

Abstract
NO synthase (NOS) inhibitors partially block bradykinin (BK)-mediated vasorelaxation. Here we investigated whether this is due to incomplete NOS inhibition and/or NO release from storage sites. We also studied the mechanism behind ACE inhibitor-mediated BK potentiation.
Porcine coronary arteries (PCAs) were mounted in organ baths, preconstricted, and exposed to BK or the ACE-resistant BK analogue Hyp3-Tyr(Me)8-BK (HT-BK) with or without the NOS inhibitor L-NAME (100 μM), the NO scavenger hydroxocobalamin (200 μM), the Ca2+-dependent K+-channel blockers charybdotoxin+apamin (both 100 nM), or the ACE inhibitor quinaprilat (10 μM).
BK and HT-BK dose-dependently relaxed preconstricted vessels (pEC50 8.0±0.1 and 8.5±0.2, respectively). pEC50's were &10 fold higher with quinaprilat, and &10 fold lower with L-NAME or charybdotoxin+apamin. Complete blockade was obtained with hydroxocobalamin or L-NAME+ charybdotoxin+apamin.
Repeated exposure to 100 nM BK or HT-BK, to deplete NO storage sites, produced progressively smaller vasorelaxant responses. With L-NAME, the decrease in response occurred much more rapidly. L-Arginine (10 mM) reversed the effect of L-NAME.
Adding quinaprilat to the bath following repeated exposure (with or without L-NAME), at the time BK and HT-BK no longer induced relaxation, fully restored vasorelaxation, while quinaprilat alone had no effect. Quinaprilat also relaxed vessels that, due to pretreatment with hydroxocobalamin or L-NAME+charybdotoxin+apamin, previously had not responded to BK.
In conclusion, L-NAME-resistant BK-induced relaxation in PCAs depends on NO from storage sites, and is mediated via stimulation of guanylyl cyclase and/or Ca2+-dependent K+-channels. ACE inhibitors potentiate BK independent of their effect on BK metabolism.
Keywords: ACE inhibitor, bradykinin, coronary artery, nitric oxide, NO synthase
Introduction
Bradykinin induces vasodilation via endothelial bradykinin type 2 (B2) receptors. This effect can be blocked partly by inhibitors of nitric oxide synthase (NOS), suggesting a role for de-novo synthesis of nitric oxide (NO) from L-arginine by NOS (Palmer et al., 1988; Gardiner et al., 1990; Rees et al., 1990; Mombouli et al., 1992; Bjornstad-Ostensen et al., 1997). The relaxant effect of bradykinin that is not blocked by NOS inhibitors is generally attributed to so-called endothelium-derived hyperpolarizing factors, of which the exact identity has not yet been established. Several candidates have been proposed, including prostacyclin, potassium and cytochrome P-450 products of arachidonic acid (Mombouli & Vanhoutte, 1997; Edwards et al., 1998; Fisslthaler et al., 1999). However, NOS inhibitors, even at high concentrations, do not block NO release completely (Cohen et al., 1997). Moreover, in-vivo studies in the rat hindlimb (Davisson et al., 1996) and in-vitro studies in the isolated perfused rat heart (Danser et al., 1998) have shown that bradykinin also induces release of NO from stores of NO-containing factors, such as S-nitroso-thiols and dinitrosyl iron (II) thiol complexes (Ignarro, 1990; Myers et al., 1990; Verdernikov et al., 1992). Depletion of such stores occurred only after repeated exposure to bradykinin or after prolonged inhibition of NOS, treatments which themselves do not alter the response to NO (Davisson et al., 1996; Danser et al., 1998). Taken together therefore, bradykinin-induced relaxation in the presence of NOS inhibitors might also be due to NO generated by residual NOS activity or to NO released from storage sites.
Angiotensin-converting enzyme (ACE) inhibitors block bradykinin degradation. Accumulation of bradykinin is believed to contribute to the beneficial effects of ACE inhibitors in hypertension and heart failure, although elevated bradykinin levels were not always found during ACE inhibitor treatment (Miki et al., 1996; Campbell et al., 1999). Two recent studies propose that ACE inhibitors potentiate bradykinin beyond blocking its hydrolysis, by inhibiting desensitization of its receptor. These studies were performed in Chinese hamster ovary cells transfected with human B2 receptors and human ACE (Minshall et al., 1997) and in porcine aortic endothelial cells that naturally express these proteins (Benzing et al., 1999). The mechanism underlying the ACE inhibitor-induced inhibition of B2 receptor desensitization is currently unknown, but it may involve interference with the translocation of B2 receptor to caveolin-rich membrane domains (Haasemann et al., 1998; Benzing et al., 1999; Marcic et al., 1999). Dendorfer et al. (2000) however, using the isolated perfused rat Langendorff heart, found no evidence for ACE inhibitor-induced B2 receptor upregulation and suggested that the ACE inhibitor-induced potentiation of bradykinin was due to inhibition of bradykinin degradation in the vicinity of B2 receptors (e.g., in caveolae).
It was the aim of the present study to investigate, in intact porcine coronary arteries (PCAs), (1) whether the NOS inhibitor-resistant bradykinin-induced vasorelaxation involves NO, and (2) whether the ACE inhibitor quinaprilat potentiates bradykinin via blockade of bradykinin metabolism or via other mechanisms. To address the second question, we used the ACE-resistant bradykinin analogue [Hyp3-Tyr(Me)8]-bradykinin (Rhaleb et al., 1990; Minshall et al., 1997).
