

Abstract
The mechanisms of the thapsigargin (TG)-induced capacitative Ca2+ entry in in situ endothelial cells and its role in the regulation of arterial tone were investigated using front-surface fluorimetry and fura-2-loaded strips of porcine aortic valve and coronary artery.
In the presence of extracellular Ca2+, TG induced an initial rapid and a subsequent sustained elevation of cytosolic Ca2+ concentration ([Ca2+]i) in valvular strips. In the absence of extracellular Ca2+, TG induced only a transient increase in [Ca2+]i.
The TG-induced sustained elevation of [Ca2+]i in endothelial cells was inhibited completely by 1 mM Ni2+ and partly by 10 μM econazole and 30 μM ML-9, but not by 900 ng ml−1 pertussis toxin or 100 μM wortmannin. Therefore, cytochrome P450 and protein phosphorylation are suggested to be involved in the TG-induced Ca2+ influx in in situ endothelial cells.
TG induced an endothelium-dependent large relaxation consisting of an initial and a late sustained relaxation in coronary arterial strip precontracted with U46619 (a thromboxane A2 analogue). Indomethacin alone had no effect, while indomethacin plus Nω-nitro-L-arginine (L-NOARG) markedly inhibited the sustained phase and slightly inhibited the initial phase of the TG-induced relaxation.
TG induced a smaller but sustained relaxation during the 40 mM K+-induced precontraction than that seen during the U46619-induced precontraction. This relaxation was completely abolished by the pretreatment with indomethacin plus L-NOARG.
In conclusion, both nitric oxide (NO) and endothelium-derived hyperpolarizing factor were suggested to mediate the TG-induced relaxation, while NO plays a major role in the sustained relaxation. The TG-induced sustained [Ca2+]i elevation in endothelial cells was thus suggested to be mainly linked to the sustained production of NO.
Keywords: Endothelial cells, capacitative Ca2+ entry, thapsigargin, endothelium-dependent relaxation
Introduction
Endothelial cells play an important role in the regulation of vascular tone by producing both vasorelaxing and vasocontracting substances (Furchgott & Vanhoutte, 1989; Katusic & Shepherd, 1991). Endothelium-dependent relaxation has been widely reported in different species and various types of blood vessels. Endothelium-derived relaxing factors include nitric oxide (NO) (Palmer et al., 1987), endothelium-derived hyperpolarizing factor (EDHF) (Chen et al., 1988; Komori & Vanhoutte, 1990) and prostacyclin. The Ca2+ signal in endothelial cells plays a primary role in the production and release of these relaxing factors, and thereby the regulation of vascular tone (Newby & Henderson, 1990). One of the important mechanisms to induce a [Ca2+]i elevation is the Ca2+ influx from the extracellular space. In non-excitable cells such as endothelial cells, where the voltage-operated Ca2+ channels are not expressed, one of the dominant Ca2+ influx pathways is the capacitative Ca2+ entry pathways (Putney & McKay, 1999). However, the functional role of the capacitative Ca2+ entry of endothelial cells in the regulation of endothelium-dependent relaxation remains to be elucidated.
Thapsigargin (TG) is an inhibitor of the endoplasmic reticulum Ca2+-ATPase (Thastrup et al., 1990). The inhibition of Ca2+-ATPase by TG is considered to deplete the intracellular Ca2+ store and activate the capacitative Ca2+ entry (Thastrup et al., 1990). In fact, TG and cyclopiazonic acid (CPA), another inhibitor of the endoplasmic reticulum Ca2+-ATPase (Seidler et al., 1989), have been shown to induce a sustained elevation of [Ca2+]i in endothelial cells (Higuchi et al., 1996; Kawasaki et al., 1999). Since TG induces a large sustained Ca2+ influx in endothelial cells (Kawasaki et al., 1999), it could serve as a useful tool to investigate the functional role of the capacitative Ca2+ entry in the endothelium-dependent regulation of vascular tone.
Since the capacitative Ca2+ influx serves as a major Ca2+ entry pathway in endothelial cells, it is important to elucidate the mechanism of regulation for the capacitative Ca2+ influx. However, the coupling of the depletion of the intracellular stores and the activation of the Ca2+ entry has yet to be clarified. So far, two mechanisms have been proposed for such coupling; direct coupling and indirect coupling mediated by second messengers (Boulay et al., 1999; Holda & Blatter, 1997; Putney, 1999; Putney & McKay, 1999). The candidates for second messengers include tyrosine phosphorylation (Kruse et al., 1994; Pfeiffer et al., 1995; Sargeant et al., 1994), trimeric and small GTP-binding proteins (Bird & Putney, 1993; Sargeant et al., 1993), cytochrome P450 (Alvarez et al., 1992; Graier et al., 1995) and myosin light chain kinase (MLCK) (Watanabe et al., 1996). However, the precise intracellular signalling pathway of the capacitative Ca2+ entry in in situ endothelial cells has not yet been established.
In the present study, we investigated the mechanism of the TG-induced capacitative Ca2+ entry in in situ endothelial cells using front-surface fluorimetry of fura-2-loaded strips of the porcine aortic valves. We also examined the functional role of the TG-induced Ca2+ entry in the regulation of vascular tone by simultaneously monitoring the effect of TG on smooth muscle [Ca2+]i and tension in fura-2-loaded strips of the porcine coronary artery. In addition, we determined the relative contribution of NO and EDHF to the TG-induced relaxation, and evaluated the correlation between the TG-induced capacitative Ca2+ entry and the production of the endothelium-derived relaxing factors.
Methods
Tissue preparation
In order to monitor changes in [Ca2+]i of in situ endothelial cells, strips were prepared from the porcine aortic valve as previously described (Kuroiwa et al., 1993). Porcine aortic valves were obtained at a local slaughterhouse and brought to the laboratory in ice-cold normal physiological salt solution (PSS). The aortic valve leaflets were handled with extreme care in order to avoid damaging the endothelial layer. The valve leaflets were cut into strips in an axial direction (approximately 2 mm wide, 5 mm long, and 0.18 mm thick).
Arterial strips with and without endothelium were prepared from the left circumflex branch of porcine coronary arteries as previously described (Kuroiwa et al., 1993). Porcine hearts were obtained immediately after the animals had been slaughtered, and brought to the laboratory in ice-cold PSS. A segment of the left circumflex artery 2–3 cm from the origin was excised, and the adventitia of the segment was mechanically removed. The segment was opened longitudinally and cut into circular strips (approximately 1 mm wide, 5 mm long, and 0.1 mm thick). To remove the endothelium, the inner surface was rubbed off with a cotton swab.
Fura-2 loading
The valvular strips were loaded with Ca2+ indicator dye, fura-2, by incubating them in oxygenated (a mixture of 95% O2 and 5% CO2) Dulbecco's modified Eagle medium (DMEM) containing 50 μM fura-2/AM (an acetoxymethyl ester form of fura-2), 1 mM probenecid and 5% foetal bovine serum for 1.5 h at 37°C (Kanaide, 1999). Probenecid was added to prevent sequestration of fura-2 (Di Virgilio et al., 1989). Coronary arterial strips with and without endothelium were loaded with fura-2 in oxygenated DMEM containing 25 μM fura-2/AM and 5% foetal bovine serum for 4 h at 37°C (Kanaide, 1999). After loading with fura-2, both valvular and arterial strips were washed in normal PSS to remove the dye remaining in the extracellular space, and equilibrated in normal PSS for at least 1 h before starting the measurements. Loading the vascular strips with fura-2, per se, did not affect the contractility of the arterial strips (Hirano et al., 1990).
