

Abstract
In the present study we investigated a possible role for the p38 mitogen-activated protein (MAP) kinase pathway in mediating nuclear factor-kappa B (NF-κB) transcriptional activity in the erythroleukaemic cell line TF-1.
TF-1 cells stimulated with the phosphatase inhibitor okadaic acid (OA) demonstrated enhanced NF-κB and GAL4p65-regulated transcriptional activity which was associated with elevated p38 phosphorylation. However, pretreatment with the p38 MAPK specific inhibitor SB203580 (1 μM) or overexpression of kinase-deficient mutants of MKK3 or MKK6 did not affect OA-enhanced NF-κB transcriptional potency, as determined in transient transfection assays. In fact, 5 and 10 μM SB203580 enhanced rather than inhibited NF-κB-mediated promoter activity by 2 fold, which was independent of phosphorylation of the p65 subunit.
The SB203580-mediated increase in NF-κB transcriptional activity was associated with enhanced phosphorylation of extracellular signal-regulated kinase (ERK)1/2 and c-Jun N-terminal kinase (JNK), but not p38 kinase.
Overexpression of kinase-deficient mutants belonging to the ERK1/2, JNK, and p38 pathways showed that only dominant-negative Raf-1 abrogated SB203580-enhanced NF-κB activity. This would implicate the involvement of the ERK1/2 pathway in the enhancing effects of SB203580 on NF-κB-mediated gene transcription.
This study demonstrates that the p38 MAP kinase pathway is not involved in the OA-induced activation of NF-κB. SB203580 at higher concentrations activates the ERK pathway, which subsequently enhances NF-κB transcriptional activity.
Keywords: Nuclear factor kappaB, transcriptional activity, SB203580, p38 and ERK kinases, leukaemic cells
Introduction
The transcription factor nuclear factor kappa B (NF-κB) plays a crucial role in the regulation of numerous genes involved in the control of cell death (Beg & Baltimore, 1996; Wang et al., 1996; Van Antwerp et al., 1996; Beg et al., 1995; Fraser & Evan, 1996). In vitro and in vivo studies in a wide range of cell types have demonstrated that NF-κB can protect cells from programmed cell death by inducing anti-apoptotic genes and oncogenic transformation. In acute myeloid leukaemia (AML) cells and in leukaemic cell lines, constitutive or enhanced activity of NF-κB can be noticed as result of autocrine or paracrine stimulation with cytokines, such as tumour necrosis factor-α (TNF-α), interleukin-1 (IL-1β), or IL-6 (Dokter et al., 1995; Tuyt et al., 1996). Enhanced NF-κB, in turn, may activate the transcription of anti-apoptotic genes and/or of the genes for TNF-α, IL-1β and IL-6, which account for increased proliferation of leukaemic blasts (Akira & Kishimoto, 1997; Griffin et al., 1987).
NF-κB homo- or heterodimers consist of members of the rel multigene family, which is comprised of five major proteins: p50, p65 (RelA), c-rel, p52 and RelB (Akira & Kishimoto, 1997). The most abundant dimer is the p50/p65 heterodimer. Central to the activation of NF-κB are the IκB (Inhibitory κB) kinases (IKKs) (Maniatis, 1997; Stancovski & Baltimore, 1997; Zandi et al., 1997). The IKKs trigger the phosphorylation of IκB on the amino-terminal serine residues 32 and 36, upon which binding of ubiquitin then targets the phosphorylated IκB for degradation by the proteasome (Zandi et al., 1997). The released nucleophilic heterodimer then moves to the nucleus, where both p50 and p65 contribute to NF-κB DNA binding (Akira & Kishimoto, 1997). The p65 subunit is responsible for transcriptional activity, resulting in the expression of NF-κB responsive target genes. A second level of control involves phosphorylation events of NF-κB, most probably in the cell nucleus (Schmitz & Baeuerle, 1991). The kinases that regulate these processes are rather undefined. Several points of evidence suggest that mitogen-activated kinases (MAPKs) can participate in the activation of NF-κB in the cytoplasm as well as in modulation of its transactivation potential in the nucleus (Schulze-Osthoff et al., 1997). Three major subfamilies of structurally related MAPKs have been identified in mammalian cells; the extracellular signal-regulated kinase (Ras/Raf-1/MEK1/ERK1/2), the c-Jun N-terminal kinase (MEKK1/MKK4/7/JNK) and the p38 kinase (MKK3/6/p38) pathways. These MAP kinases contribute to the transmission of extracellular signals that can finally result in direct or indirect phosphorylation of various transcription factors and alterations in gene expression (Marshall, 1994).
We have recently demonstrated the involvement of the raf-1/MEK1/ERK1/2 and the MEKK1/MKK4/JNK pathways in enhancing the transcriptional activity of NF-κB in the erythroleukaemic cell line TF-1 (Tuyt et al., 1999). Other reports described the involvement of the p38 kinase pathway in mediating NF-κB-driven gene transcription. Vanden Berghe et al. (1998) found that in the mouse fibrosarcoma cell line L929s TNF-α-activated p38 kinase enhanced the transactivation of the NF-κB p65 subunit. The p38 kinase specific inhibitor SB203580 was able to suppress κB-dependent reporter gene expression without affecting the levels of NF-κB binding to DNA. The pyridinyl imidazol SB203580 binds with high affinity to p38 near the ATP binding site, thus rendering p38 inactive (Cuenda et al., 1995). In addition, Wesselborg et al. (1997) showed that inhibition of p38 by the pharmacological inhibitor SB203580 or by a dominant-negative mutant of MAP kinase kinase-6 (MKK6), an activator of the p38 pathway, interfered with NF-κB-dependent gene expression but not its DNA-binding activity.
In the present study we set out to investigate a possible role for p38 kinase in NF-κB activation in the human erythroleukaemic cell line TF-1. Pretreatment of okadaic acid (OA)- stimulated cells with the p38 specific pharmacological inhibitor SB203580, at a concentration of 1 μM, did not affect NF-κB DNA binding activity, NF-κB-driven gene transcription, nor GAL4p65-mediated promoter activity. Similarly, overexpression of kinase deficient mutants of MKK3 or MKK6, the most relevant upstream kinases from p38, did not influence NF-κB-mediated gene transcription. Rather, SB203580 at concentrations of 5 and 10 μM significantly enhanced NF-κB-driven promoter activity, which was preceded by enhanced phosphorylation of the ERK and JNK proteins as a result of treatment with high concentrations of SB203580.