Methods
Drugs
Bradykinin (acetate salt), prostaglandin F2α (PGF2α), 9,11-dideoxy-11α,9α-epoxymethano-prostaglandin F2α (U46619), substance P (acetate salt), L-arginine HCl, Nω-nitro-L-arginine methyl ester HCl (L-NAME), aminoguanidine, 7-nitroindazole (7-NI), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), hydroxocobalamin (acetate salt), indomethacin, glibenclamide, charybdotoxin, apamin, sulphaphenazole and captopril were from Sigma-Aldrich Chemie (Zwijndrecht, The Netherlands). [Hyp3-Tyr(Me)8]-bradykinin was from Calbiochem/Novabiochem AG, Läufelfingen, Switzerland. D-Arg[Hyp3,Thi5,D-Tic7,Oic8]-bradykinin (Hoe140) was a kind gift of Dr W. Linz, Hoechst, Frankfurt, Germany. Quinaprilat was a kind gift of Dr H. van Ingen, Parke-Davis, Hoofddorp, The Netherlands. 7-NI, indomethacin, glibenclamide and quinaprilat were dissolved in dimethylsulphoxide. Sulphaphenazole was dissolved in ethanol. Hydroxocobalamin was dissolved in methanol. All other chemicals were dissolved in saline.
Tissue collection
Porcine coronary arteries were obtained from 31 2–3 month-old pigs (Yorkshire×Landrace, weight 10–15 kg). The pigs had been used in in-vivo experiments studying the effects of α-adrenoceptor and serotonin receptor agonists and antagonists under pentobarbital (600 mg, i.v.) anaesthesia (de Vries et al., 1999; Willems et al., 1999). The Ethics Committee of the Erasmus University Rotterdam dealing with the use of animals for scientific experiments approved the protocol for this investigation. Hearts were explanted at the end of the experiment, and the coronary arteries were removed immediately and stored overnight in a cold, oxygenated Krebs bicarbonate solution of the following composition (mmol l−1): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25 and glucose 8.3; pH 7.4. Vessels were then cut into segments of approximately 4 mm length, suspended on stainless steel hooks in 15 ml organ baths containing Krebs bicarbonate solution, aerated with 95% O2/5% CO2, and maintained at 37°C.
Organ bath studies
All vessel segments were allowed to equilibrate for at least 30 min and the organ bath fluid was refreshed every 15 min during this period. Changes in tissue contractile force were recorded with a Harvard isometric transducer (South Natick, MA, U.S.A.). The vessel segments, stretched to a stable force of about 15 mN, were exposed to 30 mmol l−1 K+ twice. The functional integrity of the endothelium was verified by observing relaxation to 1 nM substance P after preconstriction with 1 μM PGF2α. Subsequently, the tissue was exposed to 100 mmol l−1 K+ to determine the maximal contractile response to K+. The segments were then allowed to equilibrate in fresh organ bath fluid for 30 min. Thereafter, the following experiments were performed.
First, possible mediators of the relaxant effect of bradykinin with and without NOS inhibition were investigated. Vessels were pre-incubated for 30 min in the absence or presence of the non-selective NOS inhibitor L-NAME (100 μM), the inducible NOS inhibitor aminoguanidine (1 mM), the neuronal NOS inhibitor 7-NI (10 μM), the guanylyl cyclase inhibitor ODQ (10 μM), the NO scavenger hydroxocobalamin (200 μM), the cyclooxygenase inhibitor indomethacin (10 μM), the ATP-sensitive K+-channel inhibitor glibenclamide (1 μM), the large-conductance voltage and Ca2+-activated K+-channel (BKCa) blocker charybdotoxin+the small-conductance Ca2+-activated K+-channel (SKCa) blocker apamin (both 100 nM), the cytochrome P-450 inhibitor sulphaphenazole (10 μM) or the B2 receptor antagonist Hoe140 (1 μM). Vessels were then preconstricted with 10 μM PGF2α or 1 μM U46619 and concentration-response curves (CRCs) to bradykinin (0.1 nM–1 μM) were constructed.
Second, to test whether desensitization of the bradykinin-induced relaxation occurs more rapidly with NOS inhibition (due to more rapid depletion of NO storage sites), preconstricted vessel segments were exposed three times to a concentration of bradykinin (0.1 μM) that is capable of inducing maximal relaxation. Each subsequent exposure was started as soon as the effect of the previous exposure had disappeared, i.e., after approximately 15 min. To investigate whether L-arginine could reverse the effect of L-NAME, the repetitive exposure experiments were repeated in the presence of L-arginine (10 mM), using a concentration of bradykinin (10 nM) that induces submaximal relaxation. To rule out NOS inhibitor-related differences in B2 receptor desensitization, we constructed two consecutive bradykinin CRCs in a preconstricted vessel segment in the presence or absence of L-NAME.
Third, the effect of ACE inhibition on bradykinin-induced relaxation was investigated. Vessel segments were preconstricted with 10 μM PGF2α or 1 μM U46619 and CRCs were constructed to quinaprilat (1 nM–10 μM) to verify the presence of endogenous bradykinin. Next, in the presence of the highest concentration of quinaprilat, CRCs were constructed to bradykinin and the ACE-resistant bradykinin analogue [Hyp3-Tyr(Me)8]-bradykinin (0.1 nM–1 μM). In addition, quinaprilat (10 μM) or captopril (100 μM) were added to preconstricted vessels with desensitized B2 receptors (i.e., vessels that had been exposed three times to a concentration of bradykinin (0.1 μM) that is capable of inducing maximal relaxation; see above). For comparison, quinaprilat (10 μM) was also added to preconstricted vessels that had been exposed three times to another ACE substrate, substance P, at a concentration (1 nM) that is capable of inducing maximal relaxation.
Statistical analysis
Data are given as mean±s.e.mean and expressed as a percentage of the contraction in response to PGF2α or U46619. CRCs were analysed using the logistic function described by de Lean et al., (1978) to obtain pEC50 (−10log EC50) values. The addition of L-NAME, ODQ, hydroxocobalamin, L-NAME+ODQ or L-NAME+hydroxocobalamin caused an increase in basal tone of 6±1 mN (n=31), 11±2 mN (n=6), 8±1 mN (n=6), 8±1 mN (n=6) and 9±2 mN (n=6), respectively. The PGF2α- and U46619-induced preconstrictions were corrected for this increase in baseline. Statistical analysis was by ANOVA, followed by post hoc evaluation (according to Tukey or Dunnett where appropriate). P values <0.05 were considered significant.
