

Abstract
The aims of this study were to (a) determine whether inducible nitric oxide synthase (iNOS) is expressed in small mesenteric arteries from rats with chronic heart failure (CHF), (b) investigate the functional significance of this potential source of nitric oxide (NO) on vascular responsiveness and (c) investigate the role that superoxide plays in modulating vascular function in these arteries.
CHF was induced in male Wistar rats by coronary artery ligation (CAL). In sham-operated rats the ligature was not tied but pulled under the artery. Six weeks after surgery CAL rats had left ventricular (LV) infarctions and elevated LV end-diastolic pressures.
Immunoreactive iNOS was found in endothelial cells, vascular smooth muscle cells and in the adventitia of small mesenteric arteries from CAL rats but not those from sham-operated rats.
Third order mesenteric arteries (300–350 μm) were mounted in a small vessel pressure myograph. Endothelium-intact arteries from CAL rats were more responsive to phenylephrine (PE) than arteries from sham-operated rats (pD2 value, CAL, 6.2±0.1; sham-operated, 5.9±0.1, P<0.05).
Both the selective iNOS inhibitor, N-(3-(Aminomethyl) benzyl) acetamidine dihydrochloride (1400W; 10−6 M) and the superoxide dismutase mimetic, Mn [III] tetrakis [1-methyl-4-pyridyl] porphyrin, (MnTMPyP; 10−4 M) reversed the hyperesponsiveness (pD2 values, 1400W, 5.9±0.1; MnTMPyP, 5.81±0.1, P<0.05). The NOS substrate, L-arginine (10−3 M), reduced responsiveness of endothelium-denuded small mesenteric arteries from CAL rats (P<0.01). None of these drugs altered responses to PE in arteries from sham-operated rats.
In summary, this study demonstrates that iNOS is expressed in mesenteric arteries from rats with CHF. However, instead of generating large quantities of NO, iNOS appears to be generating superoxide, perhaps because of a deficiency in its substrate, L-arginine. Increased superoxide generation from iNOS contributes to the hyperesponsive nature of endothelium-intact small mesenteric arteries from rats with CHF.
Keywords: Nitric oxide, inducible nitric oxide synthase, superoxide, L-arginine, endothelium, rat mesenteric arteries, heart failure
Introduction
Endothelium-derived nitric oxide (NO) plays a vital role in the regulation of vasomotor tone and arterial blood pressure (for review see Moncada et al., 1991). Evidence is accumulating to suggest that impaired NO-mediated vasodilatation contributes to increased peripheral vascular resistance associated with chronic heart failure (CHF). In clinical and experimental CHF it has been established that endothelium-dependent relaxation in response to acetylcholine is impaired (Drexler et al., 1992; Ramsey & Jones, 1994; Bauersachs et al., 1999). In contrast, data on basal release of NO in CHF is controversial. Some studies suggest that basal release of NO is preserved or may even be enhanced in CHF (Drexler et al., 1992; Habib et al., 1994; Winlaw et al., 1994), while other studies show that it is impaired (Elsner et al., 1991; Teerlink et al., 1994). The aetiology of endothelial dysfunction associated with CHF is unclear and is likely to be complex.
The inducible isoform of nitric oxide synthase (iNOS) is expressed in the vasculature in response to inflammatory cytokines and pathogens (Moncada et al., 1991). Inflammatory cytokines such as tumour necrosis factor-α are elevated in the plasma of patients with CHF (Katz et al., 1994). Furthermore, iNOS has been reported by some investigators to be present in the myocardium (Fukuchi et al., 1998; Vejlstrup et al., 1998) and skeletal muscle (Adams et al., 1997) of patients with CHF. The role of iNOS in the vasculature has not been directly addressed in CHF. It has been suggested, however, that iNOS may be responsible for increased basal production of NO in CHF (Carville et al., 1998).
Irrespective of its source, increased basal production of NO should counteract compensatory constrictor mechanisms and thus reduce peripheral vascular resistance. Increased superoxide production is associated with CHF (Belch et al., 1991). Superoxide scavenges NO (Gryglewski et al., 1986), therefore, a reduction in NO bioavailability may well explain why peripheral vascular resistance remains elevated in CHF despite a preserved or enhanced basal release of NO.
The aims of the present study were to determine if iNOS is expressed in isolated small mesenteric arteries from a rat model of CHF, to determine the functional significance of this potential source of NO on vascular responsiveness and to investigate the role that superoxide plays in modulating vascular function in these arteries.
Methods
Coronary artery ligation model of CHF
All experiments were carried out in accordance with the Animals (Scientific Procedures) Act 1986. Myocardial infarction was induced in 5 week old male Wistar rats (250–300 g, Charles River) by coronary artery ligation (CAL), as previously described (Pfeffer et al., 1979). In brief, rats were anaesthetized (sodium pentobarbitol; 60 mg kg−1, i.p.), the thoracic cavity was opened and the heart exteriorized. Using a silk ligature, the left anterior descending coronary artery was tied. In control sham-operated rats, the ligature was not tied but pulled through under the coronary artery. After closure of the chest, animals were allowed to recover and were maintained on standard chow and water ad libitum.
Six weeks after surgery, rats were anaesthetized (sodium pentobarbitol; 60 mg kg−1, i.p.), the right carotid artery was isolated and a Millar pressure transducer-tipped catheter (2F, model SPR 407, Millar Instruments, Inc., Houston, TX, U.S.A.) introduced into the artery for measurement of arterial blood pressure. The catheter was then advanced into the left ventricle (LV) for measurement of left ventricular end-diastolic pressure (LVEDP) as an index of left ventricular function.
