

Abstract
The centrally acting analgesic tramadol has recently been reported to cause seizures at re-commended dosages in patients, whereas animal experiments had indicated that seizures only occur in high, toxic doses. Tramadol has a dual mechanism of action that includes weak agonistic effects at the mu-opioid receptor as well as inhibition of monoamine (serotonin, norepinephrine) re-uptake. Its major (M1) metabolite mono-O-desmethyltramadol, which is rapidly formed in vivo, has a markedly higher affinity for mu receptors and may thus contribute to the effects of the parent compound. Furthermore, the pharmacological effects of tramadol appear to be related to the different, but complementary and interactive pharmacologies of its enantiomers. In the present study, we evaluated (±)-tramadol, its enantiomers, and its M1 metabolite ((+)-enantiomer) in the amygdala kindling model of epilepsy in rats. Adverse effects determined in kindled rats were compared to those in nonkindled rats.
At doses within the analgesic range, (±)-tramadol and its enantiomers induced anticonvulsant effects in kindled rats. However, at only slightly higher doses seizures occurred. With (±)-tramadol, generalized seizures were observed at 30 mg kg−1 in most kindled but not in nonkindled rats. The (−)-enantiomer induced myoclonic seizures at 30 mg kg−1 in most kindled but not in nonkindled rats, although myoclonic seizure activity was observed in some nonkindled rats at 10 or 20 mg kg−1. Seizures were also observed after the (+)-enantiomer and the (+)-enantiomer of the M1 metabolite, but experiments with higher doses of these compounds were limited by marked respiratory depression.
The data demonstrate that kindling enhances the susceptibility of rats to convulsant adverse effects of tramadol and its enantiomers, indicating that a preexisting lowered seizure threshold increases the risk of tramadol-induced seizures.
Keywords: Opioids, analgesics, seizures, noradrenaline, serotonin
Introduction
The centrally acting analgesic tramadol possesses weak opioid agonist properties and inhibits norepinephrine (NE) and serotonin (5-hydroxytryptamine, 5-HT) uptake (Dayer et al., 1994; Raffa et al., 1995; Lewis & Han, 1997). Tramadol is an effective and relative safe analgesic that has been prescribed for almost two decades in Europe, and was approved for marketing in the United States in 1995 for the treatment of moderate to moderately severe pain (Lee et al., 1993; Gibson, 1996; Lewis & Han, 1997; Bamigbade & Langford, 1998). However, in the years after its release in the United States, an increased risk of seizures associated with tramadol was reported (Kahn et al., 1997). This adverse drug effect occurred at recommended dosages, although an overdose may increase the risk of tramadol-related seizures (Kahn et al., 1997). It is long known that opioid analgesics such as morphine and related drugs can produce convulsions, but with most opioids convulsions occur only in doses far in excess of those required to produce profound analgesia (c.f., Frenk, 1983). With respect to tramadol, animal experiments also indicated that convulsions only occur in high, toxic doses (Friderichs et al., 1978; Osterloh et al., 1978; Matthiesen et al., 1998). This apparent paradox between preclinical data and the risk of seizures associated with tramadol in patients may be related to the fact that most clinical reports were from patients receiving concomitant treatment with other drugs which may increase the risk of tramadol-related seizures (Khan et al., 1997; Jick et al., 1998).
In the present study, we used the kindling model of temporal lobe epilepsy to evaluate the proconvulsant activity of tramadol in more detail. Because a recent study indicated that tramadol exerts anticonvulsant effects in the maximal electroshock seizure (MES) test in mice (Manocha et al., 1998), we also assessed whether tramadol possesses anticonvulsant activity in the kindling model. In view of our previous finding that kindling-induced epileptogenesis may enhance the adverse effect potential of drugs (Löscher & Hönack, 1991; Hönack & Löscher, 1995; Löscher, 1998; Wlaz & Löscher, 1998), adverse effects of tramadol in kindled rats were compared to those in nonkindled rats.