Front-surface fluorimetry
The changes in [Ca2+]i of in situ endothelial cells were monitored in the fura-2-loaded valvular strips as described previously (Aoki et al., 1991; Kuroiwa et al., 1995). The measurements were carried out at 25°C to prevent any sequestration of the fluorescence dye, because in situ endothelial cells actively sequestrated fura-2 at 37°C (Kuroiwa et al., 1993; 1995). In brief, the valvular strips were mounted vertically in the quartz organ bath and changes in fluorescence intensity of the fura-2-Ca2+ complex in endothelial cells were monitored with a front-surface fluorimeter specifically designed for fura-2 fluorimetry (CAM-OF2; Japan Spectroscopic Co., Tokyo, Japan). The ratio of the fluorescence intensity (500 nm) at 340 nm excitation to that at 380 nm excitation was monitored as an indicator of [Ca2+]i. The response to 10 μM ATP was recorded as a reference response at the beginning of each experimental protocol. The changes in [Ca2+]i of in situ endothelial cells were expressed in per cent, assigning the level of [Ca2+]i at rest and that at the peak response to 10 μM ATP to be 0 and 100%, respectively. The absolute value of [Ca2+]i was estimated in the same manner as previously described (Grynkiewicz et al., 1985) with an apparent Kd value of 162 nM at 25°C (Aoki et al., 1991). The [Ca2+]i levels of endothelial cells in situ at rest (0%) and at the peak level (100%) obtained with 10 μM ATP were determined in separate experiments and found to be 63.9±7.4 and 176.7±16.5 nM, respectively (n=5).
Front-surface fluorimetry of vascular strips was carried out at 37°C, since fura-2 fluorescence was stable in smooth muscle cells for more than 1 h at 37°C (Kanaide, 1999; Kuroiwa et al., 1993). The response to 118 mM K+-depolarization was recorded as a reference response before starting each experimental protocol. Changes in the fluorescence ratio in the smooth muscle cells were expressed in per cent, assigning the value at rest (5.9 mM K+ PSS) and that obtained in 118 mM K+ PSS to be 0 and 100%, respectively. The absolute value of [Ca2+]i of vascular strips was estimated with the Kd of 224 nM at 37°C (Grynkiewicz et al., 1985). The [Ca2+]i levels corresponding to 0 and 100 % fluorescence ratios in smooth muscle were determined in separate experiments and found to be 108±27 and 715±103 nM, respectively (Hirano et al., 1990; Kanaide, 1999). We previously demonstrated that the fura-2 signal of the arterial strips arose exclusively from smooth muscle cells even in the presence of an intact endothelium, because endothelial cells of arterial strips excluded fura-2 under the present experimental conditions (37°C and without probenecid) (Kuroiwa et al.; 1993; 1995).
Measurement of tension development in the coronary arterial strips
Using a force-transducer TB-612T (Nihon Koden, Tokyo, Japan), the tension development of the fura-2-loaded arterial strip was simultaneously monitored during the fura-2 fluorimetry. During the equilibration period, the strips were stimulated with 118 mM K+ every 15 min, and the resting load was increased stepwise and then finally adjusted to 250 mg. The developed tension was expressed in per cent, assigning the value at rest (5.9 mM K+) and that at a steady state of contraction induced by 118 mM K+ to be 0 and 100%, respectively.
Drugs and solutions
The composition of normal PSS was as follows (mM): NaCl 123, KCl 4.7, NaHCO3 15.5, KH2PO4 1.2, MgCl2 1.2, CaCl2 1.25 and D-glucose 11.5. High K+ PSS was prepared by replacing NaCl with equimolar KCl. PSS was aerated with a mixture of 95% O2 and 5% CO2, with the resulting pH being 7.4. The Ca2+-free PSS contained no CaCl2 but 2 mM ethylenglycol-bis (β-aminoethylether)-N,N,N′,N′-tetra acetic acid (EGTA). Fura-2/AM and EGTA was purchased from Dojindo (Kumamoto, Japan). Indomethacin was purchased from Wako (Osaka, Japan). Nω-nitro-L-arginine (L-NOARG) was from the Aldrich Chemical Company, Inc. (Milwaukee, WI, U.S.A.). ATP was purchased from Boehringer Mannheim (Germany). Thapsigargin, probenecid, wortmannin and econazole were purchased from Sigma (St. Louis, MO, U.S.A.). Foetal bovine serum was purchased from GIBCO (Rockville, MD, U.S.A.). U46619 (a thromboxane A2 analogue 9, 11-Dideoxy-11α, 9α-epoxymethano-prostaglandin F2α) was purchased from Funakoshi (Tokyo, Japan). ML-9 (1-(5-Chloronaphthalene-1-sulphonyl)-1H-hexahydro-1,4-diazepine) and IAP (pertusis toxin) were purchased from Seikagaku Co. (Tokyo, Japan).
Data analysis
All data were expressed as the mean±standard error of the mean (s.e.mean). Student's t-test was used to determine statistical significance. P values of less than 0.05 were considered to be significant. All data were collected using a computerized data acquisition system (MacLab; Analog Digital Instruments, Australia, Macintosh; Apple Computer, U.S.A.).
Results
Effects of TG on [Ca2+]i in endothelial cells of the porcine aortic valve
In the presence of extracellular Ca2+, TG induced a large sustained increase in [Ca2+]i in the endothelial cells of aortic valvular strips (Figure 1). As shown in Figure 1a, 1 μM TG induced a rapid increase in [Ca2+]i, which reached a peak (the first component) at 8.4±0.2 min (n=6), followed by a sustained increase at a slightly lower level (the second component). The levels of [Ca2+]i elevation observed at the peak and at 30 min (the second component) were 510.4±31.3 and 389.7±29.1% (n=6) of the peak response to 10 μM ATP, respectively. When TG was applied in a cumulative manner (0.001 through 10 μM), the [Ca2+]i level increased in a concentration-dependent manner (Figure 1c). A significant elevation was observed at concentrations of higher than 0.01 μM TG and the maximum elevation of [Ca2+]i was obtained at a concentration of 1 μM TG.
Figure 1.
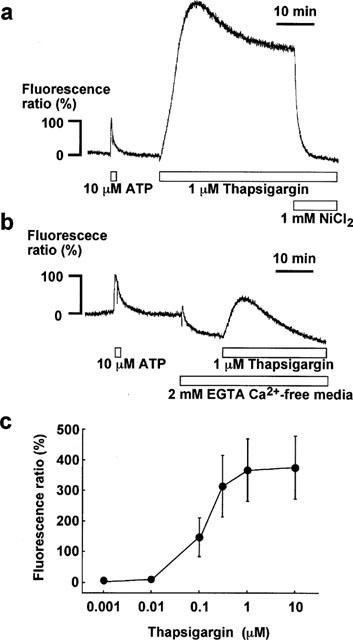
Effect of thapsigargin on [Ca2+]i in in situ endothelial cells of the porcine aortic valve. (a,b) Representative recordings of changes in [Ca2+]i induced by 10 μM ATP and 1 μM thapsigargin (TG) in endothelial cells in the presence (a) and absence (b) of extracellular Ca2+. In (a), 1 mM NiCl2 was applied during a sustained phase of the TG-induced elevation of [Ca2+]i. In (b), the valvular strip was exposed to the 2 mM EGTA-containing Ca2+-free media for 10 min before and during the application of 1 μM TG. The level of [Ca2+]i elevation induced by 10 μM ATP was assigned to be 100%. (c) Concentration-dependent effect of TG on [Ca2+]i in endothelial cells in situ. TG was applied in a cumulative manner. Data are the mean±s.e.mean (n=5).