Following exsanguination, the mesenteric bed was excised, the gut removed and the remaining vascular bed placed into cold, oxygenated (95% O2, 5% CO2), Krebs-Henseleit solution (composition, in mM: NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2 PO4 1.2, NaHCO3 25 and glucose 5.5) for functional studies. Sections of the mesenteric bed from both CAL and sham-operated rats were placed in 10% neutral phosphate buffered formalin for fixation. The hearts and lungs were removed and weighed. Hearts were bisected, fixed in formalin as above for detection of mature collagen in the infarcted zone and measurement of infarct size.
Immunohistochemistry
Formalin fixed mesenteric arteries from both CAL and sham-operated rats were processed in ethanol and embedded in paraffin wax. Three-μm sections were taken from blocks, dewaxed in xylene (10 min), rehydrated through a descending alcohol series (100, 90 and 70% alcohol; 3 min each) and washed in water (15 min). To expose antigenic sites, arteries were treated with 0.1% trypsin (diluted in Tris-buffered saline; pH 7.8; 15 min). Arteries were then treated with anti-iNOS monoclonal antibody for 24 h (4°C), washed in phosphate buffered saline (PBS; pH 7.6; 10 min) and treated with alkaline phosphatase-conjugated goat anti-rabbit IgG for 30 min (room temperature). To detect iNOS within arteries, sections were treated with an alkaline phosphatase substrate and red fuchsin as a chromogen (∼20 min). Negative controls were treated with an antibody of the same immunoglobulin class but not directed against the iNOS epitope. Tissues were counterstained with Harris' haematoxylin (BDH, Poole, U.K.).
Measurement of infarct size
Hearts were processed as above and 3 μm sections taken from paraffin blocks. After dewaxing and rehydration, hearts were treated with Van Gieson's stain for detection of collagen formation in infarctions. Infarct size was measured in sections as previously described (Mulder et al., 1998). Sections were placed under a CCD video camera module (Sony, U.K.) attached to a microscope with a ×20 lens. The endocardial and epicardial circumferences of the infarcted tissue and of the LV were determined with image analysis software (Imaging Associates, U.K.). Infarct size was calculated as a percentage [endocardial+epicardial circumference of the infarcted LV (mm)/endocardial+epicardial circumference (mm)] of the whole LV.
Preparation of arteries from mesenteric bed
Third order branches of the mesenteric artery (∼3 mm length, internal diameter; 300–350 μm) were dissected free of the bed and mounted between two microcannulae in a small vessel pressure myograph (Living Systems Instrumentation Inc., Burlington, U.S.A.). Arteries were constantly superfused with warm (37°C), oxygenated (95% O2, 5% CO2) Krebs-Henseleit solution. The intraluminal pressure of the vessel was raised to 60 mmHg and maintained at this level with a pressure servo unit without further intraluminal perfusion. A pressure of 60 mmHg was chosen because it has been estimated that vessels of this size would experience pressures approximately 50% of mean arterial pressure in vivo (Halpern & Kelley, 1991). Luminal diameter was measured using a video dimension analyser (Living Systems Instrumentation Inc., Burlington, U.S.A.) and a calibrated hand micrometer, when the optical dimension analyser was unable to detect differences in optical density at smaller lumen diameters.
After an equilibration period of 60 min, arteries were exposed twice in succession, separated by washout, to modified Krebs-Henseleit solution containing 60 mM KCl (equimolar replacement of NaCl by KCl) to obtain maximal constriction. Arteries were then exposed twice to a supramaximal concentration of phenylephrine (PE; 10−5 M). In all arteries the endothelial integrity was tested by measuring ACh-induced relaxation (10−5 M) of PE-induced tone (10−5 M), with maximal relaxation indicating the presence of an intact endothelium. In some arteries, the endothelium was removed by passing an air bubble through the vessel lumen by a method previously described (Falloon et al., 1993), and successful denudation verified by loss of ACh-induced relaxation (10−5 M). After sufficient washout and re-equilibration, arteries were constantly superfused in a closed system with a total volume of 30 ml warm (37°C), oxygenated (95% O2, 5% CO2) Krebs-Henseleit solution. All drugs used throughout this study were applied to this 30 ml reservoir, removing 1 ml of Krebs-Henseleit solution and replacing with the drug being added (as previously described, Mickley et al., 1997). Responses were recorded 5 min after addition of drug in order to allow time for responses to reach equilibrium. Four arteries were isolated from each CAL and sham-operated rat and the following experiments carried out in a random order.
Experimental protocols
Vascular responsiveness of arteries from coronary artery ligation and sham-operated rats
To assess vascular responsiveness of small mesenteric arteries from CAL and sham-operated rats to adrenoceptor stimulation, cumulative concentration response curves (CRC) to PE (1×10−9 to 3×10−5 M) were constructed in endothelium-intact (n=8) and endothelium-denuded (n=6) arteries. Following completion of CRCs all vessels were repeatedly washed and allowed to re-equilibrate before further experiments.
Effect of iNOS inhibition on vascular responsiveness
To investigate the role of iNOS in modulating vascular responsiveness, endothelium-intact mesenteric arteries were exposed to the iNOS inhibitor, N-(3-(Aminomethyl) benzyl) acetamidine dihydrochloride (1400W; 10−6 M, n=8) for 30 min prior to repeating cumulative CRCs to PE (1×10−9 to 3×10−5 M). At this concentration, 1400W has been shown to inhibit the activity of iNOS in isolated rat aortic rings treated with lipopolysaccharide, without modifying the release of NO from endothelial NOS (eNOS) (Garvey et al., 1997). 1400W was present in the superfusate throughout the CRC.