Tramadol is a racemic 1:1 mixture of two enantiomers, (+)-tramadol and (−)-tramadol which differ in their potencies at opioid receptors and monoamine uptake sites (Raffa et al., 1993). Furthermore, (±)-tramadol is rapidly metabolized to mono-O-desmethyltramadol (M1 metabolite; see Figure 1) which also binds to opioid receptors (Raffa et al., 1995; Gibson, 1996). In addition to testing tramadol, we therefore evaluated the extent to which the enantiomers and the M1 metabolite contribute to tramadol's anticonvulsant and proconvulsant effects in rats.
Figure 1.
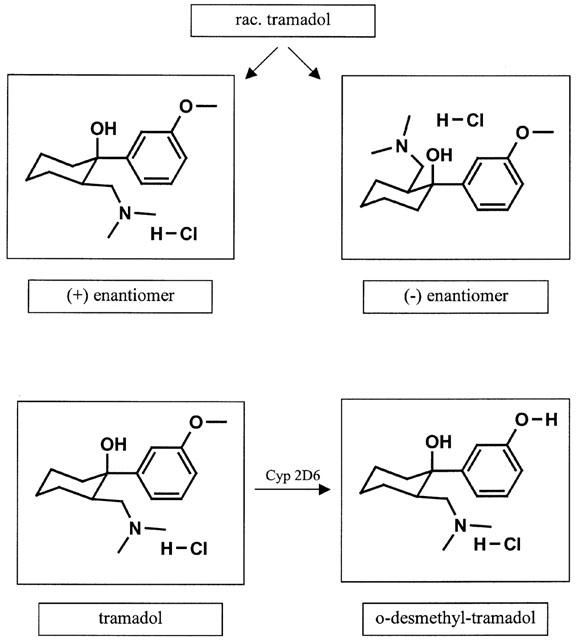
Stereochemistry of tramadol and metabolic activation to the M1-metabolite O-desmethyltramadol.
Methods
Animals
Female Wistar rats (Harlan-Winkelmann, Borchen, Germany), weighing 200–300 g, were used. The animals were purchased from the breeder at a body weight of 180–220 g. Following arrival in the animal colony, the rats were kept under controlled environmental conditions (ambient temperature 24–25°C, humidity 50–60%, 12/12 h light/dark cycle, light on at 0700) for at least 1 week before being used in the experiments. Standard laboratory chow (Altromin 1324 standard diet) and tap water were allowed ad libitum. All animal care and handling was conducted in compliance with the German Animal Welfare Act and was approved by the responsible governmental agency in Hannover. With respect to the use of females, it is important to note that we previously showed that neither seizure susceptibility nor anticonvulsant drug effects are affected by the estrous cycle in fully kindled female rats as used in the present study (Rundfeldt et al., 1990; Wahnschaffe & Löscher, 1992).
Kindling
A bipolar electrode for electrical stimulation and electroencephalogram (EEG) recording was stereotaxically implanted into the basolateral nucleus of the right amygdala as previously described (Löscher & Hönack, 1995). After a postoperative period of 2 weeks, constant current stimulations (500 μA, 1 ms, monophasic square-wave pulses, 50 s−1 for 1 s) were delivered to the amygdala once per day (five stimulations per week) until 10 sequential fully kindled (i.e., focal and secondarily generalized clonic) seizures were elicited.