When the strips were exposed to the Ca2+-free PSS containing 2 mM EGTA, [Ca2+]i level gradually decreased reaching a new steady level (−50.8±4.6%, n=3) at 10 min (Figure 1b). The endothelial cells were then stimulated with 1 μM TG in the absence of extracellular Ca2+, which caused only a transient [Ca2+]i elevation with no sustained [Ca2+]i elevation. The transient elevation of [Ca2+]i in the Ca2+-free PSS reached its peak slightly earlier (5.1±0.1 min, n=3) than that seen in the normal PSS. The peak level obtained with 1 μM TG in the Ca2+-free PSS was 47.3±8.0% (n=3) and much lower than that observed in the presence of extracellular Ca2+. After [Ca2+]i returned to the pre-stimulation level, EGTA was removed and the extracellular Ca2+ was replenished to a concentration of 1.25 mM, which restored the sustained increase in [Ca2+]i (data not shown). As a result, the second component of the TG-induced [Ca2+]i elevation observed in the presence of extracellular Ca2+ was dependent on the extracellular Ca2+.
Mechanism of the TG-induced Ca2+ influx in endothelial cells of the porcine aortic valve
To elucidate the mechanisms of the TG-induced Ca2+ influx in in situ endothelial cells, effects of possible inhibitors of Ca2+ signalling pathway on the sustained elevation of [Ca2+]i were investigated. The sustained elevation of [Ca2+]i was completely inhibited by 1 mM NiCl2 (Figure 1a), while 10 μM diltiazem, an L-type Ca2+ channel blocker had no effect (data not shown).
Figure 2 summarizes the effects of other inhibitors on the TG-induced sustained increase in [Ca2+]i. First, the effects of econazole, an inhibitor of cytochrome P450 mono-oxygenase, was examined (Figure 2a). The endothelial cells were pretreated with 10 μM econazole 5 min before and during the application of TG. This treatment inhibited the elevation of [Ca2+]i induced by the subsequent application of TG (Figure 2a, pre). Econazole significantly decreased the level of [Ca2+]i both in the first and second component induced by TG (first component: control, 510.4±31.3% vs econazole, 246.1±14.5%, n=5; second component (30 min): control, 389.7±29.1% vs econazole, 183.2±13.0%, n=5). When 10 μM econazole were applied after the TG-induced [Ca2+]i elevation reached the sustained phase, the [Ca2+]i level gradually decreased to 175.0±14.4% (n=5) at 15 min after the application (Figure 2a, post). The effect of the post-treatment with econazole was thus comparable to that of pretreatment.
Figure 2.
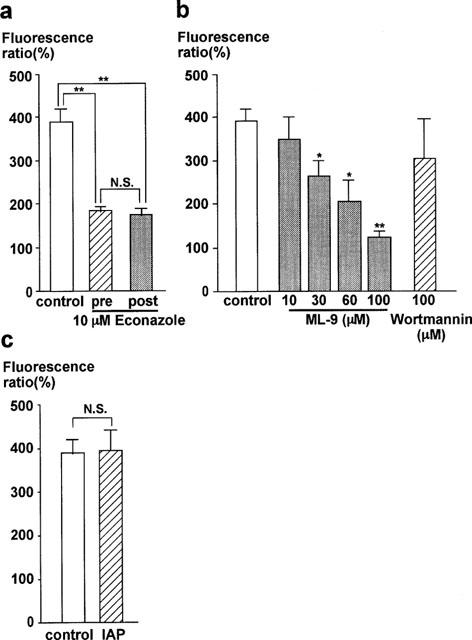
Characterization of the thapsigargin-induced Ca2+ entry in in situ endothelial cells of the porcine aortic valve. (a) Effect of econazole on the thapsigargin (TG)-induced sustained [Ca2+]i increase, as evaluated by the [Ca2+]i level obtained at 30 min after the application of 1 μM TG. Econazole (10 μM) was applied 5 min before the application of TG (pre) or 15 min after the application of TG (post). (b) Effects of pretreatment with myosin light chain kinase inhibitors, ML-9 and wortmannin on the [Ca2+]i level obtained at 30 min after the application of TG. Inhibitors were applied 10 min before and during the stimulation with 1 μM TG. (c) Effect of pertussis toxin (IAP) on the TG-induced sustained increase in [Ca2+]i. The valvular strips were treated with 900 ng ml−1 IAP for 3 h. All data are the mean±s.e.mean (n=5). The level of the control [Ca2+]i elevation induced by 10 μM ATP was considered to be 100%. *P<0.05, **P<0.01 compared with the control values. N.S., not significantly different.
Next, the effects of the inhibitors of MLCK, ML-9 and wortmannin, were examined (Figure 2b). Pretreatment with ML-9 10 min before and during the application of TG inhibited the elevation of [Ca2+]i in a concentration-dependent manner. A significant inhibition was seen at 30 μM and higher concentrations (Figure 2b). However, even with 100 μM ML-9, a residual small sustained [Ca2+]i elevation (122.3±13.6%, n=5) was observed. As in the case with econazole, the application of ML-9 during the sustained phase of the TG-induced [Ca2+]i elevation decreased [Ca2+]i to a similar level as that observed with pretreatment (data not shown). We also examined the effect of wortmannin, another MLCK inhibitor. In contrast to ML-9, pretreatment with wortmannin for 30 min prior to the application of TG had no significant effect on the TG-induced [Ca2+]i elevation even at a concentration of 100 μM wortmannin (Figure 2b; control, 389.7±29.1% vs wortmannin, 304.0±90.5%, n=5).
In our previous study, we showed that the endothelin-1-induced Ca2+-influx in endothelial cells of the porcine aortic valve was inhibited by treatment with 300 ng ml−1 IAP for 3 h (Aoki et al., 1994). However, IAP had no significant effect on the TG-induced elevation of [Ca2+]i, even after 3 h treatment at 900 ng ml−1 (Figure 2c). The levels of the TG-induced elevation of [Ca2+]i with and without treatment by IAP were 396.2±46.7% and 389.7±29.1% (n=5), respectively.
Effects of TG on smooth muscle [Ca2+]i and tension of porcine coronary arterial strips
To determine the functional significance of the TG-induced [Ca2+]i elevation in endothelial cells, the effects of TG on [Ca2+]i and tension of smooth muscle were simultaneously determined in porcine coronary arterial strips with and without endothelium. In the strips without endothelium, TG gradually elevated [Ca2+]i in smooth muscle, which reached a sustained phase within 10 min (Figure 3a). The level of [Ca2+]i elevation induced by TG was 20.0±2.1% of that seen with 118 mM K+ depolarization (n=5). However, this increase in [Ca2+]i did not produce any tension development (Figure 3a). A thromboxane A2 analogue, U46619, induced sustained increases in both [Ca2+]i and tension of smooth muscle in the strips without endothelium (Figure 3b). The level of [Ca2+]i and tension induced by 100 nM U46619 was 71.4±3.2 and 84.8±1.1% (n=3) at 10 min after the application, respectively. When applied during the sustained phase of this contraction, TG induced a slight increase, if any, in [Ca2+]i and had no effect on the U46619-induced tension development (Figure 3b). As a result, TG had no direct effect on the tension of smooth muscle.
