINGAL P.K. Right and left myocardial antioxidant responses antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. 1997;96:2414–2420. doi: 10.1161/01.cir.96.7.2414. [DOI] [PubMed] [Google Scholar]
- HIRSCH A.T., PINTO Y.M., SCHUNKERT H., DZAU V.J. Potential role or the tissue renin-angiotensin system in pathophysiology of congestive heart failure. Am. J. Cardiol. 1990;66:22D–32D. doi: 10.1016/0002-9149(90)90473-e. [DOI] [PubMed] [Google Scholar]
- HORNIG B., DREXLER H. Endothelial function and bradykinin in humans. Drugs. 1997;54 Suppl.5:42–47. doi: 10.2165/00003495-199700545-00007. [DOI] [PubMed] [Google Scholar]
- HUTCHESON I.R., CHAYTOR A.T., EVANS W.H., GRIFFITH T.M. Nitric oxide-independent relaxations to acetylcholine and A23187 involve different routes of heterocellular communication. Role of gap junctions and phospholipase A2. Circ. Res. 1999;84:53–63. doi: 10.1161/01.res.84.1.53. [DOI] [PubMed] [Google Scholar]
- JESERICH M., PAPE L., JUST H., HORNIG B., KUPFER M., MUNZEL T., LOHMANN A., OLSCHEWSKI M., DREXLER H. Effect of long-term angiotensin-converting enzyme inhibition on vascular function in patients with chronic congestive heart failure. Am. J. Cardiol. 1995;76:1079–1082. doi: 10.1016/s0002-9149(99)80305-1. [DOI] [PubMed] [Google Scholar]
- KATZ S.D. Mechanisms and implications of endothelial dysfunction in congestive heart failure. Curr. Opin. Cardiol. 1997;12 3:259–264. doi: 10.1097/00001573-199705000-00007. [DOI] [PubMed] [Google Scholar]
- KEITH M., GERANMAYEGAN A., SOLE M.J., KURIAN R., ROBINSON A., OMRAN A.S., JEEJEEBHOY K.N. Increased oxidative stress in patients with congestive heart failure. J. Am. Coll. Cardiol. 1998;31:1352–1356. doi: 10.1016/s0735-1097(98)00101-6. [DOI] [PubMed] [Google Scholar]
- KUBO S.H., RECTOR T.S., BANK A.J., WILLIAMS R.E., HEIFETZ S.M. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation. 1991;84:1589–1596. doi: 10.1161/01.cir.84.4.1589. [DOI] [PubMed] [Google Scholar]
- MALMSJÖ M., BERGDAHL A., ZHAO X.-H., SUN X.-Y., HEDNER T., EDVINSSON L., ERLINGE D. Enhanced acetylcholine and P2Y-receptor stimulated vascular EDHF-dilatation in congestive heart failure. Cardiovasc. Res. 1999;43:200–209. doi: 10.1016/s0008-6363(99)00062-0. [DOI] [PubMed] [Google Scholar]
- MAZOR D., GOLAN E., PHILIP V., KATZ M., JAFE A., BEN ZVI Z., MEYERSTEIN N. Red blood cell permeability to thiol compounds following oxidative stress. Eur. J. Haematol. 1996;57:241–246. doi: 10.1111/j.1600-0609.1996.tb01370.x. [DOI] [PubMed] [Google Scholar]
- MEYERS P.R., MINOR R.L., GUERRA R., BATES J.N., HARRISON D.G. Vasorelaxant properties of the endothelium-derived relaxing factor more closely resemble S-nitrocysteine than nitric oxide. Nature. 1990;345:161–163. doi: 10.1038/345161a0. [DOI] [PubMed] [Google Scholar]
- MOSHAGE H., KOK B., HUIZINGA J.R., JANSEN P.L.M. Nitrite and nitrate determinations in plasma: a critical evaluation. Clin. Chem. 1995;41:892–896. [PubMed] [Google Scholar]
- MULDER P., ELFERTAK L., RICHARD V., COMPAGNON P., DEVAUX B., HENRY J.P., SCALBERT E., DESCHE P., MACE B., THUILLEZ C. Peripheral artery structure and endothelial function in heart failure: effect of ACE inhibition. Am. J. Physiol. 1996;271:H469–H477. doi: 10.1152/ajpheart.1996.271.2.H469. [DOI] [PubMed] [Google Scholar]
- ONTKEAN M.T., GAY R., GREENBERG B. Diminished endothelium-derived relaxing factor activity in an experimental model of chronic heart failure. Circ. Res. 1991;69:1088–1096. doi: 10.1161/01.res.69.4.1088. [DOI] [PubMed] [Google Scholar]