Effect of superoxide quenching on vascular responsiveness
To investigate the role that superoxide plays in modulating vascular responsiveness, endothelium-intact mesenteric arteries were exposed to the superoxide dismutase (SOD) mimetic, Mn [III] tetrakis [1-methyl-4-pyridyl] porphyrin (MnTMPyP; 10−4 M, n=8) for 30 min prior to repeating cumulative CRCs to PE (1×10−9 to 3×10−5 M). At a concentration of 10−4 M, MnTMPyP has been shown to protect endothelial-derived NO from destruction by superoxide in an in vitro model of oxidative stress (MacKenzie & Martin, 1998). MnTMPyP was present in the superfusate throughout the CRC.
Effect of combined iNOS inhibition and superoxide quenching on vascular responsiveness
To further investigate the role of iNOS and superoxide in modulating vascular responsiveness, endothelium-intact mesenteric arteries were exposed to the iNOS inhibitor, 1400W (10−6 M, n=8) and the SOD mimetic, MnTMPyP (10−4 M, n=8) for 30 min prior to repeating cumulative CRC's to PE (1×10−9 to 3×10−5 M). Both drugs were present in the superfusate throughout the CRC.
Effect of NOS substrate on vascular responsiveness
To investigate the effect of the NOS substrate, L-arginine, on vascular responsiveness, endothelium-denuded mesenteric arteries were exposed to a supramaximal concentration (Schott et al., 1993) of L-arginine (10−3 M, n=6) for 30 min prior to repeating cumulative CRCs to PE (1×10−9 to 3×10−5 M). L-arginine was present in the superfusate throughout the CRC.
Assessment of endothelium-dependent relaxations in arteries from coronary artery ligation and sham-operated rats in the presence and absence of the SOD-mimetic, MnTMPyP
To assess endothelium-dependent relaxations in mesenteric arteries from CAL and sham-operated rats ACh-mediated relaxations were measured in PE-constricted arteries. A submaximal concentration of PE (∼EC50), as determined from PE CRCs, was used to induce arterial tone. Cumulative CRCs to ACh (1×10−10 to 3×10−6 M, n=7) were then obtained in the absence and presence of the SOD-mimetic MnTMPyP (10−4 M, n=6).
Materials
Anti-iNOS monoclonal antibodies were obtained from Affiniti Laboratories (Exeter, U.K.) and alkaline phosphatase-conjugated goat anti-rabbit IgG from Vector Laboratories (Peterborough, U.K.). IgG antibodies were purchased from Vector Laboratories (Peterborough, U.K.). New Fuchsin Substrate System was purchased from Dako Corporation (Carpinteria, U.S.A.).
Phenylephrine hydrochloride, acetylcholine chloride and L-arginine were obtained from Sigma (Poole, U.K.), were diluted in Krebs-Henseleit solution to give a stock solution of 10−1 M and frozen (−20°C) in aliquots until use on day of experiment. 1400W (N-(3-(Aminomethyl) benzyl acetamidine dihydrochloride) was obtained from Calbiochem (Nottingham, U.K.) and subsequently diluted in saline (0.9% NaCl) under argon to give a stock solution of 10−3 M. Aliquots were then frozen (−20°C) and stored under argon. Further dilutions were made in Krebs-Henseleit solution. MnTMPyP (Mn [III] tetrakis [1-methyl-4-pyridyl] porphyrin) was obtained from Alexis (Nottingham, U.K.) and diluted in Krebs-Henseleit solution immediately prior to experimentation.
Analysis of data
All results were expressed as means±s.e.mean. Where maximal values were obtained for cumulative CRCs, the negative logarithm of the agonist concentration that results in a half-maximal constriction or relaxation (pD2) was calculated. Two-tailed t-tests, one-way analysis of variance (ANOVA) with Bonferroni multiple comparisons post-test and two-way repeated measures ANOVA were used as indicated in the text. P<0.05 was considered to be statistically significant.
Results
Effect of left coronary artery ligation
All the animals that survived the first 24 h after CAL surgery (76%) were still alive 6 weeks later. There were no significant differences between rat weights for CAL and sham-operated groups at 6 weeks after surgery (Table 1). Heart weights (corrected for body weight) for CAL rats were significantly greater than those of sham-operated rats despite the presence of large infarcts (P<0.05, two-tailed unpaired t-test, Table 1), indicating that hearts from CAL rats had undergone compensatory hypertrophy as a result of left ventricular infarction. There were no significant differences between groups with respect to lung weights and arterial blood pressures (Table 1). LVEDPs measured from CAL rats were significantly greater than those for sham-operated rats (P<0.05, two-tailed unpaired t-test, Table 1). CAL rats had left ventricular infarctions averaging 60±3% of the LV free wall. Additionally there was histological evidence for mature collagen scar formation within the infarcted zone (not shown).
Table 1.
Summary of all parameters measured from coronary artery ligation (CAL) and sham-operated (SHAM) rats

Immunohistochemistry
Immunoreactive iNOS was found in endothelial cells, vascular smooth muscle (VSM) cells and in the adventitia of small mesenteric arteries from CAL rats (Figure 1). No immunoreactivity was found in mesenteric arteries from sham-operated rats (Figure 1) or in negative controls using IgG antibodies (not shown).
Figure 1.
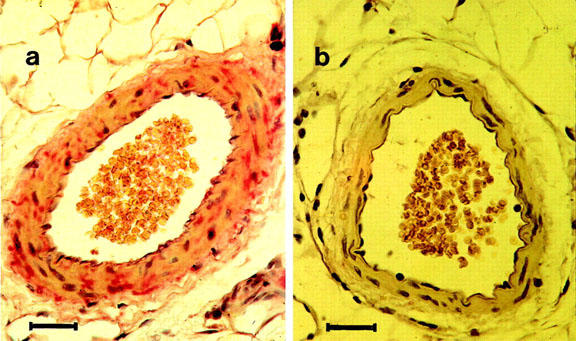
Localization of iNOS by immunohistochemical staining in mesenteric arteries from coronary artery ligation and sham-operated rats. (a) Showing immunoreactive iNOS (pink staining) in endothelial cells, vascular smooth muscle cells and in the adventitia of mesenteric arteries from rats following coronary artery ligation. (b) Showing absence of immunoreactive iNOS in arteries from sham-operated rats. Nuclei are stained with Harris' Haematoxylin (purple staining). Magnification ×400, scale bar=30 μm.