Figure 3.
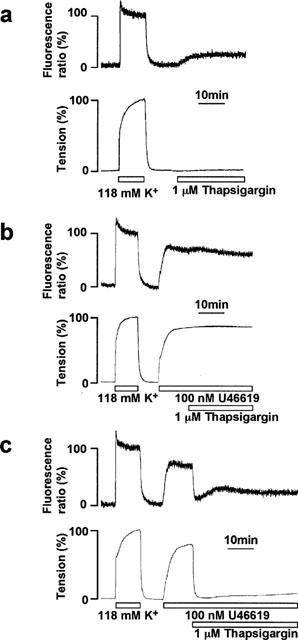
Effect of thapsigargin on smooth muscle [Ca2+]i and tension in the porcine coronary arterial strips with and without endothelium. (a) A representative recording of the changes in [Ca2+]i (upper trace) and tension (lower trace) induced by 1 μM thapsigargin (TG) under resting conditions in the strips without endothelium. (b, c) Representative recordings of changes in [Ca2+]i and tension induced by 1 μM TG during contraction induced by 100 nM U46619 in coronary arterial strips without (b) and with (c) endothelium. TG was applied 10 min after the initiation of the precontraction. The responsiveness to 118 mM K+-depolarization was recorded at the beginning of each measurement. The levels of [Ca2+]i and tension seen in normal (5.9 mM K+) PSS and 118 mM K+-PSS were designated to be 0 and 100%, respectively.
In the strips with an intact endothelium, U46619 also induced a rapid increase in [Ca2+]i and tension of smooth muscle, which reached the sustained phase within 10 min (Figure 3c). The levels of [Ca2+]i and tension were 71.5±1.5 and 83.5±5.2% (n=4) at 10 min, and 67.8±3.2 and 82.9±3.5% (n=4) at 25 min after the induction of contraction, respectively (Figure 5). When 1 μM TG was applied during the sustained contraction induced by U46619, both [Ca2+]i and tension of smooth muscle decreased rapidly until reaching their lowest levels (the initial relaxation) at 4.8±0.4 min (n=4), followed by sustained decreases at a level slightly higher than the resting level (Figure 3c). The levels of [Ca2+]i and tension decreased to 10.2±1.4 and 3.7±0.8% (n=4) at the initial relaxation, and 33.4±8.0 and 8.8±0.9% (n=4) at 15 min after the application of TG (25 min after the initiation of the precontraction), respectively (Figure 5).
Figure 5.
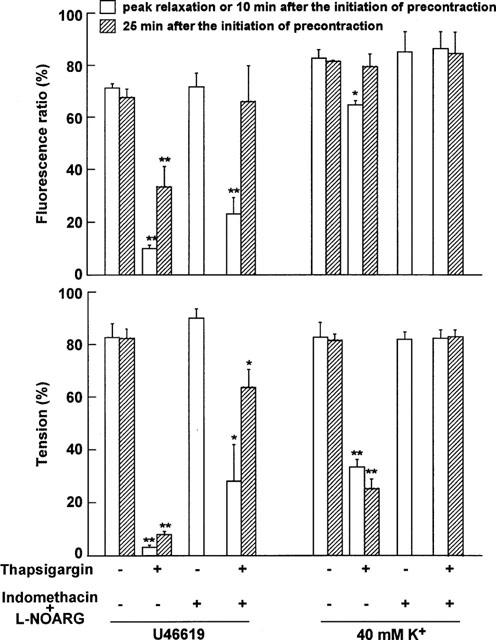
Summary of the effect of thapsigargin on [Ca2+]i and tension during the contraction induced by U46619 or 40 mM K+, either in the presence or absence of indomethacin and L-NOARG. The levels of [Ca2+]i (upper panel) and force (lower panel) were obtained at peak relaxation and at 25 min (sustained phase of relaxation) after the initiation of precontraction by U46619 and 40 mM K+, either in the presence or absence of 10 μM indomethacin+100 μM L-NOARG. During the control precontraction obtained in the absence of indomethacin and L-NOARG (thapsigargin (−), indomethacin+L-NOARG (−)), the level of [Ca2+]i and force sustained for more than 25 min. For the precontraction obtained in the presence of indomethacin and L-NOARG (thapsigargin (−), indomethacin+L-NOARG (+)), therefore, only the data obtained at 10 min after the initiation of precontraction are shown. The levels of [Ca2+]i and tension seen in normal (5.9 mM K+) PSS and 118 mM K+-PSS were considered to be 0 and 100 %, respectively. When the effects of indomethacin and L-NOARG were evaluated, the 0 and 100% levels were determined in their presence. The data are the mean±s.e.mean (n=3–4). *P<0.05, **P<0.01 compared with the value obtained at the corresponding time point without thapsigargin.
When TG was applied in a cumulative manner (0.001 through 10 μM) during the sustained phase of U46619-induced contraction, both [Ca2+]i and tension decreased in a concentration-dependent manner (data not shown). Significant decreases in both [Ca2+]i and tension were observed with 0.1 μM TG. The maximum response was obtained with 1 μM TG. The concentrations of TG required to induce the endothelium-dependent relaxation was thus correlated with those required to induce the sustained [Ca2+]i elevation in valvular strips (Figure 1c).
Mechanisms of the TG-induced endothelium-dependent relaxation
The relative contribution of prostacyclin, NO, and EDHF in the TG-induced relaxation was determined by either using specific inhibitors such as indomethacin and L-NOARG or by using high K+-depolarization to inhibit hyperpolarization as shown in Figure 4. The results are summarized in Figure 5. Treatment with 10 μM indomethacin had no significant effect on [Ca2+]i and tension of either the U46619-induced precontraction or the subsequent TG-induced relaxation (data not shown). On the contrary, the treatment with 10 μM indomethacin plus 100 μM L-NOARG significantly attenuated the decreases in [Ca2+]i and tension, especially in the sustained phase of the TG-induced relaxation (Figure 4a). Under this condition, TG induced only a large transient decrease in [Ca2+]i without a sustained decrease, while it induced an initial large relaxation followed by a small sustained relaxation (Figure 4a). Since treatment with indomethacin and L-NOARG augmented the 118 mM K+- and U46619-induced contraction, the level of [Ca2+]i and tension obtained at rest and by 118 mM K+ in the presence of indomethacin and L-NOARG were considered to be 0 and 100%, respectively (Figure 5). Based on these criteria, the levels of [Ca2+]i and tension obtained at 10 min after the initiation of the U46619-induced precontraction were 71.9±5.4 and 90.6±3.1% (n=3), respectively (Figure 5). In the presence of indomethacin and L-NOARG, the levels of [Ca2+]i at the initial phase of the TG-induced relaxation and at 15 min after the application of TG (25 min after the initiation of precontraction) were 23.4±6.2 and 66.1±14.0% (n=3), respectively (Figure 5). The [Ca2+]i level seen at 15 min in the presence of indomethacin and L-NOARG did not differ significantly from that obtained during the precontraction induced by 100 mM U46619 (67.8±3.2%, n=3) (Figure 5). The levels of tension seen at the initial relaxation and at 15 min after the application of TG were 28.5±13.8 and 64.3±6.7% (n=3), respectively (Figure 5). Both levels were significantly higher than those obtained with TG in the absence of inhibitors (control relaxation), and significantly lower than those obtained during the precontraction (Figure 5). Treatment with L-NOARG alone had the same effect as that seen with the combination of indomethacin and L-NOARG (data not shown).
