

Abstract
The relationship between persistent ERK (extracellular signal-regulated kinase) activity, cyclin D1 protein and mRNA levels and cell cycle progression in human cultured airway smooth muscle was examined in response to stimulation by ET-1 (endothelin-1), thrombin and bFGF (basic fibroblast growth factor).
Thrombin (0.3 and 3 u ml−1) and bFGF (0.3 and 3 nM) increased ERK activity for more than 2 h and increased cell number, whereas ET-1 (100 nM) transiently stimulated ERK activity and was non-mitogenic.
The MEK1 (mitogen-activated ERK kinase) inhibitor, PD 98059 (30 μM), inhibited both ERK phosphorylation and activity, and either prevented (thrombin 0.3 and 3 u ml−1, bFGF 300 pM) or attenuated (bFGF 3 nM) DNA synthesis.
Thrombin and bFGF increased both cyclin D1 mRNA and protein levels. PD 98059 decreased cyclin D1 protein levels stimulated by the lower but not higher thrombin concentrations. Moreover, increases in cyclin D1 mRNA levels were unaffected by PD 98059 pretreatment, irrespective of the mitogen or its concentration, suggesting that inhibition of cyclin D1 protein levels occurred by a post-transcriptional mechanism.
These findings indicate that the control of cyclin D1 protein levels may occur independently of the MEK1/ERK signalling pathways. The inhibition of S phase entry by PD 98059 at higher thrombin concentrations appears to result from effects on pathways downstream or parallel to those regulating cyclin D1 protein levels. These findings suggest heterogeneity in the signalling of DNA synthesis in human cultured airway smooth muscle.
Keywords: Airway smooth muscle, thrombin, basic fibroblast growth factor, endothelin-1, MEK1, ERK, cyclin D1, PD98059, mitogenesis
Introduction
Chronic asthma is characterized by an eosinophilic inflammation and airway obstruction of variable intensity, airway hyperresponsiveness and airway wall structural changes. The cellular changes responsible for the airway wall remodelling include infiltration of leukocytes, glandular hypertrophy, goblet cell metaplasia, thickening of the basement membrane and increases in the volume of airway smooth muscle (Dunnill & Massarella, 1969; Hossain, 1973; Ebina et al., 1993). Smooth muscle makes the most important contribution to the increase in airway wall volume that results from all of these cellular changes (James et al., 1989). The mechanisms underlying the increase in airway wall volume occupied by smooth muscle may include increased deposition of extracellular matrix (Bramley et al., 1995), hypertrophy and hyperplasia of smooth muscle (Ebina et al., 1993). The increase in airway wall volume contributes to airway hyperresponsiveness (AHR), a phenomenon that comprises an increased airway narrowing in response to chemically and physically diverse stimuli that closely relates to the severity of asthma (Wiggs et al., 1992). Mitogenic responses of airway smooth muscle may be suitable targets for anti-asthma drugs. However, a more detailed understanding of the stimuli and the heterogeneity of the signalling pathways is required to guide the choice of drug targets.
Inflammatory mediators including histamine (Maruno et al., 1995), interleukins 1β and 6 (De et al., 1993, 1995) and tyrosine kinase receptor-linked growth factors, such as epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) (Stewart et al., 1995a), are mitogenic for human airway smooth muscle. Thrombin, which activates a protease-activated receptor (PAR), also stimulates human airway smooth muscle proliferation (Tomlinson et al., 1994; Panettieri et al., 1995). Airway smooth muscle cells release autocrine pro-inflammatory mediators, thereby regulating the production of growth factors and further growth and proliferation (Johnson & Knox, 1997; Vlahos & Stewart, 1999).
A complex signal transduction cascade regulates the cell cycle progression that controls the growth and division of airway smooth muscle cells. The phosphorylation of the restriction protein, retinoblastoma (pRb), is a central event controlling progression of the cell cycle to S phase (Sherr, 1995). The activated cyclin D1/cyclin-dependent kinase4 (Cdk4) complex phosphorylates pRb in mid-to-late G1 phase to promote the dissociation of pRb from E2F (Chellappan et al., 1991), and permit the progression from G1 to S phase of the cell cycle (Lundberg & Weinberg, 1998). A sustained increase in the levels of cyclin D1, and assembly with Cdk4 are dependent on continuous mitogenic stimulation (Sherr, 1995; Matsushime et al., 1994). Withdrawal of mitogenic stimulation leads to a rapid decline in cyclin D1 levels (Sherr, 1993). The degradation of cyclin D1 protein is mediated by ubiquitin-dependent proteolysis, which is triggered by the phosphorylation of a threonine residue of cyclin D1 (Diehl et al., 1997). Proteosomal degradation of cyclin D1 appears to be activated by increases in cyclic AMP levels (Stewart et al., 1999).
Several studies in transformed cell lines have demonstrated the requirement of cyclin D1 for S phase entry (Quelle et al., 1993). Recent studies by Xiong et al. (1997) demonstrated that mitogenic stimulation of bovine airway smooth muscle induced activation of both the cyclin D1 promoter and cyclin D1 protein expression, followed by phosphorylation of pRb (Xiong et al., 1997). Conversely, the microinjection of an anti-cyclin D1 antibody reduced DNA synthesis indicating a requirement for cyclin D1 in S phase entry and cell cycle progression (Xiong et al., 1997).
The p44/p42MAPK isoforms (ERK1 and 2, respectively) of the mitogen-activated protein kinase (MAPK) family play a major role in the signalling of cell proliferation. The phosphorylation of ERK by MEK1 results in catalytic activation of ERK (Posada & Cooper, 1992; Whelchel et al., 1997). Within a few minutes of mitogenic stimulation, activated ERK isoforms translocate to the nucleus in most cell types (L'Allemain et al., 1991) thereby transmitting mitogenic signals to the cell nucleus (Chen et al., 1992; Lenormand et al., 1993) such as phosphorylation of specific transcription factors that are required for cell cycle progression (Gille et al., 1995; Janknecht et al., 1995; Kato et al., 1995). Only those agents able to induce sustained ERK activation are mitogenic for CCL39 hamster lung fibroblasts (Meloche et al., 1992). ERK activity then rapidly declines during G1 to S phase transition (Meloche, 1995). Persistent ERK activation has also been specifically implicated in mitogenic responses of bovine airway smooth muscle (Kelleher et al., 1995). Links between persistent ERK activation and cyclin D1 expression have been demonstrated in Chinese hamster lung (Lavoie et al., 1996) and embryo fibroblast cell lines (Weber et al., 1997). Catalytic activation of ERK appears to be required for the induction of cyclin D1 expression in primary bovine airway smooth muscle cells (Ramakrishnan et al., 1998).
The relationship between ERK activation, cyclin D1 transcription and translation and S phase entry in human cultured airway smooth muscle has not been fully established. There is much species variation in the responses to different mitogenic agents. For example, endothelin-1 (ET-1), which is linked to a G-protein-coupled receptor, stimulates mitogenesis in rabbit (Noveral et al., 1992), ovine (Glassberg et al., 1994), guinea-pig (Stewart et al., 1994) and rat airway smooth muscle (Whelchel et al., 1997), but does not directly stimulate proliferation in human (Panettieri et al., 1995) or bovine airway smooth muscle (Panettieri et al., 1996). In contrast, bradykinin, which also activates a G-protein-coupled receptor, stimulates ERK activity that leads to an increase in PGE2 levels and negatively feeds back to inhibit ERK, thereby restricting mitogenesis in guinea-pig ASM (Pyne et al., 1997). However, thrombin, which activates another G-protein-coupled receptor, is mitogenic for bovine (Walker et al., 1998), rat (Shapiro et al., 1996) and human (Tomlinson et al., 1994) airway smooth muscle. Growth factors, including platelet-derived growth factor (PDGF) and EGF, which activate tyrosine kinase-linked receptors, also demonstrate species dependent mitogenic potentials. Both PDGF and EGF are mitogenic for human airway smooth muscle (Tomlinson et al., 1994; Kelleher et al., 1995), but only PDGF is active on bovine airway smooth muscle. The mechanism for the strong mitogenic response observed in guinea-pig ASM has been partly elucidated in that activation of the PDGF receptor evokes a powerful stimulation of the ERK signalling pathway by utilizing the G protein, Gi, in addition to the PDGF tyrosine kinase signalling pathway to promote mitogenesis (Conway et al., 1999). Furthermore, the effect of ERK activation can vary markedly between different strains of cells of the same species. ERK has either a stimulatory or inhibitory effect on smooth muscle proliferation, depending on whether the arterial smooth muscle strain expresses COX-2. Expression of COX-2 leads to the production of PGE2, which inhibits DNA synthesis via an elevation of PKA (Bornfeldt et al., 1997). Moreover, in cytokine-stimulated ASM glucocorticoids increase rather than decrease growth factor-induced DNA synthesis by repression of COX-2 induction (Vlahos & Stewart, 1999). Species differences in functional (mitogenic) responses to growth factors that do not result from lack of receptor expression imply the existence of differential activation of cell cycle progression signalling pathways. Thus, there is a clear need to establish both the functional and biochemical mitogen responsiveness of human cultured airway smooth muscle.