Evaluation of drug effects on seizure threshold
For evaluation of anticonvulsant drug effects on focal seizures, the afterdischarge threshold (ADT), i.e., the most sensitive measure of anticonvulsant activity against focal seizure activity in kindled rats, was recorded after kindling acquisition (with an interval of at least 4 days after the 10th stage 5 seizure) using an ascending stairstep procedure (Freeman & Jarvis, 1981). The initial current intensity was 10 μA, and the current intensity was increased in steps of about 20% of the previous current at intervals of 1 min until an afterdischarge of at least 3 s duration was elicited in the EEG recorded via the implanted electrode from the amygdala. In addition to ADT, the following parameters of kindled seizures were measured at ADT current: Seizure severity was classified according to Racine (1972): (1) immobility, eye closure, twitching of vibrissae, sniffing, facial clonus; (2) head nodding associated with more severe facial clonus; (3) clonus of one forelimb; (4) rearing, often accompanied by bilateral forelimb clonus; (5) rearing with loss of balance and falling accompanied by generalized clonic seizures. Almost all rats showed focal (stage 1 and 2) and secondarily generalized (stage 3–5) seizures at focal seizure threshold (ADT) currents. Seizure duration was the duration of limbic (stage 1 and 2) and/or motor seizures (stage 3–5). Motor seizures were followed by a period characterized by immobility and occasional facial clonus or head nodding. This period from the end of motor seizures to onset of normal behaviour was termed postictal behavioural depression. Afterdischarge duration was the total time of spikes in the EEG recorded from the site of stimulation. After all rats showed reproducible ADTs, the effects of test compounds on ADT and severity and duration of seizures recorded at ADT were determined in groups of 7–9 fully kindled rats 30 min after i.p. administration. The control ADT was determined 2–3 days prior to and after each drug treatment, and the next drug experiment was only undertaken if the post-drug ADT was not significantly different from the pre-drug ADT. For control determinations, rats received i.p. administration of vehicle with the same pretreatment time as in the respective drug experiment. For all drug experiments, at least 7 days were interposed between two drug injections in the same group of rats in order to avoid alterations in drug potency due to cumulation or tolerance.
Evaluation of adverse effects
For examination of behavioural drug effects, the animals were removed from their home cages and placed singly in plastic cages. The animals were continuously observed for alterations in behaviour after i.p. drug injection up to the time of amygdala stimulation. Control experiments with vehicle injection were done in the same way. For all observations, rigorous observational protocols described elsewhere were used (Löscher and Hönack, 1992; 1995). Ataxia and sedation were scored using a rating system as described previously (Hönack & Löscher, 1995). In addition to rating motor impairment by observational scores, impaired motor function was quantitated by the rotarod test 15 min after drug injection and shortly before amygdala stimulation as described previously (Hönack & Löscher, 1995). Except at the highest doses, rats passed the rotarod test in all drug experiments, so that data are not illustrated in the Results section.
For comparison with the kindled rats, age-matched non-kindled (and non-implanted) rats were used in some experiments on drug adverse effects. In all experiments in kindled and nonkindled rats, rectal body temperature was recorded by an electronic thermometer immediately before and 15 and 28 min after drug or vehicle administration. Since none of the various drug treatments significantly affected body temperature, no detailed data are given in the Results section.
All rats were habituated to the various manipulations prior to onset of the drug experiments. Vehicle injection did not induce any behavioural alterations or rotarod failures, but sometimes significantly increased body temperature, most likely due to the stress associated with handling of the animals.
Statistics
Significance of differences between seizure readings (ADT and severity and duration of seizures) in the same group of rats (e.g., the difference between control and drug trial) was calculated by the Wilcoxon signed-rank test for paired replicates. With respect to body temperature, significance of difference to predrug values in the same group of rats was determined by Student's t-test for paired data. Fisher's exact test was used to compare seizure incidence between groups.
Drugs
All compounds (racemic tramadol, (−)-tramadol, (+)-tramadol, and the (+)-enantiomer of O-desmethyltramadol; see Figure 1) were synthesized at Grünenthal and were freshly dissolved in distilled water prior to each experiment. Controls received the same volume of saline. Administration volume was 3 ml kg−1 i.p.