KCl, PE maximal constrictions and endothelial integrity
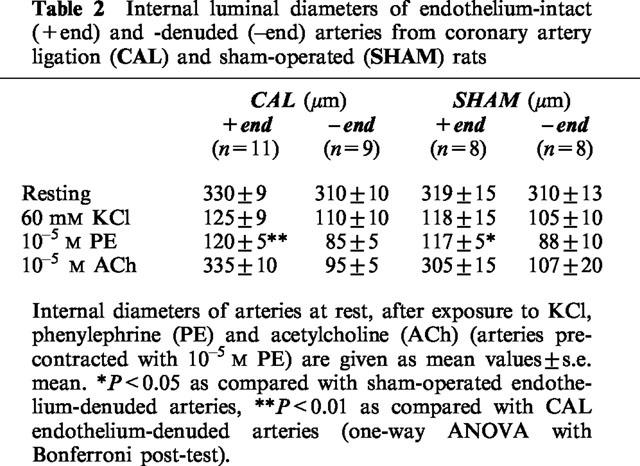
There were no significant differences between endothelium-intact and endothelium-denuded small mesenteric arteries from CAL and sham-operated groups with respect to internal diameters at rest and in response to KCl (60 mM; Table 2). However, maximal constriction to PE was significantly greater in endothelium-denuded than in endothelium-intact mesenteric arteries from both CAL and sham-operated rats (P<0.05, one-way ANOVA with Bonferroni post-test, Table 2). All endothelium-intact small mesenteric arteries studied had functional endothelium (CAL, 101.3±4%; sham-operated, 95.9±3% relaxations to ACh) and no significant differences in maximal relaxations were found between CAL and sham-operated groups (Table 2).
Table 2.
Internal luminal diameters of endothelium-intact (+end) and -denuded (–end) arteries from coronary artery ligation (CAL) and sham-operated (SHAM) rats
Assessment of vascular responsiveness to adrenoceptor stimulation in coronary artery ligation and sham-operated rats
In both endothelium-intact and endothelium-denuded small mesenteric arteries from CAL and sham-operated rats, cumulative additions of PE (1×10−9 to 3×10−5 M) resulted in concentration-dependent constrictions (Figure 2a,b). In endothelium-intact arteries from CAL rats, cumulative CRCs to PE were shifted significantly to the left when compared with responses in sham-operated groups, indicating that arteries from CAL rats were more responsive to PE than those from sham-operated rats (P<0.05, two-way ANOVA, Figure 2a). Indeed, pD2 values for PE CRCs in arteries from CAL rats were significantly greater than those in sham-operated rats (CAL, 6.2±0.1; sham-operated, 5.9±0.1, P<0.05, two-tailed unpaired t-test). There was no significant difference between maximal constrictions observed in arteries from CAL and sham-operated rats (Figure 2a).
Figure 2.
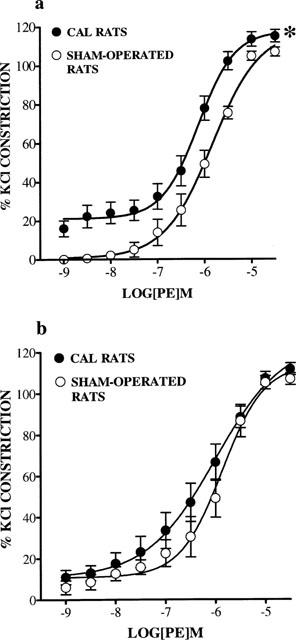
Cumulative concentration response curves (CRC) showing contractile responses to phenylephrine (PE; 1×10−9 to 3×10−5 M) in (a) endothelium-intact (n=8) and (b) endothelium-denuded (n=6) small mesenteric arteries from coronary artery ligation (CAL) and sham-operated rats 6 weeks post-surgery. Values are given as means±s.e.mean. *P<0.01 as compared with CRCs in sham-operated arteries (two-way ANOVA).
In endothelium-denuded arteries no significant differences were found between CAL and sham-operated rats with respect to PE CRCs (Figure 2b). In addition, there were no significant differences between pD2 values (CAL, 6.3±0.3; sham-operated, 5.9±0.2) and maximal % constrictions (Figure 2b).
Effect of 1400W and MnTMPyP on vascular responsiveness
Treatment of endothelium-intact arteries from CAL rats with 1400W (10−6 M) or MnTMPyP (10−4 M) shifted PE CRCs to the right with respect to responses in untreated arteries (P<0.05 for both, two-way ANOVA, Figure 3a), indicating a reduction in responsiveness to PE. pD2 values were significantly decreased in the presence of 1400W or MnTMPyP when compared with untreated arteries (untreated, 6.3±0.1; 1400W, 5.9±0.1; MnTMPyP, 5.8±0.1, P<0.05, one-way ANOVA with Bonferroni post-test). Responses to PE in arteries from CAL rats treated with 1400W or MnTMPyP were not significantly different from those in sham-operated arteries (Figure 3a). Responses to PE in endothelium-intact arteries from sham-operated rats were unaffected by 1400W (10−6 M) or MnTMPyP (10−4 M) (Figure 3b).
Figure 3.