The present study examines the relationship of persistent ERK activity to cyclin D1 protein and mRNA levels and S phase entry in human airway smooth muscle cells. This relationship has been investigated using the MEK1-inhibitor, PD 98059 (Dudley et al., 1995), a selective tool that has been widely used to elucidate the role of the MEK1/ERK pathway (Grammer & Blenis, 1997; Karpova et al., 1997; Weber et al., 1997; Whelchel et al., 1997). Since the primary mitogens for human airway wall remodelling are not known, we have compared the signals initiated by thrombin with those in response to bFGF and the potent bronchoconstrictor ET-1. These putative mitogens activate different classes of receptors and have been identified in inflamed airways (Redington et al., 1995; Gabazza et al., 1998; Springall et al., 1991). In addition, as the prevailing mitogen concentrations associated with variable intensities of inflammation are not known, the ERK-dependence of near maximal and supramaximal concentrations of bFGF and thrombin have been contrasted.
Our findings suggest that persistent ERK activation makes a substantial contribution to the signalling of mitogenesis of human airway smooth muscle, but at supramaximal mitogen concentrations other pathways may additionally be recruited to signal mitogenesis.
Methods
Culture of human airway smooth muscle
Human airway smooth muscle was cultured from macroscopically normal bronchi (0.5–2 cm diameter) obtained from lung resection or heart–lung transplant specimens provided by the Alfred Hospital (Melbourne). Cultures were prepared as previously described in detail (Tomlinson et al., 1995) and maintained in Dulbecco's Modified Eagle's Medium (DMEM) (supplemented with 0.25% v v−1 BSA, 2 mM L-glutamine, 100 u ml−1 penicillin G, 100 μg ml−1 streptomycin and 2 μg ml−1 amphotericin B) and containing foetal calf serum (FCS) (10% v v−1). Cells were maintained in Falcon culture flasks and incubated (37°C, 5% CO2) until monolayer confluence was reached and harvested weekly by a 10 min exposure to 0.5% (w v−1) trypsin, 1 mM EDTA in PBS and passaged at a 1 : 3 split ratio. Cells at passage numbers 4–15 were used for experiments, over which range of passage number there is no detectable relationship between passage number and responsiveness to growth factors or inhibitors, or to the expression of α-actin (Stewart et al., 1997b).
Immunohistochemistry
The cellular composition of the cultures was determined using expression of smooth muscle specific α-actin and myosin as described previously (Panettieri et al., 1989; Stewart et al., 1997a; Fernandes et al., 1999).
Measurement of cell cycle progression
DNA synthesis was measured between 24 and 28 h after mitogen stimulation, as the human airway smooth muscle cells enter S phase approximately 22 h after the addition of thrombin (Stewart et al., 1995b). A longer stimulation time is required for the measurement of changes such as cell proliferation or increases in cellular protein (Stewart et al., 1995b). In this study, we have measured both increases in cell number and the DNA profiles of the cells 48 h after mitogen stimulation, and have demonstrated both cell division and cycling at this time.
Measurement of DNA synthesis
Cells were subcultured into 24 well plates in a 1 : 3 split ratio at a density of approximately 1.5 × 101.5 × 104 cells cm−2 and grown to monolayer confluence in DMEM containing 10% FCS, over a 72–96 h period (5% CO2 in air, 37°C). Quiescence was induced 24 h prior to stimulation by removing the medium, washing with PBS and replacing the medium with serum-free DMEM (supplemented as described above). In some experiments, the cells were incubated with the MEK1-selective inhibitor, PD98059 (30 μM) (Dudley et al., 1995) for 30 min before the addition of mitogen. Cells were stimulated by the addition of endothelin-1 (human, 100 nM), thrombin (3 or 0.3 u ml−1) or bFGF (3 nM or 300 pM). Monomed A (1% v v−1), a serum-free medium supplement containing insulin (100 ng ml−1), transferrin (50 ng ml−1), and selenium (1.5 pg ml−1) was added to all cells to provide progression factors that are essential for mitogenic activity. Although the higher concentrations of bFGF and thrombin were used predominantly in this study, one-tenth of these concentrations were also used to contrast differences in the recruitment of signalling pathways in response to near maximal and supramaximal mitogen concentrations. Cells were incubated with the indicated agent for 24 h (5% CO2 in air, 37°C) then pulsed with [3H]-thymidine (1 μCi ml−1) for 4 h to measure the incorporation of the radiolabel into newly synthesized DNA, according to our previous study (Stewart et al., 1997b).
Flow cytometry
Cells were seeded into six-well plates at a density of 1.5 × 104 cells cm−2 and were made quiescent as described previously, then were stimulated with the indicated mitogen for 48 h. Monomed A was added to all cells (including control cells) at the time of mitogen addition. During the stimulation period, the medium was removed after 24 h and replaced with warmed fresh medium and stimulants. Cells were detached from the culture plates by incubation with trypsin for 30 min at 37°C (0.5% w v−1). The resulting suspension was washed in PBS twice before resuspension in 1 ml of 70% ethanol for storage for up to 3 weeks at −20°C. Prior to staining, cells were washed twice (2% FCS in PBS) to remove the ethanol. Fixed cells were stained with propidium iodide (50 μg ml−1) in Triton X-100 (0.1% v v−1) with Rnase II (180 mu ml−1). The cell suspension was passed through an 18-gauge needle to facilitate the separation of cell clumps. Cells were stored for 24 h before analysis. Cell cycle status was analysed using a Becton-Dickinson FACScan instrument (Becton Dickinson, NJ, U.S.A.). Ten thousand events from each sample were counted and analysed using a ModFitLT V2.0 analysis package (Verity Software House, ME, U.S.A.).
Measurement of cell proliferation
Cells were seeded onto six-well plates at a density of 1.5 × 104 cells cm−2, made quiescent as previously described and then stimulated for 48 h with the appropriate mitogen. During the stimulation period, the medium was removed after 24 h and replaced with warmed fresh medium and stimulants. Cells were detached from the culture plate by the addition of trypsin (0.5% w v−1 in PBS containing 1 mM EDTA), washed twice (2% FCS in PBS) isolated by centrifugation (12,000 × g, 5 min) and resuspended in 200 μl 2% FCS in PBS for counting in an haemocytometer chamber.
Western blot analyses
Cells were subcultured onto six-well plates and stimulated under conditions identical to those used for estimation of DNA synthesis. Thrombin, bFGF or ET-1 was added to each well for 20 h. A stimulation period of 20 h was chosen in order to measure ERK phosphorylation and cyclin D1 levels late in G1 before S phase entry at approximately 22 h (Stewart et al., 1995b). The cell monolayer was washed twice with ice-cold PBS, lysed on ice and prepared for electrophoresis exactly as described previously (Fernandes et al., 1999). Aliquots were removed for protein assay (BioRad reagent, BioRad, Sydney, Australia). Identical amounts of protein (60–100 μg) from separate samples were loaded onto lanes in the SDS-polyacrylamide gel. The samples were resolved on PAGE, transferred to nitrocellulose membranes and Western blotted for phospho-ERK and cyclin D1 according to the methods described previously (Fernandes et al., 1999). To visualize the antigen, ECL reagents were added for 1 min and the membrane was then apposed to Kodak X-omat AR film before development. Exposure levels were quantitated by laser scanning densitometry (Molecular Dynamics Personal Densitometer, Molecular Dynamics, U.S.A.) and volumes normalized to levels of protein detected in control cells (fold increment over baseline), or in cells incubated with thrombin.
Analysis of ERK activation
ERK activity was measured by two independent methods. The Biotrak™ ERK (p42/p44 MAP kinase) enzyme assay kit (Amersham, Cardiff, U.K.) was used to measure the ERK kinase activity in response to mitogenic and non-mitogenic stimulation. Cells were seeded onto 24-well plates at a density of 1.5 × 104 cells cm−2 and made quiescent as previously described, then incubated with either ET-1, bFGF or thrombin for 5 min, 2 h or 20 h as indicated. Monomed A was added to all cells at the time of mitogen addition. The stimulation period was terminated and the cell lysates were prepared for the assay as previously described in detail (Fernandes et al., 1999). The ERK activity in the cell lysate was assayed by incubation for 30 min (30°C) in 15 ml buffer containing 6 nmol ATP (1 μCi [γ-32P]-ATP) and a synthetic substrate of a sequence of the epidermal growth factor receptor comprising a single phosphorylation site which is recognized by ERK. The peptide was separated on P81 filter paper and the non-incorporated [32P]-ATP was removed by extensive washing in 1% acetic acid then water. The paper disks were dried, Packard Microscint-40 added to each disk, and then the amount of radioactivity was counted in a Packard Topcount Scintillation Counter. Data are presented as absolute ERK 1/2 activity (nmol 32P transferred min−1 mg−1 protein).
ERK activity was also determined by immunoprecipitation of ERK using a specific anti-ERK antibody (goat polyclonal IgG). Kinase activity was measured by the amount of [γ-32P]-ATP incorporated into a non-specific protein substrate, myelin basic protein. Cells were seeded onto six-well plates as previously described, grown to confluence, then serum starved for 24 h. The cells were incubated for 30 min with PD98059 (30 μM) as indicated and all cells were stimulated with bFGF and/or Monomed A for 5 min, 2 h or 20 h. The stimulation period was terminated and the immunoprecipitation and kinase assay performed as previously described in detail (Fernandes et al., 1999). The phosphorylation of MBP by the immunoprecipitated ERK was assessed by phosphorimaging and densitometry (Molecular Dynamics Phosphorimager, Molecular Dynamics, U.S.A.).