Results
Precontractions
The PGF2α- and U46619-induced precontractions in control vessels did not differ and amounted to approximately 30% (13±1 mN, n=31) of the maximal contraction induced by 100 mmol l−1 K+. Precontractions were not affected by aminoguanidine, 7-NI, indomethacin, glibenclamide, charybdotoxin+apamin, sulphaphenazole or Hoe140. In the vessel segments pretreated with L-NAME, ODQ, hydroxocobalamin, L-NAME+ODQ, or L-NAME+hydroxocobalamin, the precontractions (23±1 mN, n=31; 20±2 mN, n=6; 28±2 mN, n=6; 21±3 mN, n=6; and 24±2 mN, n=6, respectively) were approximately 2 fold higher than in control vessel segments (P<0.01), which illustrates the importance of endogenous NO generation by endothelial NOS in this preparation.
Mediators of the relaxant effect of bradykinin
Bradykinin caused complete relaxation of preconstricted vessel segments in a concentration-dependent manner (pEC50= 8.03±0.05, n=31; Figure 1). The bradykinin CRC was not affected by aminoguanidine (pEC50=8.21±0.11, n=5), 7-NI (pEC50=7.98±0.22, n=5), indomethacin (pEC50=7.58±0.22, n=5), glibenclamide (pEC50=8.35±0.28, n=5) or sulphaphenazole (pEC50=7.63±0.07, n=6). L-NAME (pEC50=6.93±0.07, n=25; P<0.01 vs control) and ODQ (pEC50=7.19±0.30, n=5; P<0.05 vs control) shifted the CRC of bradykinin to the right, while in the presence of hydroxocobalamin (n=6) relaxation was only observed at the highest concentration of bradykinin (Figure 1). Charybdotoxin+apamin also shifted the CRC of bradykinin to the right (pEC50=6.47±0.10, n=6; P<0.01 vs control, Figure 1). Complete blockade of the response was obtained with Hoe140 (Figure 1). The addition of indomethacin (pEC50=7.19±0.16, n=4) or sulphaphenazole (pEC50=7.08±0.04, n=6) on top of L-NAME did not cause a further rightward shift of the bradykinin CRC as compared to L-NAME alone (data not shown), nor did the addition of ODQ on top of L-NAME (pEC50=7.24±0.12, n=5; Figure 2). Charybdotoxin+apamin in combination with L-NAME completely blocked the response to all bradykinin concentrations tested (Figure 2), whereas in the presence of L-NAME+hydroxocobalamin (n=6) relaxation was again observed at the highest concentration of bradykinin only (Figure 2).
Figure 1.
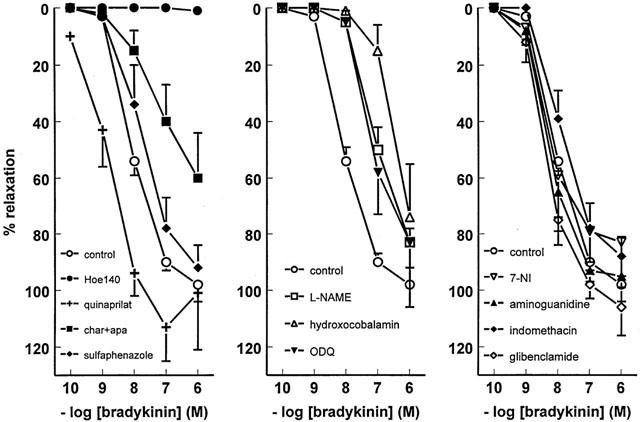
Relaxations of PCAs, preconstricted with 10 μM PGF2α or 1 μM U46619, to bradykinin in the absence (control) or presence of 1 μM Hoe140, 10 μM quinaprilat, 100 nM charybdotoxin (char)+100 nM apamin (apa), 10 μM sulphaphenazole, 100 μM L-NAME, 200 μM hydroxocobalamin, 10 μM ODQ, 10 μM 7-NI, 1 mM aminoguanidine, 10 μM indomethacin or 1 μM glibenclamide. For the sake of clarity, data have been divided across three panels, and the control curve is shown in each panel. Data (mean±s.e.mean of five to 31 experiments) are expressed as a percentage of the contraction induced by PGF2α or U46619.
Figure 2.

Relaxations of PCAs, following preconstriction with 10 μM PGF2α or 1 μM U46619, to bradykinin in the presence of 100 μM L-NAME without (control) or with 10 μM ODQ, 10 μM quinaprilat, 100 nM charybdotoxin (char)+100 nM apamin (apa) or 200 μM hydroxocobalamin. Data (mean±s.e.mean of five to 25 experiments) are expressed as a percentage of the contraction induced by PGF2α or U46619.
Desensitization of the bradykinin-induced effect
The construction of a bradykinin CRC resulted in B2 receptor desensitization, as evidenced by the approximate 10 fold rightward shift that was observed when constructing a second bradykinin CRC in the same vessel segment (Figure 3). A similar rightward shift was observed in the presence of L-NAME.
Figure 3.
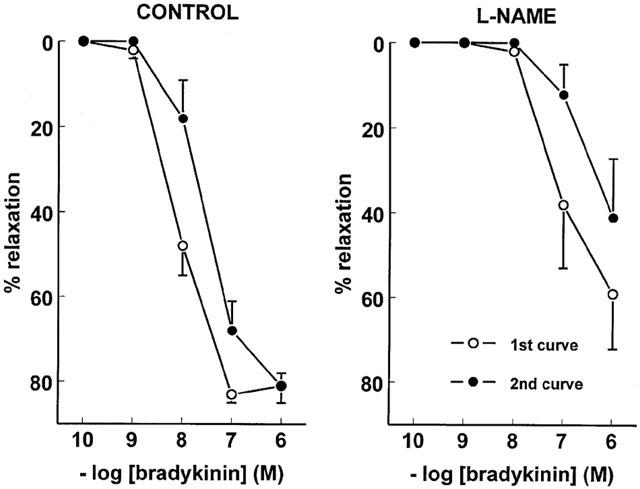
Two consecutive concentration-response curves, obtained in the same PCA vessel segment following preconstriction with 10 μM PGF2α, to bradykinin in the absence (left panel) or presence (right panel) of 100 μM L-NAME. Data (mean±s.e.mean of five experiments) are expressed as a percentage of the contraction induced by PGF2α.