Methods
Materials
RPMI 1640 media was purchased from Flow (Rockville, MD, U.S.A.), foetal bovine serum from Hyclone (Logan, UT, U.S.A.) and Lymphoprep was obtained from Nycomed (Oslo, Norway). Okadaic acid was obtained from Sigma (St. Louis, MO, U.S.A.). Radionucleotides were obtained from Amersham (Buckinghamshire, U.K.). Antibodies against phospho-p38, phospho-JNK and phospho-ERK were purchased from New England Biolabs, Inc. (Beverly, MA, U.S.A.), whereas antibodies against p38, JNK, and ERK were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). The anti-CD14 was obtained from Becton Dickinson (Sunnyville, CA, U.S.A.). The p38 MAPK inhibitor SB203580 was obtained from SmithKline Beecham Pharmaceuticals (King of Prussia, PA, U.S.A.). cDNA probe for IL-6 was kindly provided by Dr L. Aarden (Central Laboratory of the Netherlands Red Cross Blood Transfusion Service, Amsterdam, The Netherlands). Luciferase detection kit was purchased from Promega Corporation (Madison, WI, U.S.A.), the CAT Elisa kit from Boehringer Mannheim GmbH (Mannheim, Germany), and IL-3 was received from Genetics Institute (Cambridge, MA, U.S.A.). All IL-6 promoter constructs, containing various portions of the human IL-6 promoter cloned into the pGL2 luciferase reporter vector (Promega Corporation, Madison, WI, U.S.A.), were generated in our laboratory. pGAL4p65 was kindly provided by Dr P.A. Baeuerle (Tularik Inc., San Francisco, CA, U.S.A.); pGAL4dbd and pGAL4tkluc were kindly provided by Dr S. Wissink (NIOB, Utrecht, The Netherlands). pRSV-NΔRaf1 was provided by Dr P.J. Coffer (Department of Pulmonary Diseases, University Hospital Utrecht, The Netherlands). pcDNA3-Flag-JNK, pRSV-MKK3(Ala), pcDNA3-MKK4(Ala), and pcDNA3-MKK6(K82A) encoding catalytically inactive mutants of respectively JNK, MKK3, MKK4, and MKK6 were provided by Dr R.J. Davis (Howard Hughes Medical Institute, University of Massachusetts Medical School, Worchester, MA, U.S.A.). p(TRE)5CAT was a generous gift from Dr H. Van Dam (Department of Molecular Carcinogenesis, University of Leiden, The Netherlands).
Cell culture
The cell line employed in these studies is TF-1, a human erythroleukaemia cell line (Kitamura et al., 1989). TF-1 cell line was cultured in RPMI 1640 supplemented with 5% FBS and 10 ng ml−1 IL-3. Toxicity of OA was assessed by trypan blue exclusion assays.
Electrophoretic mobility shift assay (EMSA)
After overnight culture in RPMI 1640 supplemented with 5% FBS and 10 ng ml−1 IL-3, TF-1 cells were stimulated and nuclear extracts were prepared according to the mini-scale procedure described (Schreiber et al., 1989). Nuclear extracts were divided into small aliquots and stored at −80°C. Double-stranded synthetic oligonucleotide probes containing the NF-κB (NF-κB: 5′-AGCTGCGGGGATTTTCCCTG-3′) and NF-IL6 (NF-IL6: 5′-AGCTCGCGTTGTGCAATCTG-3′) consensus sequences were used in the gel retardation assay. The consensus sequences for binding of the respective nuclear factors are underlined. Fifty ng of HPLC-purified single-stranded oligonucleotide were labelled with T4-polynucleotide kinase and [γ32P]-ATP (3000 Ci mmol−1, Amersham), separated from non-incorporated radiolabel by sephadex G50 chromatography, ethanol precipitated, dried, and dissolved in 20 μl of Tris-HCl (pH 7.5) 10 mM, NaCl 50 mM, MgCl2 10 mM, EDTA 1 mM, and DTT 1 mM, containing a 4 fold excess of the opposite strand. Annealing of the two strands was performed by heating the mixture for 2 min at 90°C and slow cooling to room temperature. Five μg nuclear extract and 0.1 ng double-stranded labelled oligonucleotide were incubated in HEPES (pH 7.9) 20 mM, KCl 60 mM, EDTA 0.06 mM, DTT 0.6 mM, spermidine 2 mM, 10% glycerol, supplemented with 2 μg poly(dI-dC). The binding reaction was performed at 26°C for 25 min. Competition experiments were performed by adding a 100 fold molar excess of unlabelled self or non-self double-stranded oligonucleotides. Supershift experiments were performed by incubating the nuclear extracts and labelled oligonucleotide with polyclonal antibodies (1 μg) against the p50, p65, c-rel and p52 subunits of NF-κB (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). The samples were loaded on pre-run (30 min, 100 V) 4% (30:1) polyacrylamide gels and run for 1 h at 150 V in 0.5× TBE at room temperature. Gels were dried and exposed to Kodak XAR films at −80°C with an intensifying screen. Quantification of protein binding was performed by densitometry using a PhosphoImager (Molecular Dynamics, Sunnyvale, CA, U.S.A.).
Transient transfection assays
The luciferase reporter plasmid pIL6luc(-122) and the CAT reporter plasmid p(TRE)5CAT were transfected into TF-1 cell line by means of electroporation. Prior to transfection, cells were cultured for 16 h at a density of 0.5×106 cells ml−1 in the appropriate medium, washed twice and resuspended in RPMI 1640 at a density of 10×106 in 200 μl. When transfected with a single plasmid, 25 μg of DNA was added and the mixture was left at room temperature for 15 min. Cotransfections were performed with 15 μg of the reporter plasmid pIL6luc(-122) together with 15 μg of the dominant-negative expression plasmids (pRSV-MKK3(Ala), pcDNA3-MKK6(K82A), pRSV-NΔRaf1, pcDNA3-MKK4(Ala), pcDNA3-Flag-JNK1, or pcDNA3 (empty vector). Cotransfections of pGAL4tkluc (5 μg) with either pGAL4p65 (5 μg) or pGAL4dbd (5 μg) were performed under similar conditions. In addition, cells were cotransfected with 2 μg of a CMV-CAT plasmid, to normalize for transfection efficiency. Electroporation, in 0.4 cm electroporation cuvettes, was performed at 240 V and 960 μF with Gene Pulser electroporator (Bio-Rad Laboratories, Richmond, VA, U.S.A.). After electroporation, the cells were replated in RPMI 1640 containing 2% FBS. Six hours after transfection cells were stimulated for 24 h with medium or OA (30 ng ml−1) or SB203580 for 30 min prior to OA stimulation. The cells were then harvested and lysed by commercially available luciferase lysis buffer. One-hundred μl of lysis product was added to 100 μl of luciferase assay reagents and luciferase activity was measured with the Anthos Lucy1 luminometer (Anthos Labtec Instruments, GmbH, Salzburg, Austria). CAT reporter activity of 100 μl lysis product plus 100 μl CAT dilution buffer was determined with a commercially available CAT Elisa kit.
Western blotting for p38, JNK1/2, and ERK1/2
Phosphorylation of p38, JNK1/2, and ERK1/2 was analysed by Western blotting. Briefly, TF-1 cells were cultured for 16 h in RPMI 1640 containing 0.1% FBS and subsequently stimulated for various periods of time with medium or OA (30 ng ml−1) or SB203580 plus OA. After harvesting, total cell extracts were prepared by resuspending the cells in 500 μl 1× sample buffer (containing 2% SDS, 10% glycerol, 2% β-mercaptoethanol, 60 mM Tris-HCl (pH 6.8) and bromophenol blue) and lysing the cells by passing them through a 23G1 needle (three times). Cell extracts were directly boiled for 10 min and stored at −20°C. Before loading, samples were again boiled for 5 min and cell extracts were resolved by running 1/10th volume on a SDS/12.5%PAGE gel (acryla-mide:bisacrylamide is 173:1) and transferred to cellulosenitrate membrane (Schleicher & Schuell, Germany). Immunoblotting with the antibodies was performed by standard procedures and detection was performed according to manufacturer's guidelines (ECL, Amersham, Buckinghamshire, U.K.).
Statistical analysis
The student t-test for paired samples was used to determine statistical significance of the transfection data.
Results
OA enhances NF-κB DNA binding and transactivation potential
One tool in studies towards the role of phosphorylation and dephosphorylation in NF-κB activation is the use of specific pharmacologic inhibitors of protein phosphatases (PPases). Okadaic acid (OA), a polyether fatty acid, is a potent inhibitor of the phosphoserine and phosphothreonine phosphatases PP1 and PP2A. In a wide variety of cell lines OA has been proven to be a potent activator of NF-κB DNA binding (Cohen et al., 1990; Sung & Walters,1993).