Northern blot analyses
Cells were seeded into six-well plates at a density of 1.5 × 104 cells cm−2, grown to confluence, serum-starved for 24 h as previously described and stimulated for 16 h with the appropriate mitogen to coincide with the mid to late stages of G1 phase. Total RNA was extracted with 1 ml Trizol™ reagent according to the manufacturer's instructions. The mRNA was isolated from 5 μg of total RNA using Dynabeads oligo (dT)25 according to the manufacturer's instructions and was separated on a 1.2% formaldehyde denaturing gel and transferred to Immobilon-Ny+ nylon membranes using 20 × standard saline citrate (SSC). Cyclin D1 mRNA was detected by Northern hybridization (Megaprime labelling kit, Amersham, U.K.) using a 440 bp human cDNA probe (Xiong et al., 1991) labelled with α-32P-dCTP. The membranes were hybridized overnight at 65°C, washed twice with 2 × SSC+0.1% SDS at 55°C (30 min) and once with 1 × SSC+0.1% SDS at 55°C (30 min) and exposed to autoradiography film. The autoradiographs were quantitated using a Molecular Dynamics Personal Densitometer. The membranes were also probed for GAPDH using a 1.3 kbp chicken cDNA probe (Dugaiczyk et al., 1983) and hybridized as described above. To control for loading differences, cyclin D1 mRNA levels were normalized against the levels of GAPDH mRNA.
Materials
All chemicals were of analytical grade or higher. The compounds used and their sources were as follows: essentially fatty acid-free bovine serum albumin fraction V (BSA), L-glutamine, thrombin (bovine plasma), anti-smooth muscle myosin (mouse monoclonal), HEPES buffer, leupeptin, dithiothreitol, propidium iodide, Tris, sodium deoxycholate, phenylmethylsulfonylfluoride (PMSF), orthovanadate, β-mercaptoethanol (Sigma, U.S.A.); amphotericin B (fungizone), human recombinant basic fibroblast growth factor (Promega, U.S.A.); collagenase type CLS 1, elastase (Worthington Biochemical, U.S.A.); Dulbecco's Modified Eagle's Medium (Flow Laboratories, U.K.); Dulbecco ‘A' phosphate buffered saline (Oxoid, U.K.); foetal calf serum, monomed A, penicillin-G, versene, streptomycin, trypsin (CSL, Australia); Hybond™-C supernitrocellulose membranes, α-32P-dCTP (3 mCi mmol−1), [6-3H]-thymidine (5 mCi mmol−1), [γ32P]-ATP (3 mCi mmol−1), enhanced chemiluminescence reagents, Biotrak™ ERK (p42/p44 MAP kinase) enzyme assay kit, Hyperfilm MP, (Amersham, Cardiff, U.K.); anti-smooth muscle α-actin antibody (mouse monoclonal (M851)) (Dako Corporation, U.S.A.); sheep anti-rabbit IgG horseradish peroxidase-conjugated antibody (Silenus Laboratories, Melbourne, Australia); PD 98059, phospho-specific p42/p44 ERK antibody (rabbit polyclonal) (New England Biolabs, U.K); sodium hydroxide, (Biolab Scientific, Australia); RNaseII (Boehringer, Australia); dimethylsulphoxide (DMSO), EDTA, acetone, sodium dodecyl sulphate (SDS), glycine, methanol, Tween-20 (BDH, U.K.); endothelin-1 (Auspep, Australia); Aprotinin, (Bayer, Germany); X-omat AR film (Kodak, Australia); Triton X-100, sodium chloride, magnesium chloride, glycerol (Ajax, Australia); anti-cyclin D1 antibody (rabbit polyclonal IgG) (Upstate Biotechnology, NY, U.S.A.); HRP-conjugated anti-rabbit IgG secondary antibody (Silenus, Australia); P81 filter paper (Whatmann Int., U.K.); anti-ERK antibody (goat polyclonal IgG, C-16) (Santa Cruz, U.S.A.); 4-(2-aminoethyl)-benzene sulphonyl fluoride hydrochloride (Pefa Bloc) (Boehringer Mannheim, Germany); protein-G Sepharose (Pharmacia Biotech, Sweden); Trizol™ reagant, myelin basic protein (Gibco BRL, Australia); Dynabeads, oligo (dT)25 (Dynal, Oslo, Norway); Immobilon-Ny+ nylon membranes (Millipore, U.S.A).
PD 98059, initially dissolved in 100% DMSO v v−1 at 50 mM, was diluted 1 in 5 with DMSO to produce a solution of 10 mM. The final concentration of 30 μM PD 98059 in medium resulted in a final concentration of 0.3% DMSO. Cells not incubated with PD 98059 were incubated with 0.3% DMSO to account for the possible effect of DMSO on DNA synthesis. Growth factors were prepared in BSA (0.25% w v−1 in PBS).
Statistical analysis of results
All incubations for the [3H]-thymidine incorporation assays were conducted in duplicate or quadruplicate. Experiments were carried out in at least three cell lines each derived from at least three different individuals, as specified. Results are presented as the mean value±standard error (s.e.m.); n represents the number of cell lines each from a single different donor. To minimize the influence of variability between tissue donors on comparisons of data, some values have been expressed as a fold increment of the response in control cells, or as a percentage of the response in the presence of thrombin, in each experiment, with each different culture. Fold increments were calculated by dividing the response of treated cells by the response of control cells from the same 24 well plate stimulated by Monomed A (1%) alone. Grouped data were analysed by ANOVA with Dunnet's post hoc paired comparisons to identify individual differences between responses in control cells and responses in cells stimulated with thrombin, bFGF or ET-1 in the presence and absence of PD 98059. Differences were considered to be statistically significant when the 2-tailed probability was less than 0.05. All statistical analyses were performed using Graphpad Prism for Windows (Version 2.01).
Results
Thrombin and bFGF are mitogenic for human airway smooth muscle
As the concentration of mitogens in inflamed airways is likely to be variable, we have contrasted the effect of endothelin-1 (100 nM) and near-maximal or supramaximal concentrations of bFGF (300 pM and 3 nM) and thrombin (0.3 and 3 u ml−1). These concentrations were selected from concentration-response curves for the synthesis of DNA in cultured human airway smooth muscle, measured by [3H]-thymidine incorporation 24–28 h after mitogen stimulation in the presence of the insulin, transferrin and selenium-containing serum-free medium supplement (data not shown).
Basic fibroblast growth factor was the most powerful stimulant of S phase entry with a maximum fold increase of 16.8±3.1 in response to 3 nM bFGF and 16.4±3 in response to 0.3 nM bFGF (pEC50=10.5±0.2, n=11). Thrombin stimulated maximum fold increases of 9.7±2.9 in response to 3 u ml−1, and 8.6±2.7 in response to 0.3 u ml−1 (pEC50 u ml−1=1.7±0.5, n=6) and 100 nM ET-1 was the least active stimulating a fold increase of 3.6±0.1 (n=8) over control levels. Each of these stimuli elicited significantly greater [3H]-thymidine incorporation than that induced by the serum-free medium supplement alone (control cells). Control responses were 23±5% of the response to thrombin (3 u ml−1). ET-1 was used at 100 nM in this study, based on the work of Tomlinson et al. (1994) and Panettieri et al. (1996).
Flow cytometry of propidium iodide-stained cells and cell counts, measured after 48 h of mitogen stimulation, were used to examine the extent of cell cycle progression after incubation with thrombin, bFGF or ET-1. Analysis by FACS indicated that the cell population was still cycling after 48 h incubation with thrombin or bFGF, as indicated by the maintenance of an increase of the proportion of cells in S phase of the cell cycle. A small proportion of control cells are also identified in S phase probably as a consequence of Monomed A in the media. ET-1 did not stimulate detectable increases in S phase entry (Figure 1a,b). The mitogenicity of both thrombin and bFGF was confirmed by significant increases in cell number, whereas ET-1 was inactive (Figure 1c).
Figure 1.
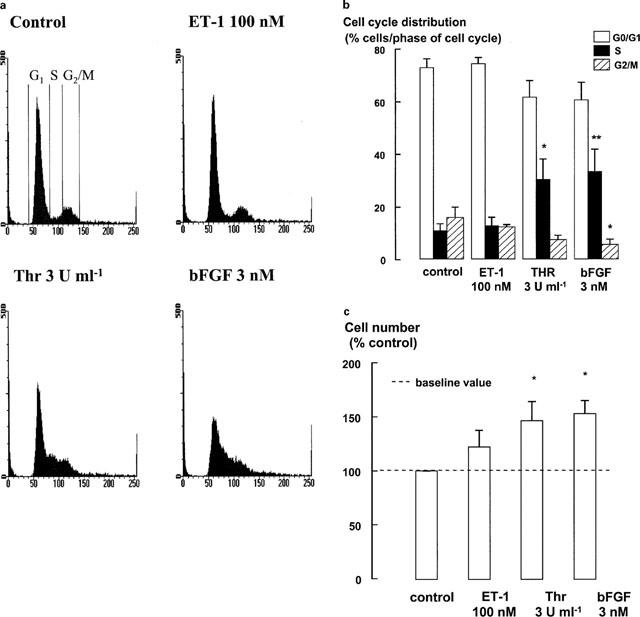
FACS analysis and cell counts were performed to measure the extent of cell cycle progression. The media was changed and bFGF, thrombin (Thr) or ET-1 were re-added after 24 h in preparation for FACS analysis and cell counts after 48 h. Monomed A was added to all cells (including control cells) at the time of mitogen addition. (a) Representative flow cytometric DNA content profiles. DNA staining by propidium iodide (DNA content) is plotted on the x-axis. The cell number is plotted on the y-axis. As indicated on the histogram profile of the control cells for each treatment, the proportion of cells in G1 is represented between 50–60 on the x-axis; the proportion of cells in the G2/M phases are represented between 100–120 on the x-axis; and the events in the area between the two peaks represent the proportion of cells in S phase. (b) FACS data are expressed as a percentage of the number of cells in each phase of the cell cycle. FACS values represent the mean and s.e.m. from at least four different cell lines. *P<0.05, **P<0.01, Responses are compared to the control response for each phase. Repeated measures ANOVA, followed by Dunnet's post-hoc test for multiple comparisons. (c) Cell number data represents the mean and s.e.m. of results from six different cell lines and are expressed as fold increments over the baseline number of cells. *P<0.05, Responses are compared to the number of cells treated with Monomed A (1%) alone (control). Repeated measures ANOVA, followed by Dunnet's post-hoc test was used for multiple comparisons. The mean number of control cells was 1.8±0.2 × 105 cells.