Results
(±)-Tramadol
The racemate of tramadol was tested in fully kindled rats at doses of 2.5, 5, 10, 20 and 30 mg kg−1. At the lowest dose, a tendency for increased ADT was observed, which, however, was not significant (Figure 2). Seizure severity (SS), seizure duration (SD), and afterdischarge duration (ADD) recorded at ADT were not significantly affected at 2.5 mg kg−1 (Figure 2). However, the duration of postictal behavioural depression was significantly increased from 133±28.9 s (mean±s.e.mean, n=9) to 262±29.1 s (P=0.0117). No behavioural adverse effects were noted at 2.5 mg kg−1. At 5 mg kg−1, a significant increase in ADT of 57% above control was obtained, i.e., tramadol exerted anticonvulsant activity on focal seizure threshold at this dose. This effect was lost at higher doses (Figure 2). However, a significant reduction of SD was determined at 10 mg kg−1 and a significant reduction in ADD at 20 mg kg−1. The duration of postictal behavioural depression was not significantly affected by 5, 10 or 20 mg kg−1 of tramadol. No behavioural adverse effects and no indication of any proconvulsant effect were seen at these doses.
Figure 2.
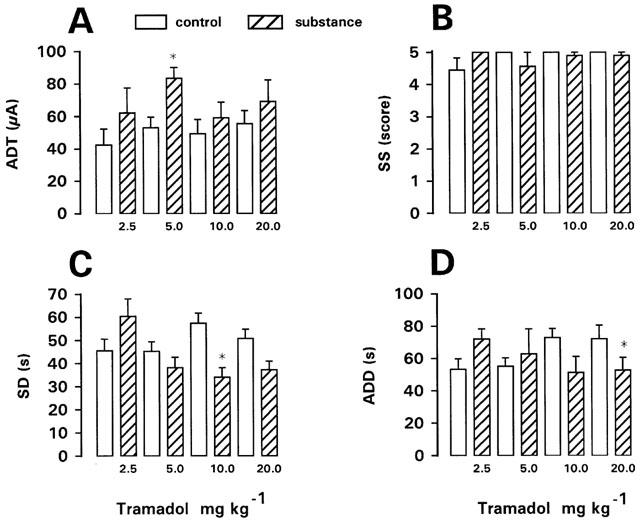
Effect of (±)-tramadol on afterdischarge threshold (ADT) and seizure severity (SS), seizure duration (SD), and afterdischarge duration (ADD) recorded at ADT in fully kindled rats. Doses in mg kg−1 i.p. are indicated below each bar. Data from predrug control trials are shown as open bars. Data are means±s.e.mean of groups of nine rats per dose. Significance of difference between a drug trial and the individual predrug control experiment in the same group of rats is indicated by asterisk (P<0.05).
However, by increasing the dosage of (±)-tramadol to 30 mg kg−1, severe adverse effects occurred. Five of seven kindled rats showed tramadol-induced seizures before amygdala stimulation (Table 1). One of these rats exhibited facial myoclonic (stage 2) seizures after 15 min; at ADT determination a stage 5 seizure occurred four steps below the predrug control ADT, indicating a markedly reduced seizure threshold. The other four rats showed generalized (stage 3, 4 or 5) seizures beginning at 15–18 min following tramadol. In one of these rats, the generalized seizures culminated into a status epilepticus, which was terminated by diazepam. In these rats with generalized seizures, no ADT was determined. In the two kindled rats without tramadol-induced seizures, ADT was either decreased or not altered compared to predrug control ADT.
Table 1.
Seizures induced by tramadol, its enantiomers, and the (+)-M1 metabolite in kindled and non-kindled rats

In five nonkindled controls, no seizures were observed after tramadol, 30 mg kg−1 (Table 1). The difference in seizure incidence between kindled and nonkindled rats was significant (Table 1). Apart from seizures, tramadol, 30 mg kg−1, induced Straub tail, moderate ataxia and sedation in both kindled and nonkindled rats.
(−)-Tramadol
The (−)-enantiomer of tramadol was tested in kindled rats at the same doses as the racemate (Figure 3). Significant increases in ADT of 79, 89 and 97% above predrug control were seen at 2.5, 5 and 10 mg kg−1, respectively. Although a tendency for increased ADT was also seen at 20 and 30 mg kg−1, this was not statistically significant, because of high interindividual variation in effects on ADT. Except a significant decrease in SD at 5 mg kg−1 and a significant decrease in ADD at 30 mg kg−1, no significant effects on SS, SD, or ADD were recorded. The duration of postictal behavioural depression was not significantly affected by the (−)-enantiomer.