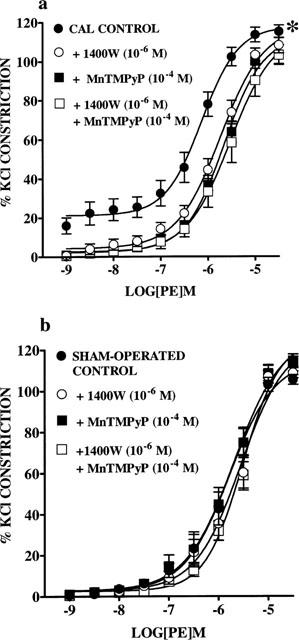
Effect of 1400W (10−6 M) and MnTMPyP (10−4 M), on their own or together on cumulative concentration response curves (CRC) to phenylephrine (PE; 1×10−9 to 3×10−5 M) in endothelium-intact small mesenteric arteries from (a) coronary artery ligation (CAL; n=8) and (b) sham-operated (n=8) rats 6 weeks post-surgery. Values are given as means±s.e.mean. *P<0.01 for CRCs in untreated arteries compared with CRCs in 1400W, MnTMPyP treated arteries from CAL rats (two-way ANOVA).
In a similar fashion to those arteries treated with 1400W (10−6 M) or MnTMPyP (10−4 M) alone, combined treatment with both drugs resulted in significant reduction in responsiveness of arteries from CAL rats to PE (P<0.01 two-way ANOVA, Figure 3a). pD2 values were significantly decreased in the presence of 1400W and MnTMPyP when compared with untreated arteries (untreated, 6.3±0.1; 1400W+MnTMPyP, 5.7±0.2, P<0.01, one-way ANOVA with Bonferroni post-test). However, no differences were found when compared with responses in arteries treated with 1400W alone. In arteries from sham-operated rats, combined treatment had no significant effect on responses to PE (Figure 3b).
Effect of L-arginine on vascular responsiveness
Treatment of endothelium-denuded arteries from CAL rats with the NOS substrate, L-arginine (10−3 M) for 30 min shifted PE CRCs to the right with respect to untreated arteries (P<0.05, two-way ANOVA, Figure 4a), indicating that in the presence of L-arginine arteries are less responsive to PE. pD2 values could not be calculated as maximal constrictions were not reached. L-arginine had no significant effect on PE responses in arteries from sham-operated rats (Figure 4b).
Figure 4.
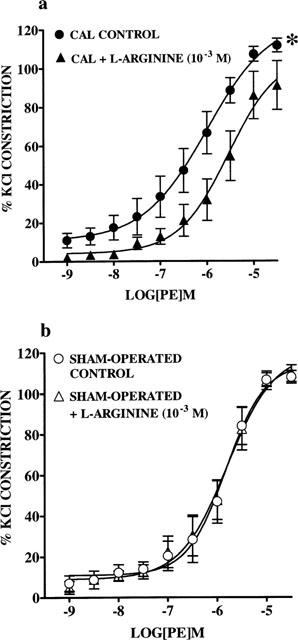
Effect of L-arginine (10−3 M) on cumulative concentration response curves (CRC) to phenylephrine (PE; 1×10−9 to 3×10−5 M) in endothelium-denuded small mesenteric arteries from (a) coronary artery ligation (CAL; n=6) and (b) sham-operated (n=6) rats 6 weeks post-surgery. Values are given as means±s.e.mean. *P<0.01 as compared with CRCs in untreated arteries (two-way ANOVA).
Endothelium-dependent relaxations: effect of superoxide quenching
Following induction of PE-induced tone (CAL, 49.1±2.3%; sham-operated, 52.6±2.4% of maximal PE constriction) in endothelium-intact arteries from CAL and sham-operated rats, cumulative additions of ACh (1×10−10 to 3×10−6 M) resulted in concentration-dependent relaxations. pD2 values for cumulative CRCs in arteries from CAL rats were significantly lower than those in sham-operated rats, indicating that endothelium-dependent relaxations were blunted in small mesenteric arteries from CAL rats (CAL, 7.4±0.1; sham-operated, 7.9±0.2, P<0.05, two-tailed unpaired t-test). Maximum relaxations to ACh were achieved in most arteries at a concentration of 3×10−6 M and no significant differences were found between CAL and sham-operated groups (CAL, 104.1±3.5%; sham-operated, 102.4±2.4%).
In the presence of MnTMPyP (10−4 M), relaxations to ACh in arteries from CAL rats were significantly enhanced, with a significant increase in pD2 values (untreated 7.4±0.1; MnTMPyP, 7.9±0.2, P<0.05, two-tailed paired t-test). Treatment of arteries from sham-operated rats with MnTMPyP had no significant effect on ACh CRCs.
Discussion
The salient finding of this study is that iNOS is expressed in mesenteric arteries from rats with CHF. However, despite the presence of iNOS, endothelium-intact small mesenteric arteries from CHF rats were found to be more responsive to phenylephrine that those from sham-operated rats. Hyperesponsiveness of endothelium-intact arteries from CAL rats was reversed by the iNOS inhibitor, 1400W and by the SOD-mimetic, MnTMPyP. Furthermore, supplementation of endothelium-denuded arteries from CAL rats with the NOS substrate resulted in a significant reduction in responsiveness to phenylephrine. These results suggest that substrate deficient, iNOS-derived superoxide may be responsible for the vascular dysfunction associated with this model of CHF.