Thrombin and bFGF, but not ET-1 induce persistent increases in ERK activity
The requirement for persistent ERK activation in mitogenic responses in human airway smooth muscle cells was examined by contrasting the duration of ERK activation in cells stimulated by bFGF, thrombin and ET-1. Western blotting of the phosphorylated form of ERK demonstrated increases in ERK phosphorylation after 20 h in response to thrombin and bFGF, but not ET-1 (Figure 2). Although ERK phosphorylation has been equated to an increased amount of ERK activity (Posada & Cooper, 1992), ERK kinase activity was also measured.
Figure 2.

Increases in ERK phosphorylation measured by Western blotting after 20 h stimulation by thrombin or bFGF and Monomed A (1%) in the presence and absence of PD 98059 (30 μM) (0.3% DMSO control). Proteins from cell lysates were separated by SDS–PAGE and transferred to a nitrocellulose membrane. Proteins were labelled by Western blotting and detected by enhanced chemiluminescence (ECL). (a) Representative blots and grouped data from four experiments of identical design showing levels of ERK phosphorylation following (b) stimulation with high mitogen concentrations and (c) stimulation with low mitogen concentrations. The immunostained panels showing levels detected in response to low and high mitogen concentrations in (a) are from separate experiments. Each histogram represents the mean and s.e.m. of results from four different cell lines. *P<0.05, **P<0.01, Mitogen responses are compared to those of cells incubated with Monomed A (1%) alone. †P<0.05, Responses in cells treated with PD 98059 are compared to those of control cells treated with PD 98059. Repeated measures ANOVA, followed by Dunnet's post-hoc test was used for multiple comparisons.
Thrombin (3 u ml−1), bFGF (3 nM) and ET-1 (100 nM) each stimulated increases in ERK activity after 5 min incubation (Figure 3). However, this increase was prolonged for 2 h or more only in response to thrombin and bFGF (Figures 3 and 4). An increase in the baseline level of ERK activity in cells stimulated with Monomed A alone (control) was also evident at 2 h, compared to the activity at 5 min and 20 h (Figure 3).
Figure 3.
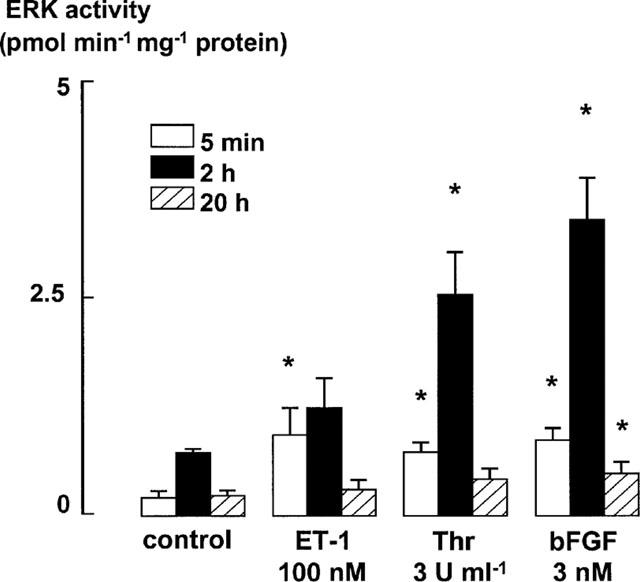
ERK activity levels in ASM incubated with bFGF (3 nM), thrombin (3 u ml−1), or ET-1 (100 nM) after 5 min, 2 h and 20 h. Monomed A (1%) was added to all cells. Increases in ERK activity in cell lysates were detected by a specific kinase assay (Amersham). Total ERK activity is expressed as nmol of 32P transferred min−1 mg−1 of protein. Each histogram represents the mean and s.e.m. of data from at least three different cell lines. *P<0.05 denotes increase in ERK activity over the control level. Repeated measures ANOVA, followed by Dunnet's post-hoc test was used to determine whether ERK activity levels in mitogen-stimulated cells were greater than those in control cells incubated with Monomed A (1%) alone at each respective time point (5 min, 2 h and 20 h). Basal ERK activities were 183±82, 704±45 and 210±58 pmol min−1 mg−1 at 5 min, 2 h and 20 h, respectively.
Figure 4.
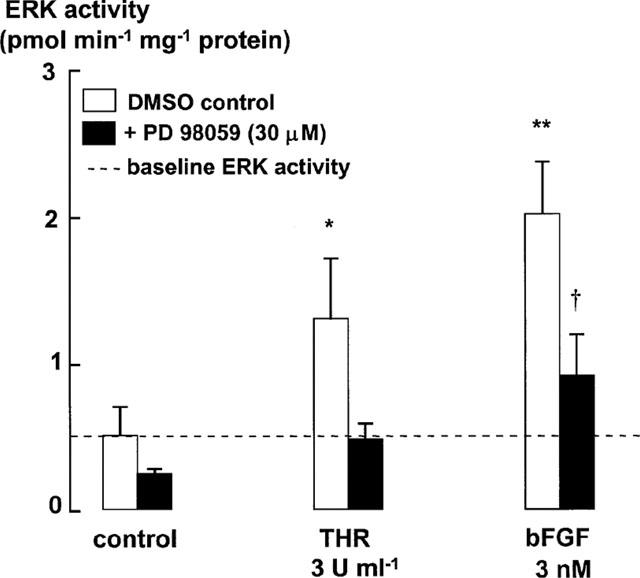
ERK activity as measured by Biotrak ERK enzyme activity assay kit (Amersham, U.K.) after 2 h stimulation by thrombin (3 u ml−1) and bFGF (3 nM). Total ERK activity is expressed as nmol of 32P transferred min−1 mg−1 protein per lysate. Each histogram represents the mean and s.e.m. of results from three different cell lines. *P<0.05, **P<0.01, mitogen-stimulated ERK activity is compared to the activity in control cells treated with Monomed A (1%) alone. †P<0.05 Mitogen-stimulated ERK activity in the presence of PD 98059 is compared to the ERK activity levels in control cells incubated with PD 98059. Repeated measures ANOVA, followed by Dunnet's post-hoc test was used for multiple comparisons. Basal ERK activity was 516±194 pmol min−1 mg−1.
MEK1 inhibition attenuates ERK activity levels and DNA synthesis
The selective MEK1 inhibitor, PD 98059 (Dudley et al., 1995), was used to further examine the role of the ERK signalling pathway for mitogen-stimulated DNA synthesis. Inhibition of MEK1 by PD 98059 decreased DNA synthesis in response to ET-1 (100 nM), thrombin (0.3 and 3 u ml−1) and the lower concentration of bFGF (300 pM). However, incubation with PD 98059 only partially inhibited DNA synthesis elicited by 3 nM bFGF (Figure 5). This partial inhibition indicated the need to confirm the extent of ERK inhibition in cells incubated with PD 98059 (30 μM).
Figure 5.

DNA synthesis stimulated by ET-1 (100 nM), thrombin (0.3 and 3 u ml−1) and bFGF (3 nM and 300 pM) for 24 h in the absence or presence of PD 98059 (30 μM), as measured by [3H]-thymidine incorporation assay. Monomed A (1%) was added to all cells. Each histogram represents the mean and s.e.m. of data from separate experiments in at least three different cell lines. *P<0.05, **P<0.01, Responses are compared to the DNA synthesis in control cells stimulated by Monomed A (1%) alone. †P<0.05, Responses are compared to the DNA synthesis in cells incubated with Monomed A and PD 98059. Repeated measures ANOVA, followed by Dunnet's post-hoc test was used for multiple comparisons.
ERK phosphorylation and activity levels were measured after incubation with ET-1, thrombin or bFGF in the presence or absence of PD 98059 (30 μM, added 30 min before the addition of mitogen). PD 98059 suppressed the phosphorylation of ERK stimulated by the higher mitogen concentrations to levels similar to those found in cells incubated with Monomed A and DMSO alone (control) (Figure 2a,b). However, these levels were higher than those measured in PD 98059-treated control cells. ERK phosphorylation stimulated by the lower concentrations of bFGF and thrombin was also prevented by PD 98059 (Figure 2a,c). The inhibition of ERK activity levels in ASM incubated with PD 98059, as measured in cell lysates (2 h) using the enzyme activity assay kit (Biotrak™ ERK enzyme assay kit, Amersham) (Figure 4), was consistent with the observed decrease in ERK phosphorylation levels (Figure 2). The Biotrak™ ERK (p42/p44 MAP kinase) enzyme assay kit uses a synthetic substrate peptide that is selective for phosphorylation by ERK. This peptide contains a single phosphorylation site (KRELVEPT669PAGEAPNALLR), which is recognized by ERK. However, it remains possible that this peptide is not phosphorylated solely by ERK. Therefore, to further confirm inhibition of ERK activity by PD 98059, we performed an immunoprecipitation of ERK, and used myelin basic protein as the ERK substrate in the subsequent kinase assay. ERK activity detected after either 5 min or 2 h incubation with bFGF (3 nM) (and Monomed A alone (1%)), was reduced to baseline levels by PD 98059 (Table 1) consistent with the results of the Biotrak™ assay kit.