Repeated exposure of preconstricted vessel segments to 0.1 μM bradykinin produced progressively smaller relaxant responses (Figure 4, top panel). The response to the third bradykinin dose was less than 50% of the response to the first bradykinin dose (Figure 4, bottom panel). A similar pattern was observed in the presence of charybdotoxin+apamin, although the relaxation to bradykinin in the presence of these drugs was always smaller than under control conditions (Figure 4, bottom panel). In the presence of L-NAME (Figure 4, top panel) or hydroxocobalamin (Figure 4, bottom panel), the relaxation observed in response to the first dose of bradykinin was significantly smaller than under control conditions, and relaxation was virtually absent in response to the second and third bradykinin dose. Results obtained with L-NAME+hydroxocobalamin were not different from those with hydroxocobalamin alone (data not shown). Charybdotoxin+apamin combined with L-NAME fully prevented all responses to bradykinin (Figure 4, bottom panel).
Figure 4.

(A) Original tracings of an experiment in which a PCA in the absence (control; top) or presence (bottom) of 100 μM L-NAME was preconstricted with 10 μM PGF2α and subsequently exposed to 0.1 μM bradykinin (BK, three times), 10 μM quinaprilat and 0.1 μM bradykinin. (B) Relaxations of PCAs, following preconstriction with 10 μM PGF2α or 1 μM U46619, to three consecutive bradykinin doses (0.1 μM; BK1, BK2, BK3), 10 μM quinaprilat (QUIN) and a fourth bradykinin dose (0.1 μM; BK4) in the absence or presence of 100 μM L-NAME, 200 μM hydroxocobalamin, 100 nM charybdotoxin (char) +100 nM apamin (apa), or 100 μM L-NAME +100 nM charybdotoxin (char) +100 nM apamin (apa). Data (mean±s.e.mean of six to 24 experiments) are expressed as a percentage of the contraction induced by PGF2α or U46619. *P<0.01 vs control; #P<0.05 vs BK1.
Exposing preconstricted vessels three times to a submaximal concentration of bradykinin (10 nM) revealed that, under control conditions, the relaxation in response to the third bradykinin dose was not significantly different from the response to the first bradykinin dose (Figure 5). In the presence of L-NAME, the first bradykinin dose induced a modest relaxation, and no further response was observed at the second and third exposure to bradykinin. L-Arginine reversed the inhibitory effect of L-NAME, and tended to enhance the effect of bradykinin in the absence of L-NAME (P=NS).
Figure 5.

Relaxations of PCAs, following preconstriction with 10 μM PGF2α, to three consecutive bradykinin doses (10 nM; BK1, BK2, BK3) in the absence or presence of 100 μM L-NAME, 100 μM L-NAME +10 mM L-arginine, or 10 mM L-arginine. Data (mean±s.e.mean of five to seven experiments) are expressed as a percentage of the contraction induced by PGF2α. *P<0.01 vs control; #P<0.05 vs BK1.
Effect of ACE inhibition on bradykinin-induced relaxation
Quinaprilat alone did not cause relaxation of preconstricted vessel segments (data not shown), thereby ruling out the presence of endogenous bradykinin. In the presence of the ACE inhibitor, the CRC to bradykinin was shifted to the left (pEC50=8.89±0.20, n=5; P<0.05 vs control, see Figure 1). This was also the case in vessel segments that had been pre-incubated with L-NAME (pEC50=8.03±0.23, n=5; P<0.05 vs L-NAME alone, see Figure 2). Quinaprilat caused a similar leftward shift of the CRC to the ACE-resistant analogue [Hyp3-Tyr(Me)8]-bradykinin (pEC50's resp. 8.50±0.18 and 9.18±0.06 without and with quinaprilat, n=4; P<0.05, Figure 6). Hoe140 fully blocked the effects of [Hyp3-Tyr(Me)8]-bradykinin, confirming that this agonist induces relaxation via stimulation of B2 receptors.
Figure 6.

Relaxations of PCAs, preconstricted with 10 μM PGF2α, to [Hyp3-Tyr(Me)8]-bradykinin in the absence or presence of 1 μM Hoe140 or 10 μM quinaprilat. Data (mean±s.e.mean of four experiments) are expressed as a percentage of the contraction induced by PGF2α.
Quinaprilat added to vessel segments that had been exposed three times to 0.1 μM bradykinin, after the effect of the last dose of bradykinin had disappeared, caused complete relaxation, even in L-NAME- or hydroxocobalamin-pretreated vessel segments that previously had not responded to bradykinin (Figure 4). Quinaprilat also induced complete relaxation of vessel segments pretreated with charybdotoxin+apamin, whereas in vessel segments pretreated with L-NAME combined with charybdotoxin+apamin, which previously had not shown any response to bradykinin, the ACE inhibitor induced a modest relaxant response. Similar results were obtained with captopril (n=5, data not shown). A fourth bradykinin dose, added after the effect of quinaprilat or captopril had disappeared, induced no further effect.
Results obtained with bradykinin and quinaprilat in the presence of aminoguanidine (n=5), 7-NI (n=5), indomethacin (n=4), glibenclamide (n=4) and sulphaphenazole (n=6) were not different from those obtained in the absence of these inhibitors (data not shown). Moreover, results obtained with [Hyp3-Tyr(Me)8]-bradykinin and quinaprilat (n=4) exactly mimicked those with bradykinin and quinaprilat (data not shown). Hoe140 completely prevented the quinaprilat-induced potentiation (data not shown). The effect of quinaprilat was specific for bradykinin, since it was not observed in combination with substance P (Figure 7).