To investigate whether OA enhances NF-κB DNA binding in the human leukaemic cell line TF1, cells were stimulated with OA for various periods of time. In unstimulated cells binding of the NF-κB complex is low (Figure 1, lane 1), whereas stimulation with OA (30 ng ml−1) for 6 h results in a distinct increase in NF-κB binding activity (Figure 1, lane 2). Exposure to OA for shorter periods of time did not enhance NF-κB binding activity (data not shown). Supershift experiments performed with antibodies specific for the p50 and p65 subunits of the NF-κB family indicated that the OA-induced NF-κB complex consisted of the p50 and p65 subunits (data not shown).
Figure 1.
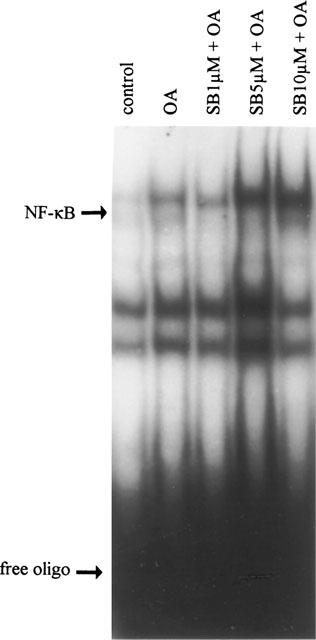
OA enhances NF-κB DNA binding. Electrophoretic mobility shift assay for the transcription factor NF-κB was performed after treatment of TF-1 cells with medium, OA (30 ng ml−1, 6 h), or SB203580 (30 min pretreatment) at 1, 5 and 10 μM plus OA (6 h). Band shift assay was performed as described in ‘Methods' utilizing 32P-labelled oligonucleotide specific for NF-κB. Excess of free probe is shown.
To confirm that NF-κB is transcriptionally active, TF-1 cells were transiently transfected with pIL6(-122)luc, a luciferase reporter construct with a minimal promoter containing a binding site for NF-κB only (Figure 2A). The pIL6 (-122)luc plasmid contains the part of the IL-6 promoter adjacent to the transcriptional start site. Results from the transfection experiments demonstrated that, in concordance with the observations at DNA binding level, OA induced activity of the NF-κB-driven reporter gene (4.5±0.6 fold, P<0.05, n=6) (Figure 2B). Transient transfection with pIL6 (-60)luc, lacking the NF-κB binding site, failed to show any induction after OA stimulation (1.2±0.1 fold) (Figure 2B).
Figure 2.
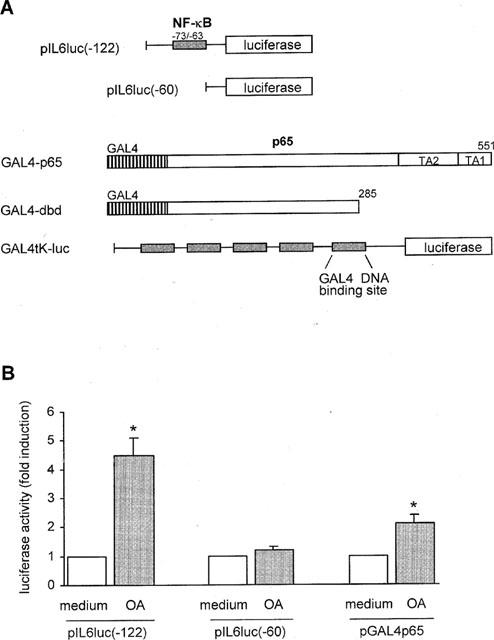
OA enhances NF-κB transcriptional activity. (A) Schematic representation of the two IL-6 promoter constructs and the pGAL4p65, pGAL4dbd, and pGAL4tkluc plasmids applied in the transfections of TF-1 cell line. Fragments of 122 (pIL6(-122)luc) and 60 (pIL6(-60)luc) basepairs, respectively, of the human IL-6 promoter are fused to the luciferase gene. Binding site for the transcription factors NF-κB is denoted above the largest construct. The Gal4-transactivator fusion proteins, pGal4p65 and pGal4dbd, are exclusively nuclear and are regulated independently of IκB. The reporter gene is under the control of five Gal4-binding sites. (B) Schematic representation of NF-κB- and p65-mediated promoter activity in the transiently transfected TF-1 cells. TF-1 cells were cultured as described in ‘Methods' and subsequently transiently transfected by means of electroporation with the respective plasmids. Six hours after transfection cells were stimulated for 24 h with medium or OA (30 ng ml−1). Twenty-four hours after stimulation cells were lysed and analysed for the amount of produced luciferase protein. Basal promoter activity for each construct when treated with medium alone is set at 1. Mean fold induction and standard error of the mean represent six or more identical experiments. *P<0.05.
The activation of NF-κB by OA is further studied by using the Gal4 ‘one hybrid' technique in eukaryotic cells. The GAL4p65 plasmid encodes a fusion of the C-terminal transactivation (TA) domain of p65 with the DNA binding domain of the yeast transcription factor GAL4 (Schmitz et al., 1995; Wissink et al., 1997). In the GAL4dbd (DNA binding domain) plasmid the TA domain of the p65 subunit is absent. The Gal4-transactivator fusion proteins, pGal4p65 and pGal4dbd, are exclusively nuclear and are regulated independently of IκB (Figure 2A). This system allows the analysis of transactivation directly mediated by the p65 subunit of NF-κB. The plasmids pGal4dbd and pGal4p65 were cotransfected with the reporter plasmid pGal4tkluc. The reporter gene is under the control of five Gal4-binding sites. The transient transfection studies demonstrated that basal transactivation by Gal4p65 can still be upregulated by OA by 2.1±0.3 fold (P<0.05, n=9) (Figure 2B), indicating that OA can activate NF-κB by directly enhancing p65 transcriptional potential. Promoter activity driven by Gal4dbd, containing a DNA binding domain but no transcriptionally active domains, was not enhanced after OA treatment.
Summarizing, these results indicate that OA is a potent activator of NF-κB, both by enhancing its translocation to the nucleus and by inducing p65-mediated transcription.
OA enhances phosphorylation of p38 kinase
The effect of OA treatment on p38 kinase activity was studied by using phospho-p38 specific antibodies. Various reports have shown that p38 phosphorylation correlates well with its kinase activity (Frantz et al., 1998). Analysis of the kinetics of p38 activation by OA revealed that enhanced phospho-p38 was first detectable after 30 min of stimulation (data not shown). Maximal phospho-p38 levels were observed at 90 min of stimulation (Figure 3).
Figure 3.