Table 1.
bFGF-induced ERK activity measured by immunoprecipitation (n=3)

Mitogen-stimulated cyclin D1 levels may be dissociated from ERK activity in human airway smooth muscle
Both the near-maximal and supramaximal concentrations of thrombin or bFGF, but not ET-1, stimulated increases in cyclin D1 protein levels over 20 h, as detected by Western blotting (Figure 6). Inhibition of ERK activity by PD 98059 completely prevented the increase in cyclin D1 protein levels in cells incubated with the lower concentration of either bFGF (300 pM) or thrombin (0.3 u ml−1). However, cyclin D1 levels in cells incubated with the higher concentrations of either bFGF (3 nM) or thrombin (3 u ml−1) remained elevated after incubation with PD 98059 (Figure 6). As these results contrast with those of studies conducted either in cell lines or in primary cells from other species (Quelle et al., 1993; Baldin et al., 1993; Xiong et al., 1997; Lavoie et al., 1996; Weber et al., 1997; Ramakrishnan et al., 1998), we investigated the effect of PD 98059 on bFGF- and thrombin-stimulated cyclin D1 mRNA levels in human ASM cells, using Northern analysis. Cyclin D1 mRNA levels were increased over those in control cells following incubation with either bFGF or thrombin (Figure 7). PD 98059 had no effect on the cyclin D1 mRNA levels in cells stimulated by either of the lower or higher concentrations of thrombin or bFGF.
Figure 6.
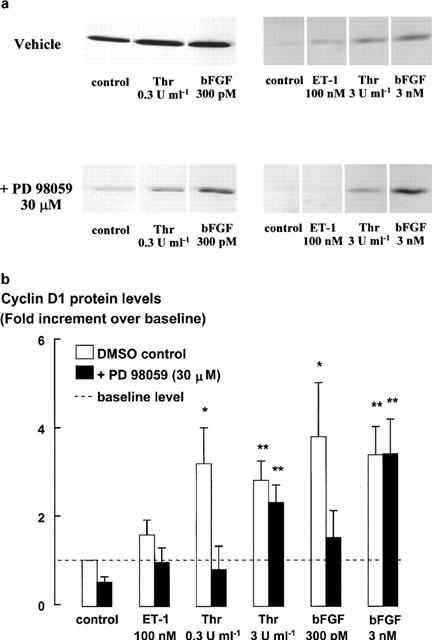
Cyclin D1 protein levels determined by Western blot in response to 20 h treatment with thrombin (0.3 and 3 u ml−1), bFGF (300 pM and 3 nM) and ET-1 (100 nM) in the absence and presence of PD 98059 (30 μM). Monomed A (1%) was added to all cells. (a) Representative blots (left-hand side and right-hand side panels are from two separate experiments) and (b) pooled data. Cyclin D1 protein levels are expressed as fold increments over the cyclin D1 levels in control cells. Each histogram is representative of the mean and s.e.m. of five different cell lines. *P<0.05, **P<0.01 Mitogen responses are compared to the cyclin D1 protein levels in control cells treated with Monomed A (1%) alone. Repeated measures ANOVA, followed by Dunnet's post-hoc test was used for multiple comparisons.
Figure 7.
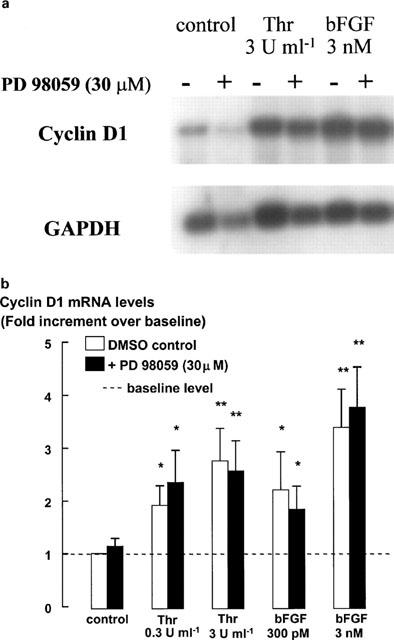
Cyclin D1 mRNA levels measured by Northern blot analysis in response to 16 h treatment with thrombin and bFGF in the absence or presence of PD 98059 (30 μM). Monomed A (1%) was added to all cells. The mRNA from cell extracts was separated on a formaldehyde denaturing gel and transferred to a nylon membrane. Cyclin D1 mRNA was hybridized with a radiolabelled DNA probe and then detected by apposition to X-ray film or phosphorimaging. (a) Representative blot of the responses to the high mitogen concentrations and (b) pooled data, expressed as the mean and s.e.m. of six experiments of identical design from at least three different cell lines. Results are expressed as a fold increment over the mRNA levels in control cells. GAPDH mRNA levels are shown as a control for loading differences (a). *P<0.05, **P<0.01 Responses are compared to the cyclin D1 mRNA levels in control cells stimulated with Monomed A (1%) alone. Repeated measures ANOVA, followed by Dunnet's post-hoc test was used for multiple comparisons (b).
Discussion and conclusion
Previous studies have demonstrated links between growth factor receptor activation and persistent ERK activation (Whelchel et al., 1997; Lenormand et al., 1993; Meloche et al., 1992; Kelleher et al., 1995), increased levels of cyclin D1 (Baldin et al., 1993; Xiong et al., 1997; Lavoie et al., 1996; Weber et al., 1997), retinoblastoma phosphorylation (Chellappan et al., 1991; Kato et al., 1993; Lundberg & Weinberg, 1998) and/or DNA synthesis in various cell types (Tomlinson et al., 1994; Panettieri et al., 1995; Karpova et al., 1997; Harris et al., 1997). We have contrasted the signalling pathways activated by the mitogens thrombin and bFGF, and the non-mitogen ET-1 in human airway smooth muscle to examine the association between persistent ERK activity and DNA synthesis. Our data suggest that pathways signalling cell cycle progression, in addition to those leading to ERK-dependent elevation of cyclin D1 protein levels, are recruited upon exposure to powerful mitogens.
Thrombin and bFGF stimulated mitogenesis (Tomlinson et al., 1995; Panettieri et al., 1995; Stewart et al., 1995a), whereas ET-1 stimulated approximately 2 fold increases in DNA synthesis in cultured human airway smooth muscle cells without increasing cell number. This modest increase in DNA synthesis was detectable by the highly sensitive [3H]-thymidine incorporation assay but not by FACS analysis. The increase in DNA synthesis without further detectable cell cycle progression could be indicative of the development of polyploidy, but no evidence for polyploidy was observed on FACS profiles. PD 98059 prevented the ET-1 stimulated increase in DNA synthesis suggesting that the increase was not due to cell damage. A small sub-population of the cells may be responsive to ET-1 and the minor stimulation of DNA synthesis may therefore reflect progression to S phase in that sub-population alone. ET-1 levels are increased in the bronchoalveolar lavage (BAL) fluid of asthmatic airways (Sofia et al., 1993). ET-1 (100 nM) stimulates approximately half-maximal constriction in isolated human airways (Hay et al., 1993; Henry et al., 1990) and therefore the concentration used in our study is expected to activate smooth muscle ET-1 receptors. The concentration of approximately 1 pM detected in BAL fluid of subjects with inflamed airways is five orders of magnitude less than the concentrations we used. Thus, even allowing for significant dilution and metabolism in BAL, our data argues against a direct role for ET-1 in the mitogenesis of human airway smooth muscle in vivo.
ERK activity and cyclin D1 protein accumulation have been unequivocally implicated in the signalling of mitogenesis in multiple cell types. In this study, ERK activity was detected by (a) phosphorylation of ERK detected by Western blotting (20 h), (b) ERK activity assay in cell extracts using a specific substrate (5 min, 2 h and 20 h) and (c) ERK activity assay in ERK immunoprecipitates using an MBP substrate (5 min and 2 h). Thrombin and bFGF increased ERK activity for at least 2 h and increased both cyclin D1 mRNA and protein levels, whereas ET-1 stimulated only a transient increase in ERK activity and did not increase cyclin D1 protein or mRNA levels or cell number (data not shown). The association between sustained ERK activity and mitogenesis is consistent with the requirement for sustained ERK activation for mitogenesis in CCL39 cells (Meloche et al., 1992), in which ERK activity was elevated 4 h after thrombin stimulation and in bovine tracheal smooth muscle cells (Kelleher et al., 1995) in which ERK activity was elevated for 6 h in response to PDGF. The addition of PD 98059 to thrombin-stimulated cells as late as 6 h after thrombin addition still reduces DNA synthesis, from which it is inferred that ERK activity is required for at least the first 6 h of mitogen signalling (Fernandes et al., 1999).
High basal levels of ERK activity, cyclin D1 protein levels and DNA synthesis result from stimulation by the serum-free medium growth supplement, Monomed A (comprising in part selenium, transferrin and insulin). Insulin activates signalling pathways, including the ERK cascade (Boulton et al., 1991; Avruch, 1998) and may therefore account for ‘basal' ERK activity. Although the exposure of cultured cells to the serum-free medium supplement decreases the apparent magnitude of the mitogen-induced increases in signalling through certain pathways by increasing the basal signalling, this supplement is used to provide an environment that more closely approximates that encountered in vivo and is required for cell proliferation and survival. The specific activity of ERK, measured by the amount of 32P transferred to MBP min−1 mg−1 protein is comparable to that measured in previous studies in response to PDGF in guinea-pig ASM (Conway et al., 1999).