- ONTKEAN M.T., GAY R., GREENBERG B. Effects of chronic captopril therapy on endothelium-derived relaxing factor in heart failure J. Am. Coll. Cardiol. 199219207A(Abstract) [Google Scholar]
- PFEFFER M.A., PFEFFER J.M., BRAUNWALD E. Myocardial infarct size and ventricular function in rats. Circ. Res. 1979;44:503–512. doi: 10.1161/01.res.44.4.503. [DOI] [PubMed] [Google Scholar]
- PINTO Y.M., BUIKEMA H., VAN GILST W.H. Hyperactive tissue renin-angiotensin systems in cardiovascular dysfunction: experimental and clinical hypotheses. Clin. Exp. Hypertension. 1995;17:441–468. doi: 10.3109/10641969509037418. [DOI] [PubMed] [Google Scholar]
- PINTO Y.M., DE SMET B.G.J.L., VAN GILST W.H., SCHOLTENS E., MONNINK S.H.J., DE GRAEFF P.A., WESSELING H. Selective and time related activation of the cardiac renin-angiotensin system after experimental heart failure: relation to ventricular function and morphology. Cardiovasc. Res. 1993;27:1933–1938. doi: 10.1093/cvr/27.11.1933. [DOI] [PubMed] [Google Scholar]
- PINTO Y.M., VAN WIJNGAARDEN J., VAN GILST W.H., DE GRAEFF P.A., WESSELING H. The effects of short- and long-term treatment with an ACE-inhibitor in rats with myocardial infarction. Bas. Res. Cardiol. 1991;86 Suppl. 2:165–172. [PubMed] [Google Scholar]
- RAJAGOPALAN S., KURZ S., MUNZEL T., TARPEY M., FREEMAN B.A., GRIEDLING K.K., HARRISON D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NAD/NADPH oxidase activiation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TEERLINK J.R., CLOZEL M., FISCHLI W., CLOZEL J.-P. Temporal evolution of endothelial dysfunction in a rat model of chronic heart failure. J. Am. Coll. Cardiol. 1993;22:615–620. doi: 10.1016/0735-1097(93)90073-a. [DOI] [PubMed] [Google Scholar]
- THUILLEZ C., MULDER P., ELFERTAK L., BLAYSAT G., COMPAGNON P., HENRY J.-P., RICHARD V., SCALBERT E., DESCHE P. Prevention of endothelial dysfunction in small and large arteries in a model of chronic heart failure. Effects of angiotensin converting enzyme inhibition. Am. J. Hypertens. 1995;8:7S–12S. doi: 10.1016/0895-7061(95)00027-m. [DOI] [PubMed] [Google Scholar]
- TSUNODA K., HODSMAN G.P., SUMITHRAN E., JOHNSTON C.I. Arterial natriuretic peptide in chronic heart failure in the rat: a correlation with ventricular dysfunction. Circ. Res. 1986;59:256–271. doi: 10.1161/01.res.59.3.256. [DOI] [PubMed] [Google Scholar]
- VAN GILST W.H., DE GRAEFF P.A., DE LEEUW M.J., SCHOLTENS E., WESSELING H. Converting enzyme inhibitors and the role of the sulfhydryl group in the potentiation of exo- and endogenous nitrovasodilators. J. Cardiovasc. Pharmacol. 1991;18:429–436. doi: 10.1097/00005344-199109000-00016. [DOI] [PubMed] [Google Scholar]
- VAN GILST W.H., VAN VELDHUISEN D.J., HEGEMAN H., BUIKEMA H., SCHOLTENS E., DE GRAEFF P.A., LIE K.I. Effect of ibopamine on ventricular remodeling after experimental myocardial infarction: a comparison with ibopamine. J. Cardiovasc. Pharmacol. 1994;24:171–174. doi: 10.1097/00005344-199407000-00026. [DOI] [PubMed] [Google Scholar]
- VAN WIJNGAARDEN J., PINTO Y.M., VAN GILST W.H., DE GRAEFF P.A., WESSELING H. Converting enzyme inhibition after experimental myocardial infarction in rats: a comparative study between spirapril and zofenopril. Cardiovasc. Res. 1991;25:936–942. doi: 10.1093/cvr/25.11.936. [DOI] [PubMed] [Google Scholar]