Figure 4.
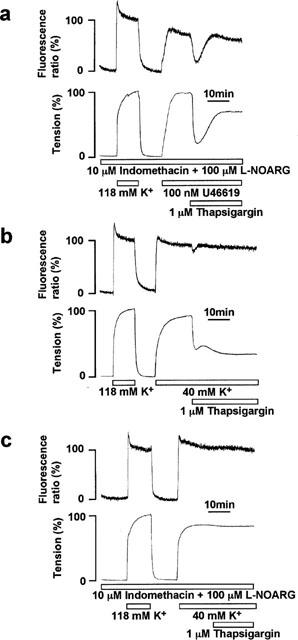
Effect of indomethacin, Nω-nitro-L-arginine (L-NOARG) and high K+-depolarization on the thapsigargin-induced endothelium-dependent relaxation. (a) A representative recording of the changes in [Ca2+]i (upper trace) and tension (lower trace) of smooth muscle induced by 1 μM thapsigargin (TG) during contraction induced by 100 nM U46619 in the strips with endothelium that were pretreated with 10 μM indomethacin and 100 μM L-NOARG. TG was applied 10 min after the initiation of the precontraction. (b) A representative recording of changes in [Ca2+]i and tension of smooth muscle induced by 1 μM TG during the 40 mM K+-induced contraction in the strip with endothelium. (c) A representative recording of changes in [Ca2+]i and tension of smooth muscle induced by 1 μM TG during the 40 mM K+-induced contraction in the presence of 10 μM indomethacin and 100 μM L-NOARG in strips with endothelium. The responsiveness to 118 mM K+-depolarization was recorded at the beginning of each measurement.
High extracellular K+ has been shown to inhibit hyperpolarization of the membrane potential induced by EDHF in smooth muscle cells (Nagao & Vanhoutte, 1992). We previously showed that the extracellular K+ concentration of 40 mM was high enough to inhibit the effects of EDHF induced by substance P in the porcine coronary artery (Kuroiwa et al., 1995) and that induced by TG in porcine renal artery (Ihara et al., 1999). Accordingly, to clarify the involvement of EDHF, the relaxation induced by TG was investigated during the contraction induced by 40 mM K+-depolarization. Stimulation with 40 mM K+-depolarization caused rapid increases in [Ca2+]i and tension, followed by the sustained increases. The levels of [Ca2+]i and tension during the sustained contraction were 83.0±3.3 and 83.0±5.5% at 10 min, and 81.6±0.5 and 82.0±2.7 % at 25 min, respectively (n=3, Figure 5). The level of tension obtained with 40 mM K+ was thus similar to that obtained with 100 nM U46619, while the level of [Ca2+]i was higher than that obtained with U46619 (Figure 5). When 1 μM TG was applied during the 40 mM K+-induced sustained contraction, it induced only a small transient decrease in [Ca2+]i and a sustained decrease in tension (Figure 4b). As a result, the sustained decrease in tension was not accompanied by a decrease in [Ca2+]i (Figure 4b). The levels of [Ca2+]i and tension at the initial relaxation were 64.8±1.8 and 34.0±2.7% (n=3), respectively (Figure 5). The levels of [Ca2+]i and tension obtained at 15 min after the application of TG were 79.6±4.8 and 25.7±3.7% (n=3), respectively (Figure 5). The [Ca2+]i level obtained at 15 min after the application of TG did not significantly differ from that obtained during the 40 mM K+-induced precontraction, while the level of tension was significantly lower than the precontraction level. Treatment with 10 μM indomethacin had no effect on the relaxation induced by TG during 40 mM K+-depolarization. However, the combination of indomethacin and L-NOARG completely abolished the decreases in both [Ca2+]i and tension (Figure 4c). The levels of [Ca2+]i and tension obtained with TG, indomethacin and L-NOARG during the 40 mM K+-induced contraction did not differ significantly from those seen during the control contraction induced by 40 mM K+ (Figure 5).
Discussion
The present study demonstrated that TG induced a large sustained elevation of [Ca2+]i in the in situ endothelial cells of the porcine aortic valve, and an endothelium-dependent large sustained relaxation in porcine coronary arterial strips. One of the most intriguing observations is that the TG-induced relaxation continued to be sustained as long as the arterial strips were exposed to TG. The endothelium-dependent relaxations induced by such receptor agonists as acetylcholine, bradykinin, endothelin-1 or substance P were originally shown to be relatively transient (Furchgott & Zawadzki, 1980; Gryglewski et al., 1986). Among the reasons for the transient nature of the endothelium-dependent relaxation are the short half life of NO (Palmer et al., 1987) and the transient nature of the [Ca2+]i elevation induced by agonists (Aoki et al., 1991; Himmel et al., 1993; Hirano & Kanaide, 1993; Kuroiwa et al., 1995). The TG-induced sustained elevation of [Ca2+]i in endothelial cells and the sustained endothelium-dependent relaxation in arterial strips were thus considered to be unique in this respect.
The TG-induced elevation of [Ca2+]i in the valvular endothelial cells consisted of the rapid peak (the first component) and the following sustained increase (the second component). In Ca2+-free PSS, the first component was partially inhibited, while the second one was completely abolished. The second component was also completely abolished by Ni2+. These findings suggested that the second component of the TG-induced [Ca2+]i elevation was due to an influx of extracellular Ca2+, and that the first component was partly due to the Ca2+ release from the intracellular store sites and partly due to the Ca2+ influx. In both the present study and a previous study (Kawasaki et al., 1999), a Ca2+ channel blocker, diltiazem, had no effect on TG-induced sustained [Ca2+]i elevation, thus indicating that L-type Ca2+ channel is not involved in the TG-induced Ca2+ influx in endothelial cells in situ. This observation is consistent with reports showing no activity of L-type Ca2+ channel in either freshly isolated (Busse et al., 1988) or cultured endothelial cells (Colden-Stanfield et al., 1987). On the other hand, a similar large sustained [Ca2+]i elevation was observed with CPA in porcine aortic valvular strips (Higuchi et al., 1996). Both TG and CPA are inhibitors of the Ca2+ pump of the intracellular Ca2+ stores (Seidler et al., 1989; Thastrup et al., 1990). The inhibition of the Ca2+ pump was considered to deplete Ca2+ in store sites and thereby induce Ca2+ entry, which is called capacitative Ca2+ entry (Putney & McKay, 1999). The Ca2+ influx induced by TG was thus suggested to be due to the capacitative Ca2+ entry, and the transient [Ca2+]i elevation seen in the absence of extracellular Ca2+ reflected the release and the resultant depletion of Ca2+ in the store sites.