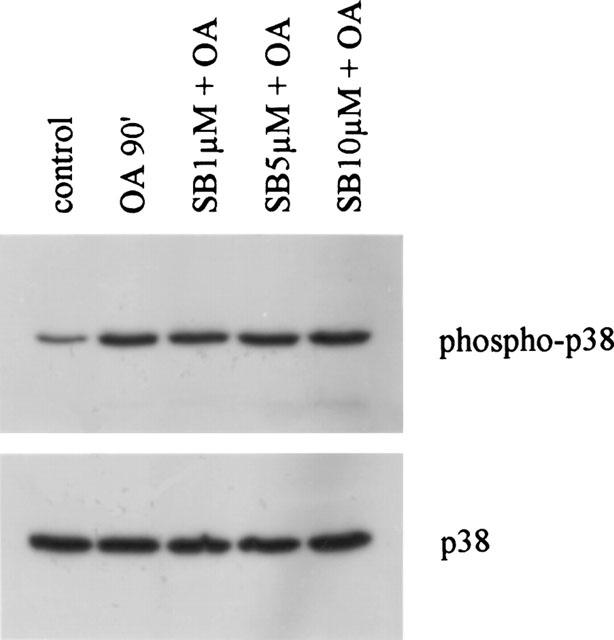
OA enhances phosphorylation of p38 MAP kinase. TF-1 cells were cultured for 16 h in RPMI 1640 containing 0.1% FBS and subsequently stimulated with medium, OA (30 ng ml−1) for 90 min, or SB203580 (30 min pretreatment) at the indicated concentrations. Cell extracts were prepared as described in detail in ‘Methods'. Phosphorylated p38 is depicted in the upper panel. Total p38 protein is shown in the lower panel and represents equal loading. Immunoblotting with anti-phospho-p38 and anti-p38 antibodies was performed by standard procedures and detection was performed using ECL immunodetection kit.
SB203580 at 1 μM and kinase-deficient MKK3 and MKK6 do not affect NF-κB transactivation potential
To analyse a possible correlation between p38 MAPK and NF-κB activation, pIL6(-122)luc-transfected TF-1 cells were pretreated with the p38 kinase specific inhibitor SB203580 prior to OA stimulation (Cuenda et al., 1995). SB203580 was utilized at a concentration of 1 μM, which was previously shown to greatly inhibit p38 kinase activity in TF-1 cells (Birkenkamp et al., 1999).
Results demonstrated that SB203580 did not affect the OA-induced NF-κB-driven promoter activity (5.1±1.1 fold for SB203580 plus OA versus 4.5±0.6 fold for OA, n=6) (Figure 4A). To support these results TF-1 cells were simultaneously transfected with pIL6(-122)luc and a plasmid overexpressing an inactive mutant of MKK3 (pRSV-MKK3(Ala)) or an inactive mutant of MKK6 (pcDNA3-MKK6(K82A)) (Raingeaud et al., 1996). Both MKK3 and MKK6 are relevant upstream kinases from the p38 MAP kinase. As a negative control cells were cotransfected with the empty plasmid pcDNA3. Blocking the p38 kinase pathway with pRSV-MKK3(Ala) or pcDNA3-MKK6(K82A) did not affect the NF-κB-mediated promoter activity (Figure 4B).
Figure 4.
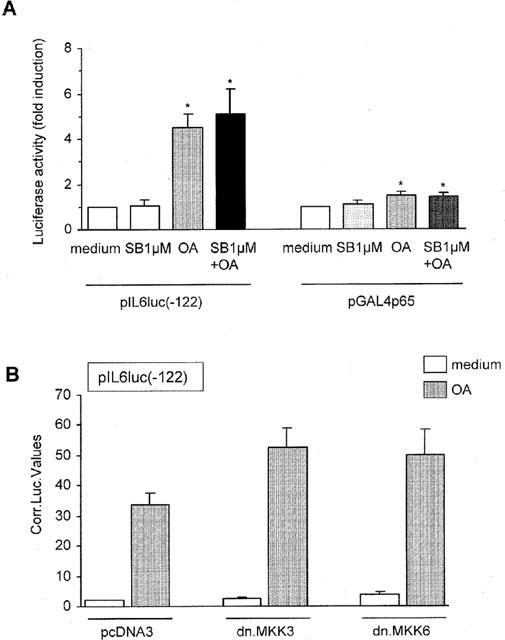
SB203580 at 1 μM and kinase deficient MKK3 and MKK6 do not affect NF-κB transactivation potential. (A) TF-1 cells were transiently transfected as described in ‘Methods' with pIL6luc(-122) or pGAL4p65. Six hours after electroporation cells were stimulated for 24 h with medium, SB203580 (1 μM), OA (30 ng ml−1), or SB203580 (1 μM, 30 min pretreatment) plus OA. Twenty-four hours after stimulation cells were lysed and analysed for the amount of produced luciferase protein. Promoter activity after treatment with medium is set at 1. Mean fold induction and standard error of the mean represent six identical experiments. *P<0.05. (B) TF-1 cells were transiently transfected as described in ‘Methods' with 15 μg pIL6luc(-122) together with 15 μg (1) pcDNA3 empty vector, (2) pRSV-MKK3(Ala), or (3) pcDNA3-MKK6(K82A) and 2 μg of a CMV-CAT construct. Six hours after electroporation cells were stimulated for 24 h with medium or OA (30 ng ml−1). Twenty-four hours after stimulation cells were lysed and analysed for the amount of produced luciferase protein. The IL-6 promoter activity is represented in arbitrary luciferase units corrected for transfection efficiency. Mean and standard error of the mean represent at least three identical experiments.
Identical results were obtained when TF-1 cells were transfected with pGal4p65 and pGal4tkluc. Blocking the p38 kinase pathway by pretreatment with SB203580 at 1 μM failed to affect the promoter activity mediated by the p65 subunit of NF-κB (Figure 4A). Effectivity of pRSV-MKK3(Ala) and pcDNA3-MKK6(K82A) was demonstrated by cotransfections of the inactive mutants with pIL6(-602)\NF-κB, a luciferase reporter driven by the full length IL-6 promoter with a mutated NF-κB binding site. OA-mediated promoter activity was suppressed 1.9 and 3.3 fold, respectively, when either pRSV-MKK3(Ala) or pcDNA3-MKK6(K82A) was introduced into the TF-1 cells (representative of two independent experiments, data not shown).
The lack of involvement of the p38 pathway in the activation of NF-κB is reflected at the level of DNA binding. Nuclear extracts from TF-1 cells treated with OA, or treated with SB203580 plus OA, did not show any difference in DNA binding activity of NF-κB (Figure 1, lane 3). Alternatively, DNA binding of the transcription factor NF-IL6 is inhibited due to suppression of p38 kinase activity (Figure 5), confirming that SB203580 at a concentration of 1 μM is sufficient for inhibiting p38 kinase activity.
Figure 5.
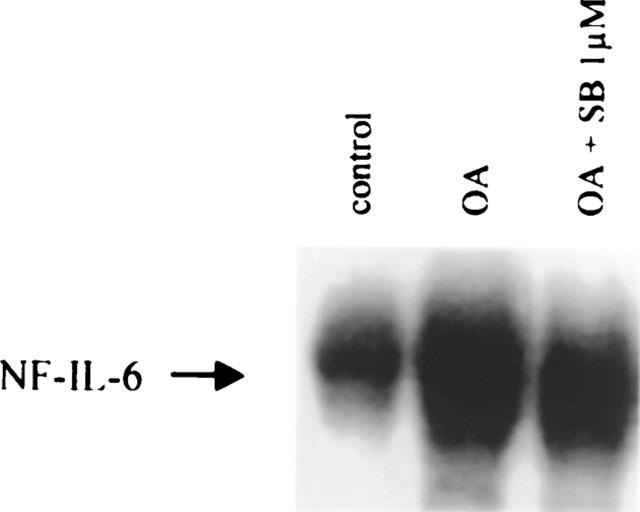
SB203580 at 1 μM inhibits NF-IL6 DNA binding activity. Electrophoretic mobility shift assay for the transcription factor NF-IL6 was performed after treatment of TF-1 cells with medium, OA (30 ng ml−1, 6 h), and SB203580 at 1 μM (30 min pretreatment) plus OA. Band shift assay was performed as described in ‘Methods' utilizing 32P-labelled oligonucleotide specific for NF-IL6.