Increased cytokine levels may serve as a stimulus for iNOS expression in CHF (Katz et al., 1994). Indeed, iNOS is expressed in the myocardium (Fukuchi et al., 1998; Vejlstrup et al., 1998) and skeletal muscle (Adams et al., 1997) in clinical CHF. In the present study, iNOS was expressed in endothelial cells, VSM cells and in the adventitia in small mesenteric arteries from rats with CHF, but not in arteries from sham-operated rats. This is the first study to demonstrate iNOS expression in the peripheral vasculature in experimental CHF. These findings are in contrast to a recent study, which failed to detect iNOS in thoracic aortae from rats 8 weeks after myocardial infarction (Bauersachs et al., 1999). Differences in vessel type may explain this discrepancy. Moreover, rats used in the present study had larger infarcts than those used in the aforementioned study, perhaps suggesting that the severity of the model influences iNOS expression.
During endotoxemia, expression of iNOS in the vasculature, and the subsequent production of NO, plays an important role in the vascular hyporeactivity to vasoconstrictors (for review see Thiemermann, 1994). Therefore, in this model of CHF, increased production of NO from iNOS would be expected to reduce vascular responsiveness to phenylephrine. Somewhat paradoxically, endothelium-intact small mesenteric arteries from CHF rats were more responsive to phenylephrine than arteries from sham-operated rats. There was no significant difference between responses to phenylephrine in endothelium-denuded arteries from CHF or sham-operated rats, demonstrating that this vascular dysfunction is dependent on the presence of the endothelium. Basal NO from eNOS serves to dampen the effects of vasoconstrictors. One could speculate, therefore, that decreased basal release of NO is responsible for the hyperesponsiveness to phenylephrine. Teerlink et al. (1994) showed increased adrenergic responsiveness of thoracic aortic rings from rats with CHF, which was dependent on the presence of the endothelium. Furthermore, their study demonstrated that impaired basal release of NO from the endothelium was responsible for this vascular dysfunction.
To investigate the role that iNOS plays in modulating vascular function in this model of CHF we used a novel selective iNOS inhibitor, 1400W, and the NOS substrate, L-arginine. 1400W is an irreversible inhibitor of iNOS that is at least 1000 fold more selective for iNOS than eNOS in rats (Garvey et al., 1997). Furthermore, 1400W is effective in abolishing iNOS activity and attenuating hypotension associated with endotoxemia in rats (Wray et al., 1998). In the present study we show, somewhat surprisingly, that inhibition of iNOS with 1400W reversed, rather than potentiated, the hyperesponsiveness of small mesenteric arteries from CHF rats. These results suggest that induction of iNOS does not result in the expected increase in NO generation. Instead, expression of the enzyme appears to be a major contributory factor in the hyperesponsive nature of endothelium-intact CHF arteries. In experimental endotoxemia addition of the NOS substrate, L-arginine, to isolated blood vessels increases NO production from iNOS and potentiates hyporesponsiveness to vasoconstrictors (Julou-Schaeffer et al., 1990). Here we show that supplementation of endothelium-denuded small mesenteric arteries from CHF rats, but not sham-operated rats, with a supramaximal concentration of L-arginine results in a reduction in responsiveness to phenylephrine, presumably through production of NO from iNOS. These results suggest that although iNOS is present in small mesenteric arteries from CHF rats, it is unable to synthesize NO because there is insufficient substrate. Recent studies have shown that iNOS, when deficient in its substrate/cofactors, produces superoxide rather than NO (Xia & Zweier, 1997; Xia et al., 1998). Therefore, in this model of CHF, it is possible that a deficiency in L-arginine will not only prevent iNOS from generating NO but will also result in the enzyme producing superoxide. Increased scavenging of NO by superoxide would result in a reduction in the bioavailability of endothelium-derived NO, which may explain the marked increase in responsiveness of endothelium-intact arteries from CHF rats to phenylephrine.
To determine if increased superoxide production plays a role in modulating vascular function in this model of CHF, we used the cell permeable metalloporphyrin SOD mimetic, MnTMPyP. In a similar fashion to 1400W, MnTMPyP reversed the hyperesponsiveness of endothelium-intact small mesenteric arteries from CHF rats. This implies that mesenteric arteries from CHF rats are indeed generating superoxide, which is responsible for the marked elevation in responsiveness to phenylephrine. Numerous studies have demonstrated increased superoxide production in CHF (McMurray et al., 1990; Belch et al., 1991; Bauersachs et al., 1999). NADH/NADPH oxidases (Bauersachs et al., 1999), autoxidation of catecholamines, increased arachidonate metabolism and inflammatory cytokines have all been suggested as possible sources of superoxide in CHF (for review see Givertz & Colucci, 1998). In the present study, however, both inhibition of iNOS and superoxide quenching reverses the hyperesponsiveness to phenylephrine suggesting that iNOS is the source of superoxide in this model of CHF. Indeed, there were no significant differences between responses to phenylephrine in arteries treated with 1400W alone and those treated with both 1400W and MnTMPyP.
Although MnTMPyP has been shown to protect SOD-null E. coli against oxidant stress (Faulkner et al., 1994), recent studies provide evidence that MnTMPyP may actually scavenge NO itself and directly inhibit the activity of eNOS (Pfeiffer et al., 1998; Mackenzie et al., 1999). Mackenzie et al. demonstrated an augmentation of phenylephrine-induced constrictions by MnTMPyP in isolated rat aortic rings that is reversed by inhibition of eNOS. However, in the present study MnTMPyP reduced responsiveness to phenylephrine in endothelium-intact small mesenteric arteries from CHF rats and had no significant effect on responses in arteries from sham-operated rats. We are convinced, therefore, that the effects of MnTMPyP in the present study are a direct consequence of its SOD-mimetic properties.