The mechanism of activation and regulation of capacitative Ca2+ entry have yet to be determined in in situ endothelial cells. In general, two mechanisms have been proposed for coupling between the store depletion and the activation of Ca2+ entry; the direct coupling mediated by direct interaction between the inositol 1,4,5- trisphosphate receptor and Ca2+ entry channel (Boulay et al., 1999; Putney, 1999) or that mediated by cytoskeletal structures such as actin filaments (Holda & Blatter, 1997), and the indirect coupling mediated by second messengers. The candidates proposed for the second messengers include Ca2+ influx factor, cyclic GMP, cytochrome P450 metabolites of arachidonic acids, heterotrimeric GTP-binding proteins, small GTP-binding proteins, tyrosine kinase, myosin light chain kinase and protein phosphatases (for review, see Parekh & Penner, (1997)).
In the present study, econazole was used as a cytochrome P450 inhibitor (Graier et al., 1995; Mason et al., 1993; Vostal & Fratantoni, 1993), and we found that it significantly inhibited the TG-induced Ca2+ influx. This finding suggested cytochrome P450 to be involved in the regulation of the TG-induced Ca2+ influx in in situ endothelial cells as well as in cultured endothelial cells (Graier et al., 1995). MLCK was suggested to be involved in the capacitative Ca2+ influx in cultured porcine aortic endothelial cells (Watanabe et al., 1996). In the present study, ML-9 inhibited the TG-induced [Ca2+]i elevation in a concentration-dependent manner. However, wortmannin had no significant effect on TG-induced [Ca2+]i elevation. The concentration of wortmannin used in the present study (100 μM) was high enough to inhibit MLCK in addition to phosphatidylinositol 3-kinase (Yano et al., 1993). Based on our results, MLCK was not suggested to play an important role in the TG-induced Ca2+ influx in in situ endothelial cells. The inhibition of the TG-induced Ca2+ influx by ML-9 may be due to an inhibition of some kinases other than MLCK. Since both ML-9 and wortmannin inhibited the TG-induced Ca2+ influx in cultured endothelial cells (Watanabe et al., 1996), the kinase involved in the TG-induced Ca2+ influx might thus have been altered by culture conditions. Several authors reported GTP-binding proteins to be involved in the activation of capacitative Ca2+ entry (Bird & Putney, 1993; Sargeant et al., 1993). We previously reported that the IAP-sensitive GTP-binding protein was involved in the endothelin-1-induced Ca2+ influx but not Ca2+ release in in situ endothelial cells (Aoki et al., 1994). However, IAP had no effect on the TG-induced [Ca2+]i elevation, thus suggesting that the IAP-sensitive GTP-binding protein did not play a role in the TG-induced Ca2+ influx in in situ endothelial cells. Collectively, the present study suggested that cytochrome P450 and ML-9-sensitive protein phosphorylation were involved in the TG-induced Ca2+ influx in in situ endothelial cells of porcine aortic valve. The mechanisms of how these two factors contribute to the TG-induced Ca2+ influx remain to be elucidated.
We previously reported that CPA induced a similar endothelium-dependent relaxation in the porcine coronary artery, and induced a large sustained [Ca2+]i elevation in in situ endothelial cells (Higuchi et al., 1996). Both TG and CPA have been reported to induce a sustained elevation of [Ca2+]i due to the activation of capacitative Ca2+ entry (Putney & McKay, 1999). It is therefore conceivable that the capacitative Ca2+ entry induced by TG or CPA was functionally linked to the sustained production of endothelium-derived relaxing factors in the porcine coronary artery.
In the present study, we evaluated the relative contribution of NO, EDHF and prostacyclin in the TG-induced relaxation by using specific inhibitors. Indomethacin did not alter the extent of the TG-induced relaxation at all, thus suggesting that prostacyclin played a negligible role in the relaxation. L-NOARG significantly but only partially inhibited the TG-induced relaxation, thus suggesting that not only NO but also other relaxing factors contributed to the relaxation. The elevation of the external K+ concentration also partially inhibited the relaxation induced by TG, but the combination of L-NOARG and high K+ completely abolished it. These observations suggested that NO and EDHF were major factors contributing to the TG-induced relaxation. L-NOARG inhibited the sustained relaxation more potently than high K+, although the [Ca2+]i decrease in smooth muscle cells during the sustained relaxation was completely inhibited by either L-NOARG or high K+. On the contrary, the initial phase of the TG-induced decreases in tension was similarly inhibited by high K+ and L-NOARG. The findings of the present study suggest that NO was the major mediator of the sustained phase of the TG-induced relaxation, while both NO and EDHF contributed to the initial phase of the relaxation. Such a time-dependent alteration of the relative contribution of NO and EDHF in the TG-induced relaxation is similar to that observed in the CPA-induced relaxation in the porcine coronary artery (Higuchi et al., 1996). Furthermore, examining the [Ca2+]i-force relationship during relaxation indicates that the Ca2+-sensitivity of the contractile apparatus decreased during sustained relaxation. The decrease in the myofilament Ca2+-sensitivity was obvious during the sustained relaxation seen with the 40 mM K+-induced precontraction, when the contribution of EDHF was blocked and NO was the only mediator of the relaxation. These observations are consistent with the involvement of NO in the late sustained relaxation, because NO was shown to increase the cytosolic cyclic GMP level (Ignarro et al., 1987), which was shown to decrease the Ca2+ sensitivity of the contractile apparatus (Nishimura & van Breemen, 1989). It could thus be concluded that the sustained [Ca2+]i elevation due to the Ca2+ entry induced by TG in endothelial cells was most closely linked to the production of NO.
The link between the [Ca2+]i elevation and NO production in endothelial cells is consistent with a biochemical study showing NO synthase to be a Ca2+/calmodulin-dependent enzyme (Fleming et al., 1997). However, NO synthase activity is considered to be regulated both in a Ca2+-dependent and Ca2+-independent manner, and the alteration of the Ca2+-sensitivity of the NO production process is considered to play an important regulatory role (Fleming et al., 1997). We previously reported that the relationship between the [Ca2+]i elevation and NO production in endothelial cells varies with the type of stimulation (Mizuno et al., 1998; 2000). The phosphorylation of NO synthase by Akt/protein kinase B was reported to activate NO synthase in a Ca2+-independent manner (Dimmeler et al., 1999; Fulton et al., 1999). The findings of the present study, using thapsigargin as the endothelial stimulus, emphasize the importance of the Ca2+ signal as a primary determinant of the NO synthase activity.
In contrast to endothelial cells, TG induced a small sustained elevation of [Ca2+]i in smooth muscle cells of the porcine coronary artery. Furthermore, such [Ca2+]i elevation was not linked to the force development. It is therefore possible that TG decreased the Ca2+-sensitivity of contractile apparatus, and therefore did not induce any force development despite of [Ca2+]i elevation. However, this is not the case because TG had almost a negligible effect on the U46619-induced contraction. On the other hand, it is possible that the level of [Ca2+]i elevation was not high enough to cause a contraction. We reported that 15 mM K+ induced a [Ca2+]i elevation (∼20% of the 118 mM K+-induced elevation) without any force development, but higher concentrations of external K+ induced both a [Ca2+]i elevation and force development (Abe et al., 1990). Therefore, the capacitative Ca2+ entry pathway is suggested to play a greater role as a major Ca2+ influx pathway in endothelial cells than it plays in smooth muscle cells.