Together, these data strongly suggest an absence of involvement of the p38 MAP kinase pathway in mediating activation of NF-κB, both at the DNA-binding level and at the transcriptional level.
SB203580 at 5 or 10 μM enhances NF-κB-mediated promoter activity, but not GAL4p65-driven promoter activity
In many reports concerning various cell types, SB203580 is applied at concentrations of 5 and 10 μM, at which it demonstrates considerable inhibitive effects (Bergmann et al., 1998; Cuenda et al., 1997). To rule out that the lack of effect of SB203580 is due to inadequate concentrations, pIL6(-122)luc-transfected cells were pretreated with 5 and 10 μM SB203580, prior to OA stimulation. Surprisingly, exposure to SB203580 at these concentrations enhanced rather than inhibited NF-κB DNA binding (Figure 1) as well as NF-κB-mediated gene transcription (Figure 6A). SB203580 at 5 μM plus OA resulted in a 8.5±1.2 fold induction of NF-κB-regulated promoter activity versus a 4.5±0.6 fold induction after stimulation with OA only (P<0.05, n=6) (Figure 6A). Similarly, when pretreated with SB203580 at 10 μM prior to OA, promoter activity was enhanced 9.1±1.6 fold (P<0.05, n=6), while SB203580 at 10 μM alone had no effect (1.0±0.2 fold) (Figure 6A).
Figure 6.
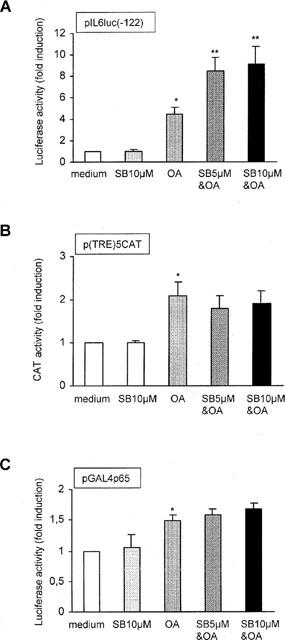
SB203580 at 5 and 10 μM enhances NF-κB activity, but not p65 activity. (A) TF-1 cells were transiently transfected as described in ‘Methods' with pIL6luc(-122). Six hours after electroporation cells were stimulated for 24 h with medium, SB203580 (10 μM), OA (30 ng ml{sF/R}−1{XF/R}), SB203580 5 μM (30 min pretreatment) plus OA, or SB203580 10 μM plus OA. Twenty-four hours after stimulation cells were lysed and analysed for the amount of produced luciferase protein. Promoter activity after treatment with medium is set at 1. Mean fold induction and standard error of the mean represent six identical experiments. *P<0.05 OA-treated cells versus medium-treated cells. **P<0.05 SB203580 plus OA-treated cells versus OA-treated cells. (B) Same as described under (A), but with p(TRE)5xCAT instead of pIL6luc(-122). The amount of produced CAT protein after medium treatment is set at 1. Mean fold induction and standard error of the mean represent three identical experiments. *P<0.05. (C) Same as described under (A), but with 5 μg pGAL4p65 together with 5 μg pGAL4tkluc. Promoter activity after treatment with medium is set at 1. Mean fold induction and standard error of the mean represent six identical experiments. *P<0.05.
TF-1 cells were subsequently transfected with p(TRE)5CAT, a CAT reporter plasmid driven by five consecutive AP-1 binding sites (Van Dam et al., 1993). Stimulation with OA alone resulted in an increase in promoter activity (2.1±0.3 fold, P<0.05, n=3) (Figure 6B). Pretreatment with SB203580, at concentrations of 5 and 10 μM, did not affect OA-mediated AP-1 promoter activity (Figure 6B). These results suggest that the activation signal triggered by SB203580 at higher concentrations may be specific for the activation of NF-κB, and not a random activation process within the TF-1 cell line.
To analyse whether the enhanced NF-κB-mediated gene transcription after higher concentrations of SB203580 could be explained by an increased transcriptional potential of the p65 subunit, TF-1 cells were transiently transfected with pGal4p65 and pGal4tkluc. The results demonstrated, however, that SB203580 at neither 5, nor 10 μM were capable of directly enhancing OA-induced p65-mediated gene transcription (1.5±0.1 fold increase for OA versus 1.6±0.1 fold and 1.7±0.1 fold for OA plus SB203580 at 5 and 10 μM, respectively, n=6) (Figure 6C). These results suggest that SB203580 at 5 and 10 μM enhances NF-κB-mediated gene transcription independently of phosphorylation on the transactivation domains of the p65 subunit.
SB203580 at 10 μM enhances phosphorylation of ERK1/2 and JNK
The above-described results would suggest that the p38 MAP kinase specific inhibitor SB203580 activates rather than inhibits signalling molecules when applied at concentrations of 5 and 10 μM. Activation of these signalling modules may then result in the activation of gene transcription regulated by NF-κB. Previously, we demonstrated the involvement of the ERK1/2 and JNK MAP kinase pathways in mediating NF-κB-regulated gene transcription in TF-1 cells and monocytes (Tuyt et al., 1999). We thus set out to investigate whether SB203580 is capable of activating the ERK and JNK pathways, when utilized at 10 μM. TF-1 cells were stimulated with OA for 90 min or pretreated with 1 or 10 μM of SB203580 for 30 min prior to OA stimulation. Total cell lysates were separated on SDS–PAGE gels and analysed for phosphorylated ERK1/2 and phosphorylated JNK1/2. Total ERK and JNK protein were visualized for equal loading. As depicted in Figure 7 stimulation with OA resulted in considerable phosphorylation of both the ERK (Figure 7A) and JNK (Figure 7B) proteins. Pretreatment with 1 μM SB203580 did not affect the level of either phospho-ERK or phospho-JNK. However, when TF-1 cells were pretreated with 10 μM SB203580 prior to OA, both phospho-ERK and phospho-JNK levels were strongly enhanced when compared with OA stimulation (Figure 7).
Figure 7.
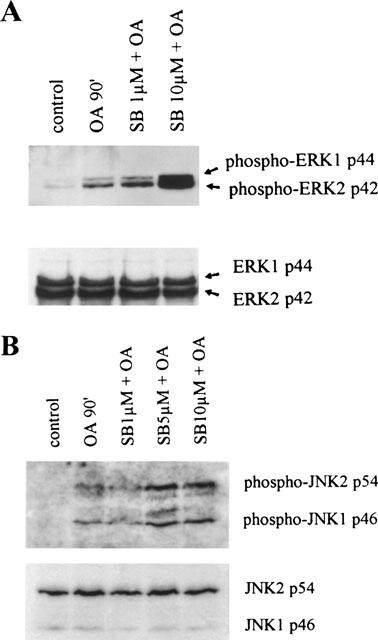
SB203580 at 10 μM enhances phosphorylation of ERK1/2 and JNK1/2. TF-1 cells were cultured for 16 h in RPMI 1640 containing 0.1% FBS and subsequently stimulated with medium, OA (30 ng ml−1) for 90 min, or SB203580 (30 min pretreatment) in the indicated concentrations. Cell extracts were prepared as described in detail in ‘Methods'. Phosphorylated ERK1 and ERK2 (A) and phosphorylated JNK1 and JNK2 (B) are shown in the upper panels. Total ERK and JNK protein are shown in the lower panels and represent equal loading. Immunoblotting with anti-phospho-ERK/JNK and anti-ERK/JNK antibodies was performed by standard procedures and detection was performed using ECL immunodetection kit.