This study presents an interesting new facet to the impact of iNOS in CHF. In this model of CHF, instead of generating large quantities of NO, iNOS appears to be generating superoxide, perhaps because of a deficiency in its substrate, L-arginine. Moreover, this increase in superoxide production is responsible for the hyperesponsive nature of endothelium-intact small mesenteric arteries from rats with CHF. The precise mechanism and time course by which superoxide results in arteries being hyperesponsive is unclear from these experiments alone. However, the fact that the hypereactivity is dependent on the endothelium suggests that increased scavenging of basal, eNOS-derived NO by superoxide is responsible. In addition to impaired basal release of NO, we also show that ACh-mediated endothelium-dependent relaxations are impaired in arteries from rats with CHF. Moreover, relaxations to ACh were improved in the presence of MnTMPyP.
In summary, the present study suggests that substrate deficient, iNOS-derived superoxide is responsible for increased responsiveness of endothelium-intact mesenteric arteries, possibly as a result of impaired release of endothelial-derived NO. While it remains to be established if this study mirrors clinical CHF, our findings may represent an important mechanism for the endothelial dysfunction and raised peripheral resistance associated with CHF.
Acknowledgments
A.A. Miller is the recipient of an MRC PhD studentship. This study was supported, in part, by the British Heart Foundation (FS/95061). We gratefully acknowledge the help of Dr Pauline McEwan and Mr Lorcan Sherry in the establishment of immunohistochemical techniques.
Abbreviations
- ANOVA
analysis of variance
- CAL
coronary artery ligation
- CHF
chronic heart failure
- CRC
concentration response curve
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- LV
left ventricle
- LVEDP
left ventricular end-diastolic pressure
- MnTMPyP
Mn [III] tetrakis [1-methyl-4-pyridyl] porphyrin; N-(3-(Aminomethyl) benzyl) acetamidine dihydrochloride
- NO
nitric oxide
- PE
phenylephrine
- SOD
superoxide dismutase
- VSM
vascular smooth muscle
- 1400W
N-(3-(Aminomethyl) benzyl) acetamidine dihydrochloride
References
- ADAMS V., YU J., MOBIUS-WINKLER S., LINKE A., WEIGL C., HILBRICH L., SCHULER G., HAMBRECHT R. Increased inducible nitric oxide synthase in skeletal muscle biopsies from patients with chronic heart failure. Biochem. Mol. Med. 1997;61:152–160. doi: 10.1006/bmme.1997.2598. [DOI] [PubMed] [Google Scholar]
- BAUERSACHS J., BOULOUMIÉ A., FRACCAROLLO D., HU K., BUSSE R., ERTL G. Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression. Role of enhanced vascular superoxide production. Circulation. 1999;100:292–298. doi: 10.1161/01.cir.100.3.292. [DOI] [PubMed] [Google Scholar]
- BELCH J.J.F., BRIDGES A.B., SCOTT N., CHOPRA M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991;65:245–248. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARVILLE C., ADNOT S., SEDIAME S., BENACERRAF S., CASTAIGNE A., CALVO F., DE CREMOU P., DUBOIS-RANDÉ J.-L. Relation between impairment of nitric oxide pathway and clinical status in patients with congestive heart failure. J. Cardiovasc. Pharmacol. 1998;32:562–570. doi: 10.1097/00005344-199810000-00008. [DOI] [PubMed] [Google Scholar]
- DREXLER H., HAYOZ D., MUNZEL T., HORNIG B., JUST H., BRUNNER H.R., ZELIS R. Endothelial function in chronic congestive heart failure. Am. J. Cardiol. 1992;69:1596–1601. doi: 10.1016/0002-9149(92)90710-g. [DOI] [PubMed] [Google Scholar]
- ELSNER D., MUNTZE A., KROMER P., RIEGGER A. Systemic vasoconstriction induced by inhibition of nitric oxide synthesis is attenuated in conscious dogs with heart failure. Cardiovasc. Res. 1991;25:438–440. doi: 10.1093/cvr/25.5.438. [DOI] [PubMed] [Google Scholar]
- FALLOON B.J., BUND S.J., TULIP J.R., HEAGERTY A.M. In vitro perfusion studies of resistance artery function in genetic hypertension. Hypertension. 1993;22:486–495. doi: 10.1161/01.hyp.22.4.486. [DOI] [PubMed] [Google Scholar]
- FAULKNER K.M., LIOCHEV S.I., FRIDOVICH I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. J. Biol. Chem. 1994;269:23471–23476. [PubMed] [Google Scholar]
- FUKUCHI M., HUSSAIN S.N., GIAID A. Heterogeneous expression and activity of endothelial and inducible nitric oxide synthases in end-stage human heart failure. Their relation to lesion site and beta-adrenergic receptor therapy. Circulation. 1998;98:132–139. doi: 10.1161/01.cir.98.2.132. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J.R., KNOWLES R.G. 1400W a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- GIVERTZ M.M., COLUCCI W.S. New targets for heart-failure therapy: endothelin, inflammatory cytokines, and oxidative stress. Lancet. 1998;352 Suppl I:34–38. doi: 10.1016/s0140-6736(98)90017-4. [DOI] [PubMed] [Google Scholar]
- GRYGLEWSKI R.J., PALMER R.M.J., MONCADA S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- HABIB F., DUTKA D., CROSSMAN D., OAKLEY C.M., CLELAND J.G.F. Enhanced basal nitric oxide production in heart failure: another failed counter-regulatory vasodilator mechanism. Lancet. 1994;344:371–373. doi: 10.1016/s0140-6736(94)91402-8. [DOI] [PubMed] [Google Scholar]