In conclusion, TG induced a large sustained elevation of [Ca2+]i consisting of an initial rapid phase and a sustained phase in in situ endothelial cells. The initial rapid [Ca2+]i elevation was due to both the Ca2+ release from the intracellular store sites and capacitative Ca2+ entry, while the sustained [Ca2+]i elevation was due to the Ca2+ entry. Cytochrome P450 and ML-9-sensitive protein phosphorylation were suggested to be involved in the TG-induced Ca2+ influx. The TG-induced Ca2+ influx was shown to be functionally linked to the production of endothelium-derived relaxing factors and thereby induce relaxation in the porcine coronary artery. This relaxation was composed of two components; an initial rapid relaxation due to NO and EDHF and a late sustained relaxation mainly due to NO. Thus, the TG-induced sustained [Ca2+]i elevation due to capacitative Ca2+ entry in endothelial cells most closely correlated with the production of NO. During the treatment with TG, the sustained Ca2+ elevation might therefore help to maintain a high activity of NO synthase thereby causing a sustained production of NO, which in turn induces the sustained relaxation of coronary artery despite the short half life of NO.
Acknowledgments
We thank Mr Brian Quinn for comments and help with the manuscript. This study was supported in part by Grants-in-Aid for Scientific Research (Nos. 10557072, 11838013, 11670687), for the Encouragement of Young Scientists (No. 10770308) from the Ministry of Education, Science, Sports and Culture, Japan, by the Research Grant for Cardiovascular Diseases (11C-1) from the Ministry of Health and Welfare, Japan, and by grants from the Vehicle Racing Commemorative Foundation, the Foundation for the Promotion of Clinical Medicine, the Suzuken Memorial Foundation and KANZAWA Medical Research Foundation.
Abbreviations
- [Ca2+]i
cytosolic Ca2+ concentration
- CPA
cyclopiazonic acid
- DMEM
Dulbecco's modified Eagle medium
- EDHF
endothelium-derived hyperpolarizing factor
- fura-2/AM
fura-2 acetoxymethyl ester
- IAP
islet activating protein, pertussis toxin
- L-NOARG
Nω-nitro-L-arginine
- MLCK
myosin light chain kinase
- NO
nitric oxide
- PSS
physiological salt solution
- TG
thapsigargin
References
- ABE S., KANAIDE H., NAKAMURA M. Front-surface fluorometry with fura-2 and effects of nitroglycerin on cytosolic calcium concentrations and on tension in the coronary artery of the pig. Br. J. Pharmacol. 1990;101:545–552. doi: 10.1111/j.1476-5381.1990.tb14118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALVAREZ J., MONTERO M., GARCIA-SANCHO J. Cytochrome P450 may regulate plasma membrane Ca2+ permeability according to the filling state of the intracellular Ca2+ stores. FASEB J. 1992;6:786–792. doi: 10.1096/fasebj.6.2.1537469. [DOI] [PubMed] [Google Scholar]
- AOKI H., KOBAYASHI S., NISHIMURA J., KANAIDE H. Sensitivity of G-protein involved in endothelin-1-induced Ca2+ influx to pertussis toxin in porcine endothelial cells in situ. Br. J. Pharmacol. 1994;111:989–996. doi: 10.1111/j.1476-5381.1994.tb14841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AOKI H., KOBAYASHI S., NISHIMURA J., YAMAMOTO H., KANAIDE H. Endothelin induces the Ca2+-transient in endothelial cells in situ. Biochem. Biophys. Res. Commun. 1991;181:1352–1357. doi: 10.1016/0006-291x(91)92087-z. [DOI] [PubMed] [Google Scholar]
- BIRD G.S., PUTNEY J.W.J. Inhibition of thapsigargin-induced calcium entry by microinjected guanine nucleotide analogues. Evidence for the involvement of a small G-protein in capacitative calcium entry. J. Biol. Chem. 1993;268:21486–21488. [PubMed] [Google Scholar]
- BOULAY G., BROWN D.M., QIN N., JIANG M., DIETRICH A., ZHU M.X., CHEN Z., BIRNBAUMER M., MIKOSHIBA K., BIRNBAUMER L. Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5- trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14955–14960. doi: 10.1073/pnas.96.26.14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUSSE R., FICHTNER H., LUCKHOFF A., KOHLHARDT M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cells. Am. J. Physiol. 1988;255:H965–H969. doi: 10.1152/ajpheart.1988.255.4.H965. [DOI] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLDEN-STANFIELD M., SCHILLING W.P., RITCHIE A.K., ESKIN S.G., NAVARRO L.T., KUNZE D.L. Bradykinin-induced increases in cytosolic calcium and ionic currents in cultured bovine aortic endothelial cells. Circ. Res. 1987;61:632–640. doi: 10.1161/01.res.61.5.632. [DOI] [PubMed] [Google Scholar]
- DIMMELER S., FLEMING I., FISSLTHALER B., HERMANN C., BUSSE R., ZEIHER A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- DI VIRGILIO F., STENBERG T.H., SILVERSTEIN S.C.Organic-anion transport inhibitors to facilitate measurement of cytosolic free Ca2+ with fura-2 Methods in Cell Biology 1989San Diego: Academic Press; 453–462.ed. Tartakoff, A.M. pp [DOI] [PubMed] [Google Scholar]
- FLEMING I., BAUERSACHS J., BUSSE R. Calcium-dependent and calcium-independent activation of the endothelial NO synthase. J. Vasc. Res. 1997;34:165–174. doi: 10.1159/000159220. [DOI] [PubMed] [Google Scholar]
- FULTON D., GRATTON J.P., MCCABE T.J., FONTANA J., FUJIO Y., WALSH K., FRANKE T.F., PAPAPETROPOULOS A., SESSA W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURCHGOTT R.F., VANHOUTTE P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GRAIER W.F., SIMECEK S., STUREK M. Cytochrome P450 mono-oxygenase-regulated signaling of Ca2+ entry in human and bovine endothelial cells. J. Physiol. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRYGLEWSKI R.J., MONCADA S., PALMER R.M. Bioassay of prostacyclin and endothelium-derived relaxing factor (EDRF) from porcine aortic endothelial cells. Br. J. Pharmacol. 1986;87:685–694. doi: 10.1111/j.1476-5381.1986.tb14586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HIGUCHI Y., NISHIMURA J., KOBAYASHI S., KANAIDE H. CPA induces a sustained increase in [Ca2+]i of endothelial cells in situ and relaxes porcine coronary artery. Am. J. Physiol. 1996;270:H2038–H2049. doi: 10.1152/ajpheart.1996.270.6.H2038. [DOI] [PubMed] [Google Scholar]
- HIMMEL H.M., WHORTON A.R., STRAUSS H.C. Intracellular calcium, currents, and stimulus-response coupling in endothelial cells. Hypertension. 1993;21:112–127. doi: 10.1161/01.hyp.21.1.112. [DOI] [PubMed] [Google Scholar]
- HIRANO K., KANAIDE H. Cytosolic Ca2+ transients in endothelium-dependent relaxation of pig coronary artery, and effects of captopril. Eur. J. Pharmacol. 1993;250:439–446. doi: 10.1016/0014-2999(93)90031-c. [DOI] [PubMed] [Google Scholar]