In order to demonstrate the specificity of the activation signal, the same cell lysates were analysed for phosphorylated p38 protein. As shown in Figure 3, SB203580 at any concentration does not affect the amount of phosphorylated p38 kinase product. Summarizing, SB203580 at high concentration activates the ERK and JNK signalling pathways with concomitant activation of NF-κB-regulated gene transcription.
Kinase-inactive Raf-1 suppresses SB203580-enhanced NF-κB activity
Since SB203580 at 10 μM considerably activated the ERK1/2 and JNK pathways, we set out to investigate whether these pathways are also involved in SB203580-enhanced NF-κB activity. For this purpose, TF-1 cells were transiently transfected with pIL6(-122)luc together with kinase-deficient mutants of molecules belonging to the ERK1/2 (pRSV-NΔRaf1) (Schaap et al., 1993), JNK (pcDNA3-MKK4(Ala), and pcDNA3-Flag-JNK) (Whitmarsh et al., 1997), and p38 (pRSV-MKK3(Ala) and pcDNA3-MKK6(K82A)) (Raingeaud et al., 1996) MAP kinase pathways. After transfection, cells were treated with medium, OA or SB203580 at 5 μM for 30 min prior to OA exposure. Possible involvement of MAP kinase pathways was identified when the induction of NF-κB activity due to SB203580 treatment was suppressed in the presence of a dominant negative construct when compared with the SB203580-mediated induction after introduction of the empty vector. The results demonstrated that only pRSV-NΔRaf1 was capable of suppressing the SB203580-enhanced NF-κB transcriptional activity (Figure 8A). NF-κB-mediated gene transcription after SB203580 plus OA treatment was suppressed down to 66±5% after the introduction of pRSV-NΔRaf1 when compared with cotransfection with pcDNA3, which was set at 100% (P<0.05, n=3).
Figure 8.
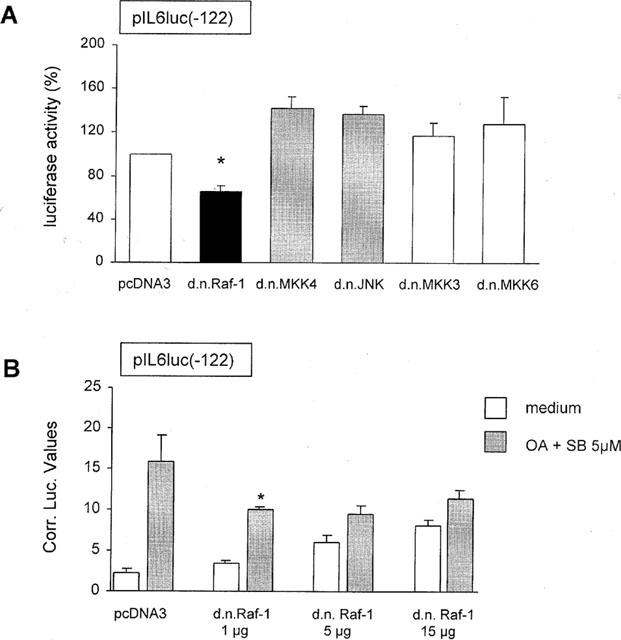
Kinase inactive Raf-1 suppresses SB203580-enhanced NF-κB activity. (A) TF-1 cells were transiently transfected as described in ‘Methods' with 15 μg pIL6luc(-122) together with (1) 15 μg pcDNA3 empty vector, (2) pRSV-NΔRaf1, (3) pcDNA3-MKK4(Ala), (4) pcDNA3-Flag-JNK1, (5) pRSV-MKK3(Ala), or (6) pcDNA3-MKK6(K82A). Six hours after electroporation cells were stimulated for 24 h with medium, OA (30 ng ml−1) or SB203580 at 5 μM (30 min pretreatment) plus OA (30 ng ml−1). Twenty-four hours after stimulation cells were lysed and analysed for the amount of produced luciferase protein. The IL-6 promoter activity for pcDNA3 construct when treated with SB203580 at 5 μM plus OA is set at 100%. Mean and standard error of the mean represent three identical experiments. *P<0.05. (B) TF-1 cells were transiently transfected as described in ‘Methods' with 15 μg pIL6luc(-122) together with (1) 15 μg pcDNA3 empty vector, (2) 1 μg pRSV-NΔRaf1 and 14 μg pcDNA3 empty vector, (3) 5 μg pRSV-NΔRaf1 and 10 μg pcDNA3 empty vector or (4) 15 μg pRSV-NΔRaf1. Six hours after electroporation cells were stimulated for 24 h with medium or SB203580 at 5 μM (30 min pretreatment) plus OA (30 ng ml−1). Twenty-four hours after stimulation cells were lysed and analysed for the amount of produced luciferase protein. Values depict the luciferase activity in arbitrary units corrected for transfection efficiency. Mean and standard error of the mean represent three identical experiments. *P<0.05.
To confirm that the transfection procedure used was efficient and that the effect of pRSV-NΔRaf1 did dominate over the endogenous protein, the effect of increasing concentrations of pRSV-NΔRaf1 (1, 5 and 15 μg) on the SB203580 plus OA-mediated pIL6luc(-122) promoter activity was investigated. As demonstrated in Figure 8B, overexpression of already 1 μg of pRSV-NΔRaf1 very potently decreased the SB203580 plus OA-induced pIL6luc(-122) promoter activity from 6.9±0.1 to 2.9±0.2 fold (P<0.05). These data demonstrate that pRSV-NΔRaf1 dominates over the endogenous protein and that the transfection procedure is very efficient. Overexpression of higher concentrations of pRSV-NΔRaf1 increased the basal luciferase activity. Overexpression of dominant-negative MKK4, JNK, MKK3, and MKK6 did not inhibit the SB203580-enhanced NF-κB transcriptional activity (Figure 8A). These results strongly suggest that SB203580-enhanced ERK phosphorylation activates NF-κB-mediated gene transcription.
Discussion
The present study demonstrates that the activated MAP kinase p38/RK is not involved in okadaic acid-mediated activation of the transcription factor NF-κB in the erythroleukaemic cell line TF-1. No effect is observed at DNA-binding nor at transcriptional level when p38 kinase activity is suppressed by either SB203580 at 1 μM or by the kinase-deficient mutants of MKK3 or MKK6, the upstream kinases from p38. The lack of involvement of the p38 kinase pathway in the activation of NF-κB appears striking, since there is also good experimental evidence for a connection between the NF-κB and p38 pathway (Schulze-Osthoff et al., 1997). In the mouse fibrosarcoma cell line L929s TNF-induced NF-κB-mediated gene transcription, but not NF-κB DNA-binding, appeared dependent on activation of the p38/RK pathway (Vanden Berghe et al., 1998). Similarly, Wesselborg et al. (1997) reported that the TNF- and phorbolester-enhanced expression of a NF-κB-controlled reporter construct was prevented by SB203580, while NF-κB DNA binding remained unaffected. Consistent with these experiments, overexpression of dominant-negative MKK6 or MKK3 mutants abolished NF-κB-mediated transactivation in these studies. However, Alpert et al. (1999) demonstrated that sustained activation of p38 by sorbitol, H2O2 or arsenite inhibits NF-κB activation, while the transient activation of p38 by TNF was associated with transactivation of NF-κB. These differences strongly indicate that the signalling cascades activating NF-κB are dependent on cell type and/or stimulus. It is, furthermore, well appreciated that the transactivating activity of the NF-κB subunit p65 is increased following phosphorylation of one of its two transactivation domains (Schmitz & Baeuerle, 1991). We demonstrated, however, that p38 MAP kinase did not affect p65 phosphorylation. This is consistent with previous findings, in which the p38 MAP kinase protein did not phosphorylate the transactivating C-terminal half of p65 in in vitro kinase assays (Wesselborg et al., 1997).