PD 98059, a specific inhibitor of MEK1 (Dudley et al., 1995), was used to characterize the role of MEK1 and ERK regulation of cyclin D1 levels in DNA synthesis. As previously shown in PDGF-stimulated bovine ASM cells (Karpova et al., 1997), PD 98059 blocked thrombin-stimulated DNA synthesis, indicating that the activation of MEK1 is required for DNA synthesis. The inhibitory effects of PD 98059 on increases in cyclin D1 protein levels stimulated by lower concentrations of thrombin are consistent with links between persistent ERK activity, increased cyclin D1 protein levels and cell cycle progression to S phase (Lavoie et al., 1996; Weber et al., 1997; Ramakrishnan et al., 1998). However, in cells incubated with the higher concentrations of thrombin, cyclin D1 protein levels increased in the absence of a detectable increase in ERK activity. The PI(3)K-dependent signalling pathway represents an alternative pathway for the maintenance of elevated cyclin D1 protein levels, since the PI(3)K pathway prolongs the half life of cyclin D1 protein via its downstream effector, PKB (Diehl et al., 1997). PKB down regulates GSK-3β which facilitates the phosphorylation of Thr-286 in cyclin D1, the residue known to be targeted by the ubiquitin-dependent proteasome degradation pathway (Diehl et al., 1997). Thus, the PI(3)K pathway may maintain cyclin D1 levels during exposure to high mitogen concentrations independently of ERK activity. However, inhibition of PI(3)K activation, which attenuates DNA synthesis, does not affect ERK activity levels, indicating the independent regulation of the ERK and PI(3)K signalling pathways (Krymskaya et al., 1999).
Thrombin-induced increases in cyclin D1 mRNA levels were unaffected by PD 98059 suggesting that there is an ERK-independent signalling pathway for the regulation of cyclin D1 mRNA in human ASM. In contrast, in transformed cell lines ERK activity leads to stimulation of cyclin D1 promoter activity (Lavoie et al., 1996; Weber et al., 1997; Ramakrishnan et al., 1998). An association between cyclin D1 promoter activity and ERK activation has been demonstrated in several different cell types including a human adrenal cell line (Watanabe et al., 1996), a Chinese hamster lung fibroblast cell line (Lavoie et al., 1996), a human trophoblast cell line, a Mink lung epithelial cell line, a Chinese ovary fibroblast cell line (Albanese et al., 1995) and in primary bovine ASM cells (Ramakrishnan et al., 1998). Mitogen-stimulated cyclin D1 mRNA levels are reduced by PD 98059 in Chinese hamster lung fibroblasts (Weber et al., 1997) and in T-47D human breast cancer cells (Fiddes et al., 1998). Glucocorticoids reduce cyclin D1 mRNA (Fernandes et al., 1999), but β2 agonists do not (Stewart et al., 1999). Increased cyclic AMP levels appear to accelerate cyclin D1 degradation by a pathway that is sensitive to inhibition by MG132 (Stewart et al., 1999), a potent, reversible inhibitor of proteasome function (Lee & Goldberg, 1996). Thus, lower mitogen concentrations may negatively regulate cyclin D1 degradation by an inhibitory action of ERK on the proteasome degradation pathway.
The nature of the ERK-independent regulation of cyclin D1 mRNA levels in human ASM remains unclear. Cyclin D1 promoter activity may be increased by c-fos and c-jun, which make up the AP-1 complex, and c-Ets-2 (Albanese et al., 1995). The transcription factor c-Myc is also known to increase cyclin D1 mRNA and protein levels through its effect on eIF-4E and eIF-2α (Rosenwald et al., 1993, 1995). In addition, a change in the synthetic rate of cyclin D1 protein synthesis, prior to changes in cyclin D1 mRNA expression, has been demonstrated in human mammary epithelial cell lines. Regulation of the translation of cyclin D1 independent of ERK activity is mediated via the PI(3)K and Akt kinase signalling pathways (Muise-Helmericks et al., 1998). Thus, the differences in mechanisms reported to regulate cyclin D1 mRNA in different cell types raise the possibility of cell-type selective pharmacological regulation.
The species-dependent effects of EGF, a potent human ASM mitogen, but a non-mitogenic agent in bovine ASM (Tomlinson et al., 1994; Kelleher et al., 1995) and the mitogenicity of ET-1 in ovine, guinea-pig, rat and rabbit, but not human ASM (Panettieri et al., 1995; Noveral et al., 1992; Glassberg et al., 1994; Stewart et al., 1994) may be explained by differential recruitment of signalling pathways in response to the same agent, in different species. Differential recruitment of signalling pathways has also been demonstrated in response to different isoforms of mitogens in the same tissue type. In the absence of serum, PDGF-BB and PDGF-AB but not PDGF-AA stimulate human ASM mitogenesis (Hirst et al., 1996).
The results of the present study indicate that the recruitment of signalling pathways is not only dependent on mitogen type but also on mitogen concentration. The activation of ERK in human ASM appears to be largely, if not exclusively MEK1-dependent during both rapid (5 min) and sustained responses (2 h). Our data support an association between persistent increases in ERK activity, cyclin D1 protein levels and DNA synthesis stimulated by the lower concentrations of thrombin (0.3 u ml−1). In the presence of high concentrations of thrombin, MEK1/ERK-independent pathways may also be important for the induction and maintenance of cyclin D1 mRNA and protein levels. Such redundancies in the signalling pathways controlling ASM mitogenesis may limit the therapeutic potential of agents targeting the MEK1/ERK pathway alone.
Acknowledgments
This work was supported by project grants from the National Health & Medical Research Council of Australia and Glaxo Wellcome (U.K.). We would like to thank Dr Darren Fernandes for assistance with the ERK immunoprecipitation and subsequent kinase assay. We also thank Associate Professor John Wilson, Dr Xun Li, Mr John Bartolo and the staff from the transplant unit at the Alfred Hospital for the provision of human lung resection specimens.
Abbreviations
- AHR
airway hyperresponsiveness
- ASM
airway smooth muscle
- BAL
bronchoalveolar lavage
- bFGF
basic fibroblast growth factor
- cdk
cyclin-dependent kinase
- EGF
epidermal growth factor
- ERK
extracellular signal-regulated kinase
- ET-1
endothelin-1
- MAPK
mitogen-activated protein kinase
- MBP
myelin basic protein
- MEK1
mitogen-activated ERK kinase
- PAR
protease-activated receptor
- PBS
phosphate buffered saline
- PD 98059
[2-(2′-amino-3′-methoxyphenol)-oxanapthalen-4-one]
- PDGF
platelet-derived growth factor
- PI(3)K
phosphinositol-3′-kinase
- pRb
retinoblastoma protein
- p70S6k
p70 ribosomal S6 kinase
- Thr
thrombin
References
- ALBANESE C., JOHNSON J., WATANABE G., EKLUND N., VU D., ARNOLD A., PESTELL RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- AVRUCH J. Insulin signal transduction through protein kinase cascades. Mol. Cell Biochem. 1998;182:31–48. [PubMed] [Google Scholar]
- BALDIN V., LUKAS J., MARCOTE M.J., PAGANO M., DRAETTA G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- BORNFELDT K.E., CAMPBELL J.S., KOYAMA H., ARGAST G.M., LESLIE C.C., RAINES E.W., KREBS E.G., ROSS R. The mitogen-activated protein kinase pathway can mediate growth inhibition and proliferation in smooth muscle cells. Dependence on the availability of downstream targets. Br. J. Pharmacol. 1997;100:875–885. doi: 10.1172/JCI119603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOULTON T.G., NYE S.H., ROBBINS D.J., IP N.Y., RADZIEJEWSKA E., MORGENBESSER S.D., DEPINHO R.A., PANAYOTATOS N., COBB M.H., YANCOPOULOS G.D. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- BRAMLEY A.M., ROBERTS C.R., SCHELLENBERG R.R. Collagenase increases shortening of human bronchial smooth muscle in vitro. Am. J. Respir. Crit. Care Med. 1995;152:1513–1517. doi: 10.1164/ajrccm.152.5.7582286. [DOI] [PubMed] [Google Scholar]
- CHELLAPPAN S.P., HIEBERT S., MUDRYI M., HOROWITZ J.M., NEVINS J.R. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- CHEN R.H., SARNECKI C., BLENIS J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONWAY A.M., RAKHIT S., PYNE S., PYNE N.J. Platelet-derived-growth-factor stimulation of the p42/p44 mitogen-activated protein kinase pathway in airway smooth muscle: role of pertussis-toxin-sensitive G proteins, c-Src tyrosine kinases and phosphoinositide 3-kinase. Biochem. J. 1999;337:171–177. [PMC free article] [PubMed] [Google Scholar]