- HIRANO K., KANAIDE H., ABE S., NAKAMURA M. Effects of diltiazem on calcium concentration in the cytosol and on force of contractions in porcine coronary arterial strips. Br. J. Pharmacol. 1990;101:273–280. doi: 10.1111/j.1476-5381.1990.tb12700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLDA J.R., BLATTER L.A. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Lett. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J., BUGA G.M., WOOD K.S., BYRNS R.E., CHAUDHURI G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IHARA E., HIRANO K., NISHIMURA J., NAWATA H., KANAIDE H. Thapsigargin-induced endothelium-dependent triphasic regulation of vascular tone in the porcine renal artery. Br. J. Pharmacol. 1999;128:689–699. doi: 10.1038/sj.bjp.0702821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANAIDE H.Measurement of [Ca2+]i in smooth muscle strips using front-surface fluorimetry Methods in Molecular Biology 1999Totowa, NJ: Humana Press Inc; 269–277.ed. Lambert, D.G. pp [DOI] [PubMed] [Google Scholar]
- KATUSIC Z.S., SHEPHERD J.T. Endothelium-derived vasoactive factors: II. Endothelium-dependent contraction. Hypertension. 1991;18:III86–III92. doi: 10.1161/01.hyp.18.5_suppl.iii86. [DOI] [PubMed] [Google Scholar]
- KAWASAKI J., HIRANO K., HIRANO M., NISHIMURA J., FUJISHIMA M., KANAIDE H. Troglitazone inhibits capacitative Ca2+ entry in endothelial cell. Eur. J. Pharmacol. 1999;373:111–120. doi: 10.1016/s0014-2999(99)00257-5. [DOI] [PubMed] [Google Scholar]
- KOMORI K., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor. Blood Vessels. 1990;27:238–245. doi: 10.1159/000158815. [DOI] [PubMed] [Google Scholar]
- KRUSE H.-J., NEGRESCU E.V., WEBER P.C., SIESS W. Thrombin-inactivated Ca2+ influx and protein tyrosine phosphorylation in endothelial cells is inhibited by herbimycin A. Biochem. Biophys. Res. Commun. 1994;202:1651–1656. doi: 10.1006/bbrc.1994.2123. [DOI] [PubMed] [Google Scholar]
- KUROIWA M., AOKI H., KOBAYASHI S., NISHIMURA J., KANAIDE H. Role of GTP-protein and endothelium in contraction induced by ethanol in pig coronary artery. J. Physiol. 1993;470:521–537. doi: 10.1113/jphysiol.1993.sp019873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUROIWA M., AOKI H., KOBAYASHI S., NISHIMURA J., KANAIDE H. Mechanism of endothelium-dependent relaxation induced by substance P in the coronary artery of the pig. Br. J. Pharmacol. 1995;116:2040–2047. doi: 10.1111/j.1476-5381.1995.tb16409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASON M.J., MAYER B., HYMEL L.J. Inhibition of Ca2+ transport pathways in thymic lymphocytes by econazole, miconazole, and SKF 96365. Am. J. Physiol. 1993;264:C654–C662. doi: 10.1152/ajpcell.1993.264.3.C654. [DOI] [PubMed] [Google Scholar]
- MIZUNO O., HIRANO K., NISHIMURA J., KUBO C., KANAIDE H. Mechanism of relaxation induced by thrombin in pig coronary arterial strips with endothelium. Eur. J. Pharmacol. 1998;351:67–77. doi: 10.1016/s0014-2999(98)00292-1. [DOI] [PubMed] [Google Scholar]
- MIZUNO O., KOBAYASHI S., HIRANO K., NISHIMURA J., KUBO C., KANAIDE H. Stimulus-specific alteration of the relationship between cytosolic Ca2+ transients and nitric oxide production in endothelial cells ex vivo. Br. J. Pharmacol. 2000;130:1140–1146. doi: 10.1038/sj.bjp.0703420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGAO H., VANHOUTTE P.M. Hyperpolarization as a mechanism for endothelium-dependent relaxations in the porcine coronary artery. J. Physiol. 1992;445:355–367. doi: 10.1113/jphysiol.1992.sp018928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWBY A.C., HENDERSON A.H. Stimulus-secretion coupling in vascular endothelial cells. Annu. Rev. Physiol. 1990;52:661–674. doi: 10.1146/annurev.ph.52.030190.003305. [DOI] [PubMed] [Google Scholar]
- NISHIMURA J., VAN BREEMEN C. Direct regulation of smooth muscle contractile elements by second messengers. Biochem. Biophys. Res. Commun. 1989;163:929–935. doi: 10.1016/0006-291x(89)92311-5. [DOI] [PubMed] [Google Scholar]
- PALMER R.M., FERRIGE A.G., MONCADA S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- PAREKH A.B., PENNER R. Store depletion and calcium influx. Physiol. Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- PFEIFFER F., SCHMID A., SCHURUZ I. Capacitative Ca2+ influx and Ca2+-dependent nonselective cation pathway are discriminated by genistein in mouse pancreatic acinar cells. Pflugers Arch. 1995;430:916–922. doi: 10.1007/BF01837405. [DOI] [PubMed] [Google Scholar]
- PUTNEY J.W., JR TRP, inositol 1,4,5-trisphosphate receptors, and capacitative calcium entry. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14669–14671. doi: 10.1073/pnas.96.26.14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUTNEY J.W., JR, MCKAY R.R. Capacitative calcium entry channels. Bioessays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- SARGEANT P., FARNDALE R.W., SAGE S. Ca2+ store depletion in dimethyl BAPTA-loaded human platelets increases protein tyrosin phosphorylation in the absence of a rise in cytosolic calcium. Exp. Physiol. 1994;79:269–272. doi: 10.1113/expphysiol.1994.sp003762. [DOI] [PubMed] [Google Scholar]
- SARGEANT P., FARNDALE R.W., SAGE S.O. ADP- and thapsigargin-evoked Ca2+ entry and protein-tyrosine phosphorylation are inhibited by the tyrosin kinase inhibitors genistein and methyl-2,5-dihydroxycinnamate in fura-2 loaded human platelets. J. Biol. Chem. 1993;268:18151–18156. [PubMed] [Google Scholar]
- SEIDLER N.W., JONA I., VEGH M., MARTONOSI A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 1989;264:17816–17823. [PubMed] [Google Scholar]
- THASTRUP O., CULLEN P.J., DROBAK B.K., HANLEY M.R., DAWSON A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOSTAL J.G., FRATANTONI J.C. Econazole inhibits thapsigargin-induced platelet calcium influx by mechanisms other than cytochrome P-450 inhibition. Biochem. J. 1993;295:525–529. doi: 10.1042/bj2950525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATANABE H., TAKAHASHI R., ZHANG X.-X., KAKIZAWA H., HAYASHI H., OHNO R. Inhibition of agonist-induced Ca2+ entry in endothelial cells by myosin light-chain kinase inhibitor. Biochem. Biophys. Res. Commun. 1996;225:777–784. doi: 10.1006/bbrc.1996.1250. [DOI] [PubMed] [Google Scholar]
- YANO H., NAKANISHI S., KIMURA K., HANAI N., SAITOH Y., FUKUI Y., NONOMURA Y., MATSUDA Y. Inhibition of histamine secretion by wortmannin through the blockade of phosphatidylinositol 3-kinase in RBL-2H3 cells. J. Biol. Chem. 1993;268:25846–25856. [PubMed] [Google Scholar]