Figure 7.

Relaxations of PCAs, following preconstriction with 10 μM PGF2α, to three consecutive doses of substance P (1 nM; SP1, SP2, SP3), 10 μM quinaprilat (QUIN) and a fourth dose of substance P (1 nM; SP4). Data (mean±s.e.mean of five experiments) are expressed as a percentage of the contraction induced by PGF2α. #P<0.01 vs SP1.
Discussion
NO is responsible for bradykinin-induced vasorelaxation in porcine coronary arteries
The results of the present study show that the B2 receptor-mediated relaxant effects of bradykinin in PCAs, at doses up to 0.1 μM, depend on NO, either synthesized de novo by endothelial NOS or derived from NO storage sites. These data fully support the interaction between endothelial NOS and B2 receptors that was recently described by Golser et al., (2000). We found no evidence for a role of NO synthases other than endothelial NOS in PCAs, since neither aminoguanidine, a preferential inhibitor of inducible NOS (Joly et al., 1994; Boulanger et al., 1998), nor 7-NI, a preferential inhibitor of neuronal NOS (Moore et al., 1993; Boulanger et al., 1998) affected basal tone or the bradykinin CRC. Furthermore, neither prostaglandins, ATP-sensitive K+-channels nor cytochrome P-450 products appeared to be involved in the bradykinin-induced vasodilation in PCAs. The latter contrasts with a recent observation in PCAs showing that bradykinin-induced relaxation in the presence of the NOS inhibitor Nωnitro-L-arginine was due to the release of 11,12-epoxyeicosatrienoic acid, an arachidonic acid metabolite formed by cytochrome P-450 (Fisslthaler et al., 1999). However, the EC50 of this effect was approximately 1 μM (Fisslthaler et al., 1999), and we did not test bradykinin concentrations above 1 μM.
The 10 fold rightward shift of the bradykinin CRC induced by both the non-selective NOS inhibitor L-NAME and the guanylyl cyclase inhibitor ODQ contrasts with the much more complete blockade observed in the presence of the NO scavenger hydroxocobalamin. Hydroxocobalamin did not block the effect of 1 μM bradykinin. This is not due to the formation of relaxant prostaglandins or cytochrome P-450 products at this concentration of bradykinin, since indomethacin and sulphaphenazole did not affect the bradykinin CRC in the presence of L-NAME. The most likely explanation is that, at the concentration used in the present study (200 μM), hydroxocobalamin did not completely scavenge all NO (Li & Rand, 1999). The maximum solubility of hydroxocobalamin in methanol (10 mg/ml) prevented us from reaching higher concentrations in the organ bath. Taken together therefore, NO release is responsible for the bradykinin-induced relaxation in PCAs, at least at bradykinin concentrations up to 0.1 μM, and NOS blockade as well as guanylyl cyclase blockade cannot prevent this relaxation completely.
The modest effect of ODQ might be due to the fact that this drug inhibits guanylyl cyclase reversibly (Garthwaite et al., 1995). Alternatively, NO-induced relaxation may occur independently of guanylyl cyclase. In support of the first possibility, we observed that ODQ fully inhibited the relaxant effects of low (<1 μM) but not of high (⩾1 μM) concentrations of the NO donor S-nitroso-N-acetylpenicillamine (Danser et al., 1999, unpublished observations). In support of the second possibility, NO has been described to induce hyperpolarization directly via activation of Ca2+-dependent K+-channels (Bolotina et al., 1994), and bradykinin is known to activate these channels through stimulation of tyrosine kinase (Lee et al., 1993; Ogiwara et al., 1995). Indeed, in the present study the BKCa blocker charybdotoxin and the SKCa blocker apamin partially blocked the bradykinin-induced relaxation when given together, and fully blocked the bradykinin-induced effects when given in combination with L-NAME. Charybdotoxin plus apamin also blocked the bradykinin-induced hyperpolarization of PCA rings (Quignard et al., 1999). Thus, the most likely explanation for our findings is that the bradykinin-induced NO release causes vasodilation via stimulation of guanylyl cyclase and/or via direct activation of Ca2+-dependent K+-channels.
Release of NO from storage sites?
The limited effect of NOS blockade on NO release has been described before in rabbit carotid arteries (Cohen et al., 1997), and may involve either residual NOS activity or release of NO from storage sites (Davisson et al., 1996; Danser et al., 1998). To investigate the latter, we exposed coronary arteries repeatedly to the same concentration of bradykinin. Previous studies have shown that repetitive exposure to bradykinin or acetylcholine will cause depletion of NO storage sites, and that this will occur more rapidly in the presence of NOS inhibitors (Davisson et al., 1996; Colombari et al., 1998; Danser et al., 1998). In the present study, exposure to bradykinin, both with and without L-NAME, resulted in a relaxant effect which lasted approximately 10–15 min. The disappearance of the relaxation is suggestive for bradykinin B2 receptor desensitization and/or bradykinin metabolism.
Desensitization was similar with and without L-NAME (Figure 3). Subsequent exposures to bradykinin initiated progressively smaller relaxant effects, and the decrease in response was much more rapid in the presence of L-NAME. L-Arginine reversed the rapid decrease in response to consecutive bradykinin doses in the presence of L-NAME. Taken together, these findings support the concept of bradykinin-coupling to NO storage sites. With NOS activity intact, the NO pools are continuously supplied with fresh NO and relaxation can be obtained multiple times, even when B2 receptors are desensitized. During NOS inhibition the NO storage sites will become depleted, especially during exposure to drugs that cause release of NO from these sites. In an earlier study in isolated perfused rat hearts, we found that a 30 min exposure to L-NAME was sufficient to cause depletion of all existing NO pools (Danser et al., 1998). Depletion may also occur during exposure to high levels of superoxide anion (Arnal et al., 1996), although in PCAs superoxide anions scavengers did not affect the response to bradykinin (Pomposiello et al., 1999).