- DE S., ZELAZNY E.T., SOUHRADA J.F., SOUHRADA M. Interleukin-1 beta stimulates the proliferation of cultured airway smooth muscle cells via platelet-derived growth factor. Am. J. Respir. Cell Mol. Biol. 1993;9:645–651. doi: 10.1165/ajrcmb/9.6.645. [DOI] [PubMed] [Google Scholar]
- DE S., ZELAZNY E.T., SOUHRADA J.F., SOUHRADA M. IL-1 beta and IL-6 induce hyperplasia and hypertrophy of cultured guinea pig airway smooth muscle cells. J. Appl. Physiol. 1995;78:1555–1563. doi: 10.1152/jappl.1995.78.4.1555. [DOI] [PubMed] [Google Scholar]
- DIEHL J.A., ZINDY F., SHERR C.J. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- DUDLEY D.T., PANG L., DECKER S.J., BRIDGES A.J., SALTIEL A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUGAICZYK A., HARON J.A., STONE E.M., DENNISON O.E., ROTHBLUM K.N., SCHWARTZ R.J. Cloning and sequencing of a deoxyribonucleic acid copy of glyceraldehyde-3-phsophate dehydrogenase messenger Ribonucleic acid isolated from chicken muscle. Biochemistry. 1983;22:1605–1613. doi: 10.1021/bi00276a013. [DOI] [PubMed] [Google Scholar]
- DUNNILL M.S., MASSARELLA G.R. A comparison of the quantitative anatomy of the bronchi in normal subjects, in status asthmaticus, in chronic bronchitis, and in emphysema. Thorax. 1969;24:176–179. doi: 10.1136/thx.24.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EBINA M., TAKAHASHI T., CHIBA T., MOTOMIYA M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am. Rev. Respir. Dis. 1993;148:720–726. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- FERNANDES D., GUIDA E., KOUTSOUBOS V., HARRIS T., VADIVELOO P., WILSON J.W., STEWART A.G. Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. Am. J. Respir. Cell Mol. Biol. 1999;21:77–88. doi: 10.1165/ajrcmb.21.1.3396. [DOI] [PubMed] [Google Scholar]
- FIDDES R.J., JANES P.W., SIVERTSEN S.P., SUTHERLAND R.L., MUSGROVE E.A., DALY R.J. Inhibition of the MAP kinase cascade blocks heregulin-induced cell cycle progression in T-47D human breast cancer cells. Oncogene. 1998;16:2803–2813. doi: 10.1038/sj.onc.1201815. [DOI] [PubMed] [Google Scholar]
- GABAZZA E.C., TAGUCHI O., KOBAYASHI T., KOBAYASHI H., HATAJI O., YASUI H., ADACHI Y.Role of thrombin in airway remodeling Am. J. Respir. Crit. Care Med. 1998157A33(Abstract) [DOI] [PubMed] [Google Scholar]
- GILLE H., KORTENJANN M., THOMAE O., MOOMAW C., SLAUGHTER C., COBB M.H., SHAW P.E. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLASSBERG M.K., ERGUL A., WANNER A., PUETT D. Endothelin-1 promotes mitogenesis in airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 1994;10:316–321. doi: 10.1165/ajrcmb.10.3.7509612. [DOI] [PubMed] [Google Scholar]
- GRAMMER T.C., BLENIS J. Evidence for MEK-independent pathways regulating the prolonged activation of the ERK-MAP kinases. Oncogene. 1997;14:1635–1642. doi: 10.1038/sj.onc.1201000. [DOI] [PubMed] [Google Scholar]
- HARRIS T., RAVENHALL C.E., SCHACHTE L.C., STEWART A.G. Relationship between airway smooth muscle mitogenesis and levels of cyclin D1 and p27Kip1. Am. J. Respir. Crit. Care Med. 1997;155:A905. [Google Scholar]
- HAY D.W., HUBBARD W.C., UNDEM B.J. Endothelin-induced contraction and mediator release in human bronchus. Br. J. Pharmacol. 1993;110:392–398. doi: 10.1111/j.1476-5381.1993.tb13822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENRY P.J., RIGBY P.J., SELF G.J., PREUSS J.M.H., GOLDIE R.G. Relationship between endothlin-1 binding site densities and constrictor activities in human and animal airway smooth muscle. Br. J. Pharmacol. 1990;110:1175–1183. doi: 10.1111/j.1476-5381.1990.tb14093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRST S.J., BARNES P.J., TWORT C.H. PDGF isoform-induced proliferation and receptor expression in human cultured airway smooth muscle cells. Am. J. Physiol. 1996;270:L415–L428. doi: 10.1152/ajplung.1996.270.3.L415. [DOI] [PubMed] [Google Scholar]
- HOSSAIN S. Quantitative measurement of bronchial muscle in men with asthma. Am. Rev. Respir. Dis. 1973;107:99–109. doi: 10.1164/arrd.1973.107.1.99. [DOI] [PubMed] [Google Scholar]
- JAMES A.L., PARE P.D., HOGG J.C. The mechanics of airway narrowing in asthma. Am. Rev. Respir. Dis. 1989;139:242–246. doi: 10.1164/ajrccm/139.1.242. [DOI] [PubMed] [Google Scholar]
- JANKNECHT R., ERNST W.H., NORDHEIM A. SAP1a is a nuclear target of signaling cascades involving ERKs. Oncogene. 1995;10:1209–1216. [PubMed] [Google Scholar]
- JOHNSON S.R., KNOX A.J. Synthetic functions of airway smooth muscle in asthma. Trends. Pharmacol. Sci. 1997;18:289–292. doi: 10.1016/s0165-6147(97)01092-4. [DOI] [PubMed] [Google Scholar]
- KARPOVA A.Y., ABE M.K., LI J., LIU P.T., RHEE J.M., KUO W.L., HERSHENSON M.B. MEK1 is required for PDGF-induced ERK activation and DNA synthesis in tracheal myocytes. Am. J. Physiol. 1997;272:L558–L565. doi: 10.1152/ajplung.1997.272.3.L558. [DOI] [PubMed] [Google Scholar]
- KATO S., ENDOH H., MASUHIRO Y., KITAMOTO T., UCHIYAMA S., SASAKI H., MASUSHIGE S., GOTOH Y., NISHIDA E., KAWASHIMA H., METZGER D., CHAMBON P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- KATO J., MATSUSHIME H., HIEBERT S.W., EWEN M.E., SHERR C.J. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Development. 1993;7:331–342. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- KELLEHER M.D., ABE M.K., CHAO T.O., JAIN M., GREEN J.M., SOLWAY J., ROSNER M.R., HERSHENSON M.B. Role of MAP kinase activation in bovine tracheal smooth muscle mitogenesis. Am. J. Physiol. 1995;268:L894–L901. doi: 10.1152/ajplung.1995.268.6.L894. [DOI] [PubMed] [Google Scholar]
- KRYMSKAYA V.P., PENN R.B., ORSINI M.J., SCOTT P.H., PLEVIN R.J., WALKER T.R., ESTERHAS A.J., AMRANI Y., CHILVERS E.R., PANETTIERI R.A. Phosphatidyl 3-Kinase mediates mitogen-induced human airway smooth muscle cell proliferation. Am. J. Physiol. 1999;277:L65–L78. doi: 10.1152/ajplung.1999.277.1.L65. [DOI] [PubMed] [Google Scholar]
- L'ALLEMAIN G., STURGILL T.W., WEBER M.J. Defective regulation of mitogen-activated protein kinase activity in a 3T3 cell variant mitogenically nonresponsive to tetradecanoyl phorbol acetate. Mol. Cell Biol. 1991;11:1002–1008. doi: 10.1128/mcb.11.2.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAVOIE J.N., L'ALLEMAIN G., BRUNET A., MULLER R., POUYSSEGUR J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- LEE D.H., GOLDBERG A.L. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- LENORMAND P., SARDET C., PAGES G., L'ALLEMAIN G., BRUNET A., POUYSSEGUR J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell. Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUNDBERG A.S., WEINBERG R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARUNO K., ABSOOD A., SAID S.I. VIP inhibits basal and histamine-stimulated proliferation of human airway smooth muscle cells. Am. J. Physiol. 1995;268:L1047–L1051. doi: 10.1152/ajplung.1995.268.6.L1047. [DOI] [PubMed] [Google Scholar]
- MATSUSHIME H., QUELLE D.E., SHURTLEFF S.A., SHIBUYA M., SHERR C.J., KATO J.Y. D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELOCHE S. Cell cycle reentry of mammalian fibroblasts is accompanied by the sustained activation of p44mapk and p42mapk isoforms in the G1 phase and their inactivation at the G1/S transition. J. Cell. Physiol. 1995;163:577–588. doi: 10.1002/jcp.1041630319. [DOI] [PubMed] [Google Scholar]
- MELOCHE S., SEUWEN K., PAGES G., POUYSSEGUR J. Biphasic and synergistic activation of p44mapk (ERK1) by growth factors: correlation between late phase activation and mitogenicity. Mol. Endocrinol. 1992;6:845–854. doi: 10.1210/mend.6.5.1603090. [DOI] [PubMed] [Google Scholar]
- MULSE-HELMERICKS R.C., GRIMES H.L., BELLACOSA A., MALSTROM S.E., TSICHRIS P.N., ROSEN N. Cyclin D1 expression is controlled post-transcriptionally via a phagohatidyl-inositol 3-kinose/Akt-dependent pathway. J. Biol. Chem. 1998;273:29864–29872. doi: 10.1074/jbc.273.45.29864. [DOI] [PubMed] [Google Scholar]
- NOVERAL J.P., ROSENBERG S.M., ANBAR R.A., PAWLOWSKI N.A., GRUNSTEIN M.M. Role of endothelin-1 in regulating proliferation of cultured rabbit airway smooth muscle cells. Am. J. Physiol. 1992;263:L317–L324. doi: 10.1152/ajplung.1992.263.3.L317. [DOI] [PubMed] [Google Scholar]