Surprisingly, SB203580 at concentrations of 5 and 10 μM enhanced rather than inhibited NF-κB-driven promoter activity. However, in view of the unaffected GAL4p65 transfections it appears that high concentrations of SB203580 enhances NF-κB-mediated promoter activity independently of phosphorylation of the transactivation domains of the p65 subunit. Enhanced NF-κB activation coincided with upregulated phosphorylation of both the ERK and JNK MAP kinases. However, only the SB203580-activated ERK pathway appeared involved in the activation of NF-κB. The distinct effect observed after the introduction of kinase-deficient Raf-1, an upstream activator of ERK, indicates that SB203580 activates the ERK pathway relatively upstream in the ERK cascade. These findings are underscored by the recent observations of Hall-Jackson et al. (1999). They demonstrated that SB203580 at 10 μM induced a remarkable activation of c-Raf in vivo. The ERK pathway may act by enhancing NF-κB DNA binding activity, but may also phosphorylate cofactors such as CBP/p300, PC1 and E1A, which in turn increase NF-κB transcriptional activity. Our previous studies in TF-1 cells showed that OA-induced ERK phosphorylation enhanced NF-κB transcriptional activity but not NF-κB DNA binding (Tuyt et al., 1999). Based on these findings it may be postulated that the ERK pathway elicits its effect by enhancing the activity of p65-associated cofactors. However, multiple studies have reported on the close association between ERK activity and phosphorylation and degradation of IκB protein, leading to NF-κB nuclear translocation and activation. Sonoda et al. (1997) reported in vitro phosphorylation of GST-IκB-α by OA-activated ERK. In the lymphoblastoid cell line CEM, overexpression of either MEK1 or ERK1 demonstrated constitutive nuclear localization of NF-κB, indicating involvement of the classical ERK pathway in NF-κB DNA binding activity (Briant et al., 1998). Future studies should reveal the exact role of the ERK cascade in mediating NF-κB transcriptional activity.
Although activation of ERK and JNK by SB203580 has not been reported before, observations in several studies may suggest the occurrence of this phenomenon. For instance, Schwenger et al. (1998) suggested that TNF-induced p38 kinase activation may exert a negative regulatory influence on the process of NFκB activation by this cytokine in COS-1 cells. At the concentration of 10 μM, SB203580 significantly prevented the ability of the drug sodium salicylate to suppress TNF-induced IκBα degradation. However, in this study it was not ruled out that SB203580 exerted its effect by activating alternate MAPK pathways and thus enhancing the degradation of IκBα.
In myeloid leukaemic cells, NF-kB expression may exert its clinically unfavourable effect by enhancing the expression of cytokine genes or by inducing the expression of anti-apoptotic genes. Insight into the regulation of NF-κB in these cells may thus lead to new clinical approaches. In the present study we showed that the p38 MAP kinase pathway does not mediate the OA-induced NF-κB activation in the TF-1 haematopoietic cell line. Moreover, SB203580 stimulation may result in adverse effects, since it enhances NF-κB and ERK.
Acknowledgments
This study was supported by grant RUG 94-788 and grant RUG 99-1944 from the Dutch Cancer Society. We would like to thank Dr L. Aarden (Central Laboratory of the Netherlands Red Cross Blood Transfusion Service, Amsterdam, The Netherlands) for providing us with the cDNA probe for IL-6, and Dr P.A. Baeuerle (Tularik Inc., San Francisco, CA, U.S.A.) for the pGAL4p65. pGAL4dbd and pGAL4tkluc were kindly provided by Dr S. Wissink (NIOB, Utrecht, The Netherlands). We are grateful to Dr P.J. Coffer (Department of Pulmonary Diseases, University Hospital Utrecht, The Netherlands) for supplying us with pRSV-NΔRaf1, and to Dr R.J. Davis (Howard Hughes Medical Institute, University of Massachusetts Medical School, Worcester, MA, U.S.A.) for pcDNA3-Flag-JNK, pRSV-MKK3(Ala), pcDNA3-MKK4(Ala), and pcDNA3-MKK6(K82A). p(TRE)5CAT was a generous gift from Dr H. Van Dam (Department of Molecular Carcinogenesis, University of Leiden, The Netherlands).
Abbreviations
- AML
acute myeloid leukaemia
- AP-1
activator protein-1
- EMSA
electrophoretic mobility shift assay
- ERK
extracellular signal-regulated kinase
- IKK
inhibitor kappa B (IκB) kinase
- IL-1
interleukin-1
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinases
- MEKK
MAP/ERK kinase kinase
- MKK
MAP kinase kinase
- NF-κB
nuclear factor kappa B
- OA
okadaic acid
References
- AKIRA S., KISHIMOTO T. NF-IL6 and NF-kappa B in cytokine gene regulation. Adv. Immunol. 1997;65:1–46. [PubMed] [Google Scholar]
- ALPERT D., SCHWENGER P., HAN J., VILCEK J. Cell stress and MKK6b-mediated p38 MAP kinase activation inhibit Tumor Necrosis Factor-induced IκB phosphorylation and NF-κB activation. J. Biol. Chem. 1999;274:22176–22183. doi: 10.1074/jbc.274.32.22176. [DOI] [PubMed] [Google Scholar]
- BEG A.A., BALTIMORE D. An essential role for NF-kappa B in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- BEG A.A., SHA W.C., BRONSON R.T., GHOSH S., BALTIMORE D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- BERGMANN M., HART L., LINDSAY M., BARNES P.J., NEWTON R. IkappaBalpha degradation and nuclear factor-kappaB DNA binding are insufficient for interleukin-1 beta and tumor necrosis factor-alpha-induced kappaB-dependent transcription. J. Biol. Chem. 1998;273:6607–6610. doi: 10.1074/jbc.273.12.6607. [DOI] [PubMed] [Google Scholar]
- BIRKENKAMP K.U., DOKTER W.H., ESSELINK M.T., JONK L.J.C., KRUIJER W., VELLENGA E. A dual function for p38 MAP kinase in hematopoietic cells: involvement in apoptosis and cell activation. Leukemia. 1999;13:1037–1045. doi: 10.1038/sj.leu.2401447. [DOI] [PubMed] [Google Scholar]
- BRIANT L., ROBERT-HEBMANN V., SIVAN V., BRUNET A., POUYSSEGUR J., DEVAUX J. Involvement of extracellular signal-regulated kinase module in HIV-mediated CD4 signals controlling activation of nuclear factor-kappaB and AP-1 transcription factors. J. Immunol. 1998;160:1875–1885. [PubMed] [Google Scholar]
- COHEN P., HOLMES C.F., TSUKITANI Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem. Sci. 1990;15:98–102. doi: 10.1016/0968-0004(90)90192-e. [DOI] [PubMed] [Google Scholar]