The nature and localization of NO pools is currently unknown. Although NO pools have been demonstrated in vascular smooth muscle cells (Venturini et al., 1993), the pools in the present study are most likely localized in endothelial cells, in view of the fact that we (Danser et al., 1999, unpublished results) and others (Mombouli et al., 1992) have found that bradykinin-induced relaxations are virtually absent following endothelium removal. The long half life of NO present at storage sites, evidenced by the fact that bradykinin was still capable of inducing relaxation after the vessels had been exposed to L-NAME for more than 30 min, is compatible with the idea that NO pools consist of stable NO-containing compounds, such as S-nitroso-thiols and dinitrosyl iron (II) thiol complexes (Ignarro, 1990; Myers et al., 1990; Verdernikov et al., 1992).
ACE inhibitors potentiate bradykinin independently of their effect on bradykinin metabolism
Finally, the ACE inhibitor quinaprilat, added at a time when the relaxant effect of bradykinin had disappeared, immediately restored the vasorelaxation, both with and without L-NAME. The effect of quinaprilat could be mimicked by captopril, and did not occur in combination with the B2 receptor antagonist Hoe 140 or without prior exposure to bradykinin. The latter finding suggests that not all bradykinin has been metabolized at the time the ACE inhibitor is added to the organ bath. When added prior to bradykinin, quinaprilat shifted the bradykinin CRC approximately 10 fold to the left, both with and without L-NAME. It is unlikely that the potentiating effects of ACE inhibition are due simply to inhibition of bradykinin metabolism, as suggested by Dendorfer et al., (2000), because (1) the effect of quinaprilat was also observed in combination with the ACE-resistant bradykinin analogue [Hyp3-Tyr(Me)8]-bradykinin, (2) the effect of quinaprilat was not observed in relationship with the vasorelaxant ACE substrate substance P, and (3) quinaprilat even induced complete relaxation in vessel segments that had previously not responded to bradykinin, i.e., vessel segments that had been preincubated with hydroxocobalamin or charbdotoxin+apamin+L-NAME. Taken together therefore, our data support the concept of ACE inhibitor-induced bradykinin potentiation independently of the effect of these drugs on bradykinin metabolism. The mechanism underlying this phenomenon is currently unknown, but it may involve the ACE inhibitor-induced resensitization of desensitized B2 receptors that has been described in isolated cells (Minshall et al., 1997; Benzing et al., 1999). Resensitized B2 receptors may still cause relaxation via coupling to remaining NO pools, even after exposure to L-NAME and hydroxocobalamin, since we do not know whether these drugs, combined with repetitive exposure to bradykinin, have resulted in complete depletion of all existing NO pools. Alternatively, non-NO-related mechanisms may have come into play. These mechanisms do not involve prostaglandins, ATP-sensitive K+-channels or cytochrome P-450 products, since indomethacin, glibenclamide and sulphaphenazole did not affect the quinaprilat-induced relaxation.
Conclusions and possible clinical implications
In conclusion, the L-NAME-resistant bradykinin-induced relaxation, at least at physiological bradykinin concentrations (i.e., concentrations up to 0.1 μM; Campbell et al., 1993), is NO-dependent, and is mediated via stimulation of guanylyl cyclase and/or Ca2+-dependent K+-channels. NO is either synthesized de novo by endothelial NOS or released from storage sites. Depletion of such sites or a decrease in their number might be involved in the impaired endothelium-dependent vasodilatory response observed in subjects with hypertension or atherosclerosis (Hirooka et al., 1992; Zeiher et al., 1993). ACE inhibitors potentiate bradykinin-induced vasorelaxation independently of their effect on bradykinin metabolism. Such potentiation has also been observed in vivo in human subjects (Hornig et al., 1997; Kuga et al., 1997). Since elevated bradykinin levels have not been found consistently during ACE inhibitor treatment (Miki et al., 1996; Campbell et al., 1999), our findings might explain, at least in part, the beneficial effects of ACE inhibitors in hypertension and heart failure, as well as the ACE inhibitor-induced reversal of the impaired endothelium-dependent vasorelaxation in hypertensive patients (Hirooka et al., 1992).
Abbreviations
- ACE
angiotensin-converting enzyme
- B2
bradykinin type 2 receptor
- BKCa
large-conductance voltage and Ca2+-activated K+-channel
- CRC
concentration-response curve
- Hoe140
D-Arg[Hyp3,Thi5,D-Tic7,Oic8]-bradykinin
- L-NAME
Nω-nitro-L-arginine methyl ester HCl
- 7-NI
7-nitroindazole
- NO
nitric oxide
- NOS
NO synthase
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- PCA
porcine coronary artery
- PGF2α
prostaglandin F2α
- SKCa
small-conductance Ca2+-activated K+-channel
- U46619
9,11-dideoxy-11α,9α-epoxymethano-prostaglandin F2α
References