- PANETTIERI R.A., GOLDIE R.G., RIGBY P.J., ESZTERHAS A.J., HAY D.W. Endothelin-1-induced potentiation of human airway smooth muscle proliferation: an ETA receptor-mediated phenomenon. Br. J. Pharmacol. 1996;118:191–197. doi: 10.1111/j.1476-5381.1996.tb15385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANETTIERI R.A., HALL I.P., MAKI C.S., MURRAY R.K. alpha-Thrombin increases cytosolic calcium and induces human airway smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 1995;13:205–216. doi: 10.1165/ajrcmb.13.2.7626288. [DOI] [PubMed] [Google Scholar]
- PANETTIERI R.A., MURRAY R.K., DEPALO L.R., YADVISH P.A., KOTLIKOFF M.I. A human airway smooth muscle cell line that retains physiological responsiveness. Am. J. Physiol. 1989;256:C329–C335. doi: 10.1152/ajpcell.1989.256.2.C329. [DOI] [PubMed] [Google Scholar]
- POSADA J., COOPER J.A. Requirements for phosphorylation of MAP kinase during meiosis in Xenopus oocytes. Science. 1992;255:212–215. doi: 10.1126/science.1313186. [DOI] [PubMed] [Google Scholar]
- PYNE N.J., TOLAN D., PYNE S. Bradykinin stimulates cAMP synthesis via mitogen-activated protein kinase-dependent regulation of cytosolic phospholipase A2 and prostaglandin E2 release in airway smooth muscle. Biochem. J. 1997;328:689–694. doi: 10.1042/bj3280689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUELLE D.E., ASHMUN R.A., SHURTLEFF S.A., KATO J.Y., BAR-SAGI D., ROUSSEL M.F., SHERR C.J. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993;7:1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- RAMAKRISHNAN M., MUSA N.L., LI J., LIU P.T., PESTELL R.G., HERSHENSON M.B. Catalytic Activation of Extracellular Signal-regulated Kinases Induces Cyclin D1 Expression in Primary Tracheal Myocytes. Am. J. Respir. Cell Mol. Biol. 1998;18:736–740. doi: 10.1165/ajrcmb.18.6.3152. [DOI] [PubMed] [Google Scholar]
- REDINGTON A.E., MADDEN J., FREW A.J., DJUKANOVIC R., ROCHE W., HOLGATE S.T., HOWARTH P. Basic fibroblast growth factor in asthma: immunolocalization in bronchial biopsies and measurement in bronchoalveolar lavage fluid at baseline and following allergen challenge. Am. J. Respir. Crit. Care Med. 1995;151:A702. [Google Scholar]
- ROSENWALD I.B., KASPAR R., ROUSSEAU D., GEHRKE L., LEBOULCH P., CHEN J.J., SCHMIDT E.V., SONENBERG N., LONDON I.M. Eukaryotic translation initiation factor 4E regulates expression of cyclin D1 at transcriptional and post-transcriptional levels. J. Biol. Chem. 1995;270:21176–21180. doi: 10.1074/jbc.270.36.21176. [DOI] [PubMed] [Google Scholar]
- ROSENWALD I.B., RHOADS D.B., CALLANAN L.D., ISSELBACHER K.J., SCHMIDT E.V. Increased expression of eukaryotic translation initiation factors eIF- 4E and eIF-2 alpha in response to growth induction by c-myc. Proc. Natl. Acad. Sci. U.S.A. 1993;90:6175–6178. doi: 10.1073/pnas.90.13.6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAPIRO P.S., EVANS J.N., DAVIS R.J., POSADA J.A. The seven-transmembrane-spanning receptors for endothelin and thrombin cause proliferation of airway smooth muscle cells and activation of the extracellular regulated kinase and c-Jun NH2- terminal kinase groups of mitogen-activated protein kinases. J. Biol. Chem. 1996;271:5750–5754. doi: 10.1074/jbc.271.10.5750. [DOI] [PubMed] [Google Scholar]
- SHERR C.J. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- SHERR C.J. D-type cyclins. Trends. Biochem. Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- SOFIA M., MORMILE M., FARAONE S., ALIFANO M., ZOFRA S., ROMANO L., CARRATU L. Increased endothelin-like immunoreactive material on bronchoalveolar lavage fluid from patients with bronchial asthma and patients with interstitial lung disease. Respiration. 1993;60:89–95. doi: 10.1159/000196180. [DOI] [PubMed] [Google Scholar]
- SPRINGALL D.R., HOWARTH P.H., COUNIHAN H., DJUKANOVIC R., HOLGATE S.T., POLAK J.M. Endothelin immunoreactivity of airway epithelium in asthmatic patients. Lancet. 1991;337:697–701. doi: 10.1016/0140-6736(91)90279-x. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., FERNANDES D., TOMLINSON P.R. The effect of glucocorticoids on proliferation of human cultured airway smooth muscle. Br. J. Pharmacol. 1995a;116:3219–3226. doi: 10.1111/j.1476-5381.1995.tb15127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEWART A.G., GRIGORIADIS G., HARRIS T. Mitogenic actions of endothelin-1 and epidermal growth factor in cultured airway smooth muscle. Clin. Exp. Pharmacol. Physiol. 1994;21:277–285. doi: 10.1111/j.1440-1681.1994.tb02513.x. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., HARRIS T., FERNANDES D.J., SCHACHTE L.C., KOUTSOUBOS V., GUIDA E., RAVENHALL C.E., VADIVELOO P., WILSON J.W. Beta2-adrenergic agonists and cAMP arrest human cultured airway smooth muscle cells in G1 phase of the cell cycle: role of proteasome degradation of cyclin D1. Mol. Pharmacol. 1999;56:1079–1086. doi: 10.1124/mol.56.5.1079. [DOI] [PubMed] [Google Scholar]
- STEWART A.G., TOMLINSON P.R., WILSON J.W.Regulation of airway wall remodelling: Prospects for the development of novel anti-asthma Drugs Advances in Pharmacology 1995bSan Diego, Academic Press; 209–254.ed. August, J.T., Anders, M.W., Murad, F. & Coyle, J.T. pp [DOI] [PubMed] [Google Scholar]
- STEWART A.G., TOMLINSON P.R., WILSON J.W. Airway wall remodelling in asthma. Anonymous Boca Raton, CRC Press inc; 1997a. Regulation of airway smooth muscle proliferation by β2-adrenoceptor agonists; pp. 295–330. [Google Scholar]
- STEWART A.G., TOMLINSON P.R., WILSON J.W. β2-adrenoceptor agonist-mediated inhibition of human airway smooth muscle cell proliferation: importance of the duration of β2-adrenoceptor stimulation. Br. J. Pharmacol. 1997b;121:361–368. doi: 10.1038/sj.bjp.0701128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMLINSON P.R., WILSON J.W., STEWART A.G. Inhibition by salbutamol of the proliferation of human airway smooth muscle cells grown in culture. Br. J. Pharmacol. 1994;111:641–647. doi: 10.1111/j.1476-5381.1994.tb14784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMLINSON P.R., WILSON J.W., STEWART A.G. Salbutamol inhibits the proliferation of human airway smooth muscle cells grown in culture: relationship to elevated cAMP levels. Biochem. Pharmacol. 1995;49:1809–1819. doi: 10.1016/0006-2952(94)00532-q. [DOI] [PubMed] [Google Scholar]
- VLAHOS R., STEWART A.G. Interleukin-1α and tumour necrosis factor-α modulate airway smooth muscle DNA synthesis by induction of cyclo-oxygenase-2: inhibition by dexamethasone and fluticasone propionate. Br. J. Pharmacol. 1999;126:1315–1324. doi: 10.1038/sj.bjp.0702424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALKER T.R., MOORE S.M., LAWSON M.F., PANETTIERI R.A.J., CHILVERS E.R. Platelet-Derived Growth Factor-BB and Thrombin Activate Phosphoinositide 3-Kinase and Protein Kinase B: Role in Mediating Airway Smooth Muscle Proliferation. Mol. Pharmacol. 1998;54:1007–1015. doi: 10.1124/mol.54.6.1007. [DOI] [PubMed] [Google Scholar]
- WATANABE G., LEE R.J., ALBANESE C., RAINEY W.E., BATLLE D., PESTELL R.G. Angiotensin II activation of cyclin D1-dependent kinase activity. J. Biol. Chem. 1996;271:22570–22577. doi: 10.1074/jbc.271.37.22570. [DOI] [PubMed] [Google Scholar]
- WEBER J.D., RABEN D.M., PHILLIPS P.J., BALDASSARE J.J. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem. J. 1997;326:61–68. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHELCHEL A., EVANS J., POSADA J. Inhibition of ERK activation attenuates endothelin-stimulated airway smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 1997;16:589–596. doi: 10.1165/ajrcmb.16.5.9160841. [DOI] [PubMed] [Google Scholar]
- WIGGS B.R., BOSKEN C., PARE P.D., JAMES A., HOGG J.C. A model of airway narrowing in asthma and in chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1992;145:1251–1258. doi: 10.1164/ajrccm/145.6.1251. [DOI] [PubMed] [Google Scholar]
- XIONG Y., CONOLLY T., FUTCHER B., BEACH D. Human D-type cyclin. Cell. 1991;65:691–699. doi: 10.1016/0092-8674(91)90100-d. [DOI] [PubMed] [Google Scholar]
- XIONG W., PESTELL R.G., WATANABE G., LI J., ROSNER M.R., HERSHENSON M.B. Cyclin D1 is required for S phase traversal in bovine tracheal myocytes. Am. J. Physiol. 1997;272:L1205–L1210. doi: 10.1152/ajplung.1997.272.6.L1205. [DOI] [PubMed] [Google Scholar]