- HALPERN W., KELLEY M. In vitro methodology for resistance arteries. Blood Vessels. 1991;28:245–251. doi: 10.1159/000158869. [DOI] [PubMed] [Google Scholar]
- JULOU-SCHAEFFER G., GRAY G.A., FLEMING I., SCHOTT C., PARRATT J.R., STOCLET J.C. Loss of vascular responsiveness induced by endotoxin involves the L-arginine pathway. Am. J. Physiol. 1990;259:H1038–H1043. doi: 10.1152/ajpheart.1990.259.4.H1038. [DOI] [PubMed] [Google Scholar]
- KATZ S.D., RAO R., BERMAN J.W., SCHWARTZ M., DEMOPOLOUS L., BIJOU R., LEJEMTEL T.H. Pathophysiology correlates of increased serum tumour necrosis factor in patients with congestive heart failure. Relation to nitric oxide-dependent vasodilation in the forearm circulation. Circulation. 1994;90:12–16. doi: 10.1161/01.cir.90.1.12. [DOI] [PubMed] [Google Scholar]
- MACKENZIE A., MARTIN W. Loss of endothelium-derived nitric oxide in rabbit aorta by oxidant stress: restoration by superoxide dismutase mimetics. Br. J. Pharmacol. 1998;124:719–728. doi: 10.1038/sj.bjp.0701899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACKENZIE A., FILIPPINI S., MARTIN W. Effects of superoxide dismutase mimetics on the activity of nitric oxide in rat aorta. Br. J. Pharmacol. 1999;127:1159–1164. doi: 10.1038/sj.bjp.0702670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCMURRAY J., MCLAY J., CHOPRA M., BRIDGES A., BELCH J.J. Evidence for enhanced free radical activity in chronic congestive heart failure secondary to coronary artery disease. Am. J. Cardiol. 1990;65:1261–1262. doi: 10.1016/0002-9149(90)90985-a. [DOI] [PubMed] [Google Scholar]
- MICKLEY E.J., GRAY G.A., WEBB D.J. Activation of endothelin ETA receptors masks the constrictor role of endothelin ETB receptors in isolated small mesenteric arteries. Br. J. Pharmacol. 1997;120:1376–1382. doi: 10.1038/sj.bjp.0701036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:109–141. [PubMed] [Google Scholar]
- MULDER P., RICHARD V., BOUCHART F., DERUMEAUX O., MUNTER K., THUILLEZ C. Selective ETA receptor blockage prevents left ventricle remodelling and deterioration of myocardial function in experimental heart failure. Cardiovasc. Res. 1998;39:600–608. doi: 10.1016/s0008-6363(98)00159-x. [DOI] [PubMed] [Google Scholar]
- PFEFFER M.A., PFEFFER J.M., FISHBEIN M.C., FLETCHER P.J., SPADARO J., KLONER R.A., BRAUNWALD E. Myocardial infarct size and ventricular function in rats. Circ. Res. 1979;44:503–512. doi: 10.1161/01.res.44.4.503. [DOI] [PubMed] [Google Scholar]
- PFEIFFER S., SCHRAMMEL A., KOESLING D., SCHMIDT K., MAYER B. Molecular actions of a Mn(III)Porphyrin superoxide dismutase mimetic and peroxynitrite scavenger: reaction with nitric oxide and direct inhibition of NO synthase and soluble guanylyl cyclase. Mol. Pharmacol. 1998;53:795–800. doi: 10.1124/mol.53.4.795. [DOI] [PubMed] [Google Scholar]
- RAMSEY M.W., JONES C.J. Large arteries are more than just passive conduits. Br. Heart J. 1994;72:3–4. doi: 10.1136/hrt.72.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHOTT C., GRAY G.A., STOCLET J.-C. Dependence of endotoxin-induced vascular hyporeactivity on extracellular L-arginine. Br. J. Pharmacol. 1993;108:38–43. doi: 10.1111/j.1476-5381.1993.tb13436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TEERLINK J.R., GRAY G.A., CLOZEL M., CLOZEL J.-P. Increased vascular responsiveness to norephinephrine in rats with heart failure is endothelium dependent. Dissociation of basal and stimulated nitric oxide release. Circulation. 1994;89:393–401. doi: 10.1161/01.cir.89.1.393. [DOI] [PubMed] [Google Scholar]
- THIEMERMANN C. The role of the L-arginine: nitric oxide pathway in circulatory shock. Adv. Pharmacol. 1994;28:45–79. doi: 10.1016/s1054-3589(08)60493-7. [DOI] [PubMed] [Google Scholar]
- VEJLSTRUP N.G., BOULOUMIE A., BOESGAARD S., ANDERSEN C.B., NIELSEN-KUDSK J.E., MORTENSEN S.A., KENT J.D., HARRISON D.G., BUSSE R., ALDERSHVILE J. Inducible nitric oxide synthase (iNOS) in the human heart: expression and localisation in congestive heart failure. J. Mol. Cell. Cardiol. 1998;30:1215–1223. doi: 10.1006/jmcc.1998.0686. [DOI] [PubMed] [Google Scholar]
- WINLAW D.S., SMYTHE G.A., KEOGH A.M., SCHYVENS C.G., SPRATT P.M., MACDONALD P.S. Increased nitric oxide production in heart failure. Lancet. 1994;344:373–374. doi: 10.1016/s0140-6736(94)91403-6. [DOI] [PubMed] [Google Scholar]
- WRAY G.M., MILLAR C.G., HINDS C.J., THIEMERMANN C. Selective inhibition of the activity of inducible nitric oxide synthase prevents the circulatory failure, but not the organ injury/dysfunction, caused by endotoxin. Shock. 1998;9:329–335. doi: 10.1097/00024382-199805000-00003. [DOI] [PubMed] [Google Scholar]
- XIA Y., ZWEIER J.L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6954–6958. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIA Y., ROMAN L.J. , MASTERS B.S.S., ZWEIER J.L. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J. Biol. Chem. 1998;273:22635–22639. doi: 10.1074/jbc.273.35.22635. [DOI] [PubMed] [Google Scholar]