- ARNAL J.F., CLAMENS S., PECHET C., NEGRE-SALVAYRE A., ALLERA C., GIROLAMI J.-P., SALVAYRE R., BAYARD F. Ethinylestradiol does not enhance the expression of nitric oxide synthase in bovine endothelial cells but increases the release of bioactive nitric oxide by inhibiting superoxide anion production. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4108–4113. doi: 10.1073/pnas.93.9.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENZING T., FLEMING I., BLAUKAT A., MÜLLER-ESTERL W., BUSSE R. Angiotensin-converting enzyme inhibitor ramiprilat interferes with the sequestration of the B2 kinin receptor within the plasma membrane of native endothelial cells. Circulation. 1999;99:2034–2040. doi: 10.1161/01.cir.99.15.2034. [DOI] [PubMed] [Google Scholar]
- BJORNSTAD-OSTENSEN A., HOLTE H.R., BERG T. Amplification of kinin-induced hypotension by nitric oxide synthesis in spontaneously hypertensive rats. Hypertension. 1997;29:53–57. doi: 10.1161/01.hyp.29.1.53. [DOI] [PubMed] [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P., COHEN R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- BOULANGER C.M., HEYMES C., BENESSIANO J., GESKE R.S., LÉVY B.I., VANHOUTTE P.M. Neuronal nitric oxidase synthase is expressed in rat vascular smooth muscle cells. Activation by angiotensin II in hypertension. Circ. Res. 1998;83:1271–1278. doi: 10.1161/01.res.83.12.1271. [DOI] [PubMed] [Google Scholar]
- CAMPBELL D.J., DUNCAN A.-M., KLADIS A. Angiotensin-converting enzyme inhibition modifies angiotensin but not kinin peptide levels in human atrial tissue. Hypertension. 1999;34:171–175. doi: 10.1161/01.hyp.34.2.171. [DOI] [PubMed] [Google Scholar]
- CAMPBELL D.J., KLADIS A., DUNCAN A.-M. Bradykinin peptides in kidney, blood, and other issues of the rat. Hypertension. 1993;21:155–165. doi: 10.1161/01.hyp.21.2.155. [DOI] [PubMed] [Google Scholar]
- COHEN R.A., PLANE F., NAJIBI S., HUK I., MALINSKI T., GARLAND C.J. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4193–4198. doi: 10.1073/pnas.94.8.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLOMBARI E., DAVISSON R.L., SHAFFER R.A., TALMAN W.T., LEWIS S.J. Hemodynamic effects of L-glutamate in NTS of conscious rats: a possible role of vascular nitrosyl factors. Am. J. Physiol. 1998;274:H1066–H1074. doi: 10.1152/ajpheart.1998.274.4.H1066. [DOI] [PubMed] [Google Scholar]
- DANSER A.H.J., DE VRIES R., SCHOEMAKER R.G., SAXENA P.R. Bradykinin-induced release of nitric oxide by the isolated perfused rat heart: importance of preformed pools of nitric oxide-containing factors. J. Hypertens. 1998;16:239–244. doi: 10.1097/00004872-199816020-00015. [DOI] [PubMed] [Google Scholar]
- DAVISSON R.L., BATES J.N., JOHNSON K., LEWIS S.J. Use-dependent loss of acetylcholine- and bradykinin-mediated vasodilation after nitric oxide synthase inhibition. Evidence for preformed stores of nitric oxide-containing factors in vascular endothelial cells. Hypertension. 1996;28:354–360. doi: 10.1161/01.hyp.28.3.354. [DOI] [PubMed] [Google Scholar]
- DE LEAN A., MUNSON P.J., RODBARD D. Simultaneous analysis of families of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am. J. Physiol. 1978;235:E97–E102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- DENDORFER A., WOLFRUM S., SCHÄFER U., STEWART J.M., INAMURA N., DOMINIAK P. Potentiation of the vascular responses to kinins by inhibition of myocardial kininases. Hypertension. 2000;35:32–37. doi: 10.1161/01.hyp.35.1.32. [DOI] [PubMed] [Google Scholar]
- DE VRIES P., WILLEMS E.W., HEILIGERS J.P.C., VILLALÓN C.M., SAXENA P.R. Investigation of the role of 5-HT1B and 5-HT1D receptors in sumatriptan-induced constriction of porcine carotid arteriovenous anastomoses. Br. J. Pharmacol. 1999;127:405–412. doi: 10.1038/sj.bjp.0702572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- GARDINER S.M., COMPTON A.M., KEMP P.A., BENNET T. Regional and cardiac haemodynamic responses to glyceryl trinitrate, acetylcholine, bradykinin and endothelin-1 in conscious rats: effects of NG-nitro-L-arginine methyl ester. Br. J. Pharmacol. 1990;101:632–639. doi: 10.1111/j.1476-5381.1990.tb14132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSEN E.B., SCHMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- GOLSER R., GORREN A.C.F., LEBER A., ANDREW P., HABISCH H.-J., WERNER E.R., SCHMIDT K., VENEMA R.C., MAYER B. Interaction of endothelial and neuronal nitric-oxide synthases with the bradykinin B2 receptor. J. Biol. Chem. 2000;275:5291–5296. doi: 10.1074/jbc.275.8.5291. [DOI] [PubMed] [Google Scholar]
- HAASEMANN M., CARTAUD J., MÜLLER-ESTERL W., DINIA I. Agonist-induced redistribution of bradykinin B2 receptor in caveolae. J. Cell. Sci. 1998;111:917–928. doi: 10.1242/jcs.111.7.917. [DOI] [PubMed] [Google Scholar]
- HIROOKA Y., IMAIZUMI T., ANDO S.-I., HARADA S., MOMOHARA M., TAKESHITA A. Captopril improves impaired endothelium-dependent vasodilation in hypertensive subjects. Hypertension. 1992;20:175–180. doi: 10.1161/01.hyp.20.2.175. [DOI] [PubMed] [Google Scholar]
- HORNIG B., KOHLER C., DREXLER H. Role of bradykinin in mediating vascular effects of angiotensin-converting enzyme inhibitors in humans. Circulation. 1997;95:1115–1118. doi: 10.1161/01.cir.95.5.1115. [DOI] [PubMed] [Google Scholar]
- IGNARRO L.J. Nitric oxide. A novel signal transduction mechanism for transcellular communication. Hypertension. 1990;16:477–483. doi: 10.1161/01.hyp.16.5.477. [DOI] [PubMed] [Google Scholar]
- JOLY G.A., AYRES M., CHELLY F., KILBOURN R.G. Effects of NG-methyl-L-arginine and aminoguanidine on constitutive and inducible nitric oxide synthase in rat aorta. Biochem. Biophys. Res. Commun. 1994;199:147–154. doi: 10.1006/bbrc.1994.1207. [DOI] [PubMed] [Google Scholar]