- CUENDA A., COHEN P., BUEE-SCHERRER V., GOEDERT M. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38) EMBO J. 1997;16:295–305. doi: 10.1093/emboj/16.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUENDA A., ROUSE J., DOZA Y.N., MEIER R., COHEN P., GALLAGHER T.F., YOUNG P.R., LEE J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- DOKTER W.H., TUYT L.M.L., SIERDSEMA S.J., ESSELINK M.T., VELLENGA E. The spontaneous expression of interleukin-1 beta and interleukin-6 is associated with spontaneous expression of AP-1 and NF-kappa B transcription factor in acute myeloblastic leukemia cells. Leukemia. 1995;9:425–432. [PubMed] [Google Scholar]
- FRANTZ B., KLATT T., PANG M., PARSONS J., ROLANDO A., WILLIAMS H., TOCCI M. J., O'KEEFE S.J., O'NEILL E.A. The activation state of p38 mitogen-activated protein kinase determines the efficiency of ATP competition for pyridinylimidazole inhibitor binding. Biochemistry. 1998;37:13846–13853. doi: 10.1021/bi980832y. [DOI] [PubMed] [Google Scholar]
- FRASER A., EVAN G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- GRIFFIN J.D., RAMBALDI A., VELLENGA E., YOUNG D.C., OSTAPOVICZ D., CANNISTRA S.A. Secretion of interleukin-1 by acute myeloblastic leukemia cells in vitro induces endothelial cells to secrete stimulating factors. Blood. 1987;70:1218–1221. [PubMed] [Google Scholar]
- HALL-JACKSON C.A., GOEDERT M., HEDGE P., COHEN P. Effect of SB203580 on the activity of c-Raf in vitro and in vivo. Oncogene. 1999;18:2047–2054. doi: 10.1038/sj.onc.1202603. [DOI] [PubMed] [Google Scholar]
- KITAMURA T., TANGE T., TERASAWA T., CHIBA S., KUWAKI T., MIYAGAWA K., PIAO Y.-F., MIYAZONO K., URABE A., TAKAKU F. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J. Cell. Physiol. 1989;140:323–334. doi: 10.1002/jcp.1041400219. [DOI] [PubMed] [Google Scholar]
- MANIATIS T. Catalysis by a multiprotein IkappaB kinase complex. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- MARSHALL C.J. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr. Opin. Gen. Develop. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- RAINGEAUD J., WHITMARSH A.J., BARRETT T., DERIJARD B., DAVIS R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAAP D., VAN DER WAL J., HOWE L.R., MARSHALL C.J., VAN BITTERSWIJK W.J. A dominant-negative mutant of raf blocks mitogen-activated protein kinase activation by growth factors and oncogenic p21ras. J. Biol. Chem. 1993;268:20232–20236. [PubMed] [Google Scholar]
- SCHMITZ M.L., BAEUERLE P.A. The p65 subunit is responsible for the strong transcription activating potential of NF-kappaB. EMBO J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMITZ M.L., DOS SANTOS SILVA M., BAEUERLE P.A. Transactivation domain 2 (TA2) of p65 NF-kappa B. Similarity to TA1 and phorbol ester-stimulated activity and phosphorylation in intact cells. J. Biol. Chem. 1995;270:15576–15584. doi: 10.1074/jbc.270.26.15576. [DOI] [PubMed] [Google Scholar]
- SCHREIBER E., MATTHIAS P., MÜLLER M.M., SCAFFNER W. Rapid detection of octamer binding proteins with ‘mini-extracts', prepared from a small number of cells. Nucl. Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHULZE-OSTHOFF K., FERRARI D., RIEHEMANN K., WESSELBORG S. Regulation of NF-kappaB activation by MAP kinase cascades. Immunobiology. 1997;198:35–49. doi: 10.1016/s0171-2985(97)80025-3. [DOI] [PubMed] [Google Scholar]
- SCHWENGER P., ALPERT D., SKOLNIK E.Y., VILCEK J. Activation of p38 mitogen-activated protein kinase by sodium salicylate leads to inhibition of tumor necrosis factor-induced IkappaB alpha phosphorylation and degradation. Mol. Cell. Biol. 1998;18:78–84. doi: 10.1128/mcb.18.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONODA Y., KASHAHARA T., YAMAGUCHI Y., KUNO K., MATSISHIMA K., MUKAIDA N. Stimulation of interleukin-8 production by okadaic acid and vanadate in a human promyelocyte cell line, an HL-60 subline. Possible role of mitogen-activated protein kinase on the okadaic acid-induced NF-kappaB activation. J. Biol. Chem. 1997;272:15366–15372. doi: 10.1074/jbc.272.24.15366. [DOI] [PubMed] [Google Scholar]
- STANCOVSKI I., BALTIMORE D. NF-kappaB activation: the I kappaB kinase revealed. Cell. 1997;91:299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- SUNG S.J., WALTERS J.A. Stimulation of interleukin-1 alpha and interleukin-1 beta production in human monocytes by protein phosphatase 1 and 2A inhibitors. J. Biol. Chem. 1993;268:5802–5809. [PubMed] [Google Scholar]
- TUYT L.M.L., DOKTER W.H.A., BIRKENKAMP K.U., KOOPMANS S.B., LUMMEN C., KRUIJER W., VELLENGA E. Extracellular-regulated kinase½, Jun N-terminal kinase, and c-Jun are involved in NF-kappa B-dependent IL-6 expression in human monocytes. J. Immunol. 1999;162:4893–4902. [PubMed] [Google Scholar]
- TUYT L.M.L., DOKTER W.H., KOOPMANS S.B., VELLENGA E. Transcriptional regulation of interleukin-6 gene expression in acute myeloid leukemia blasts with constitutive cytokine production. Blood. 1996;10 Suppl. 1:198. [Google Scholar]
- VAN ANTWERP D.J., MARTIN S.J., KAFRI T., GREEN D., VERMA I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- VAN DAM H., DUYNDAM M., ROTTIER R., BOSCH A., DE VRIES-SMITS L., HERRLICH P., ZANTEMA A., ANGEL P., VAN DER EB A.J. Heterodimer formation of cJun and ATF-2 is responsible for induction of c-jun by the 243 amino acid adenovirus E1A protein. EMBO J. 1993;12:479–487. doi: 10.1002/j.1460-2075.1993.tb05680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANDEN BERGHE W., PLAISANCE S., BOONE E., DE BOSSCHER K., SCHMITZ M.L., FIERS W., HAEGEMAN G. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- WANG C.-Y., MAYO M.W., BALDWIN A.S., JR TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappa B. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- WESSELBORG S., BAUER M.K. A., VOGT M., SCHMITZ M.L., SCHULZE-OSTHOFF K. Activation of transcription factor NF-kappaB and p38 mitogen-activated protein kinase is mediated by distinct and separate stress effector pathways. J. Biol. Chem. 1997;272:12422–12429. doi: 10.1074/jbc.272.19.12422. [DOI] [PubMed] [Google Scholar]
- WHITMARSH A.J., YANG S.-H., SU M.S., SHARROCKS A.D., DAVIS R.J. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol. Cell. Biol. 1997;17:2360–2371. doi: 10.1128/mcb.17.5.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WISSINK S., VANHEERDE E.C., SCHMITZ L.M., KALKHOVEN E., VAN DER BURG B., BAEUERLE P.A., VAN DER SAAG P.T. Distinct domains of the RelA NF-kappaB subunit are required for negative cross-talk and direct interaction with the glucocorticoid receptor. J. Biol. Chem. 1997;272:22278–22284. doi: 10.1074/jbc.272.35.22278. [DOI] [PubMed] [Google Scholar]
- ZANDI E., ROTHWARF D.M., DELHASE M., HAYAKAWA M., KARIN M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]