- KUGA T., MOHRI M., EGASHIRA K., HIRAKAWA Y., TAGAWA T., SHIMOKAWA H., TAKESHITA A. Bradykinin-induced vasodilation in human coronary arteries in vivo: role of nitric oxide and angiotensin-converting enzyme. J. Am. Coll. Cardiol. 1997;30:108–112. doi: 10.1016/s0735-1097(97)00112-5. [DOI] [PubMed] [Google Scholar]
- LEE K.-M., TOSCAS K., VILLEREAL M.L. Inhibition of bradykinin- and thapsigargin-induced Ca2+ entry by tyrosine kinase inhibitors. J. Biol. Chem. 1993;268:9945–9948. [PubMed] [Google Scholar]
- LI C.G., RAND M.J. Effects of hydroxocobalamin and carboxy-PTIO on nitrergic transmission in porcine anococcygeus and retractor penis muscles. Br. J. Pharmacol. 1999;127:172–176. doi: 10.1038/sj.bjp.0702496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARCIC B., DEDDISH P.A., JACKMAN H.L., ERDÖS E.G. Enhancement of bradykinin and resensitization of its B2 receptor. Hypertension. 1999;33:835–843. doi: 10.1161/01.hyp.33.3.835. [DOI] [PubMed] [Google Scholar]
- MIKI T., MIURA T., URA N., OGAWA T., SUZUKI K., SHIMAMOTO K., IIMURA O. Captopril potentiates the myocardial infarct size-limiting effect of ischemic preconditioning through bradykinin B2 receptor activation. J. Am. Coll. Cardiol. 1996;28:1616–1622. doi: 10.1016/s0735-1097(96)00371-3. [DOI] [PubMed] [Google Scholar]
- MINSHALL R.D., TAN F., NAKAMURA F., RABITO S.F., BECKER R.P., MARCIC B., ERDÖS E.G. Potentiation of the actions of bradykinin by angiotensin I-converting enzyme inhibitors. The role of expressed human bradykinin B2 receptors and angiotensin I-converting enzyme in CHO cells. Circ. Res. 1997;81:848–856. doi: 10.1161/01.res.81.5.848. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.-V., ILLIANO S., NAGAO T., SCOTT-BURDEN T., VANHOUTTE P.M. Potentiation of endothelium-dependent relaxations to bradykinin by angiotensin I converting enzyme inhibitors in canine coronary arteries involves both endothelium-derived relaxing and hyperpolarizing factors. Circ. Res. 1992;71:137–144. doi: 10.1161/01.res.71.1.137. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.-V., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor(s): updating the unknown. TiPS. 1997;18:252–256. [PubMed] [Google Scholar]
- MOORE P.K., BABBEDGE R.C., WALLACE P., GAFFEN Z.A., HART S.L. 7-Nitro indazole, an inhibitor of nitric oxide synthease, exhibits anti-nociceptive activity in the mouse without increasing blood pressure. Br. J. Pharmacol. 1993;108:96–297. doi: 10.1111/j.1476-5381.1993.tb12798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MYERS P.R., MINOR R.L., GUERRA R., BATES J.N., HARRISON D.G. Vasorelaxant properties of the endothelium-derived relaxing factor more closely resemble S-nitrosocysteine than nitric oxide. Nature. 1990;345:161–163. doi: 10.1038/345161a0. [DOI] [PubMed] [Google Scholar]
- OGIWARA T., MURDOCH G., CHIK C.L., HO A.K. Tyrosine kinase inhibitors enhance cGMP production in rat pinealocytes. Biochem. Biophys. Res. Commun. 1995;207:994–1002. doi: 10.1006/bbrc.1995.1283. [DOI] [PubMed] [Google Scholar]
- PALMER R.M.J., REES D.D., ASHTON D.S., MONCADA S. L-Arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 1988;153:1251–1256. doi: 10.1016/s0006-291x(88)81362-7. [DOI] [PubMed] [Google Scholar]
- POMPOSIELLO S., RHALEB N.E., ALVA M., CARRETERO O.A. Reactive oxygen species: role in the relaxation induced by bradykinin or arachidonic acid via EDHF in isolated porcine coronary arteries. J. Cardiovasc. Pharmacol. 1999;34:567–574. doi: 10.1097/00005344-199910000-00014. [DOI] [PubMed] [Google Scholar]
- QUIGNARD J.-F., FÉLÉTOU M., THOLLON C., VILAINE J.-P., DUHAULT J., VANHOUTTE P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999;127:27–34. doi: 10.1038/sj.bjp.0702493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REES D.D., PALMER R.M., SCHULZ R., HODSON H.F., MONCADA S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RHALEB N.E., DRAPEAU G., DION S., JUKIC D., ROUISSI N., REGOLI D. Structure-activity studies on bradykinin and related peptides: agonists. Br. J. Pharmacol. 1990;99:445–448. doi: 10.1111/j.1476-5381.1990.tb12947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENTURINI C.M., PALMER R.M.J., MONCADA S. Vascular smooth muscle contains a depletable store of a vasodilator which is light-activated and restored by donors of nitric oxide. J. Pharmacol. Exp. Ther. 1993;266:1497–1500. [PubMed] [Google Scholar]
- VERDERNIKOV Y.P., MORDVINTCEV P.I., MALENKOVA I.V., VANIN A.F. Similarity between the vasorelaxing activity of dinitrosyl iron cysteine complexes and endothelium-derived relaxing factor. Eur. J. Pharmacol. 1992;211:313–317. doi: 10.1016/0014-2999(92)90386-i. [DOI] [PubMed] [Google Scholar]
- WILLEMS E.W., TRION M., DE VRIES P., HEILIGERS J.P.C., VILLALÓN C.M., SAXENA P.R. Pharmacological evidence that α1- and α2-adrenoceptors mediate vasoconstriction of carotid arteriovenous anastomoses in anesthetized pigs. Br. J. Pharmacol. 1999;127:1263–1271. doi: 10.1038/sj.bjp.0702655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZEIHER A.M., DREXLER H., SAURBIER B., JUST H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J. Clin. Invest. 1993;92:652–655. doi: 10.1172/JCI116634. [DOI] [PMC free article] [PubMed] [Google Scholar]