Figure 3.
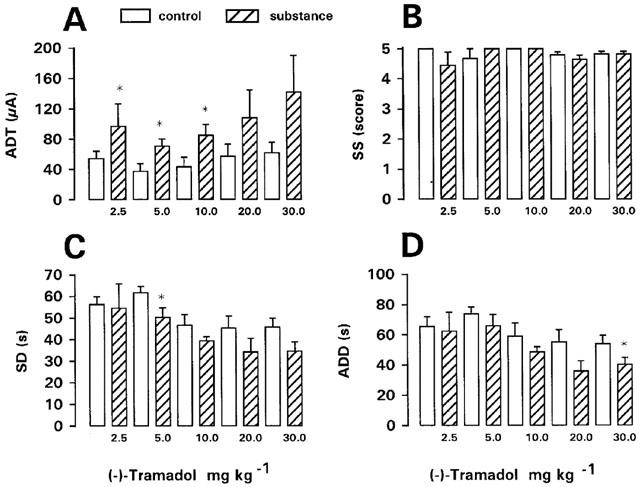
Effect of (−)-tramadol on afterdischarge threshold (ADT) and seizure severity (SS), seizure duration (SD), and afterdischarge duration (ADD) recorded at ADT in fully kindled rats. Doses in mg kg−1 i.p. are indicated below each bar. Data from predrug control trials are shown as open bars. Data are means±s.e.mean of groups of 7–9 rats per dose. Significance of difference between a drug trial and the individual predrug control experiment in the same group of rats is indicated by asterisk (P<0.05).
During the ADT determinations with the (−)-enantiomer, rats often reacted with increased susceptibility to the stimulus. Thus, all rats showed prolonged myoclonic activity after the first amygdala stimulation following 10 or 20 mg kg−1, which was not observed in these rats during determination of control ADT. At 2.5 mg kg−1, one rat developed a status with generalized seizures after the ADT determination, which had to be interrupted by diazepam.
Up to 10 mg kg−1 (−)-tramadol, no seizures were observed in kindled rats before ADT determination after 30 min (Table 1). At 20 mg kg−1, one kindled rat developed a generalized (stage 5) seizure after 28 min, which lasted for 6 min. Therefore, this rat was excluded from ADT determination. At 30 mg kg−1, seven of eight kindled rats exhibited myoclonic twitches before ADT determination (Table 1). In nonkindled rats, myoclonic seizures were observed after 10 and 20 mg kg−1 (−)-tramadol (Table 1). At 10 mg kg−1, the incidence of myoclonic activity was significantly higher in nonkindled than kindled rats. At 30 mg kg−1, nonkindled rats showed hyperexcitability but no seizure activity, resulting in a significant difference in (−)-tramadol-induced seizure activity between kindled and nonkindled rats (Table 1). Except seizures, no other adverse effects were recorded after (−)-tramadol.
(+)-Tramadol
The (+)-enantiomer was tested at doses of 1–30 mg kg−1 in kindled rats. At doses of 1, 2.5, 5 or 10 mg kg−1, ADT was not significantly altered, but SS was significantly reduced at 5 mg kg−1, SD at 2.5, 5 and 10 mg kg−1, and ADD at 10 mg kg−1 (Figure 4). The duration of postictal behavioural depression was significantly increased at 1 mg kg−1 (from 284±38.4 s to 464±48.6 s; n=8; P=0.0234) and 2.5 mg kg−1 (from 273±23.0 s to 769±96.1 s; n=8; P=0.0078), but not at higher doses. Straub tail and moderate sedation were recorded after 2.5 and 5 mg kg−1. At 10 mg kg−1, one rat displayed a generalized (stage 4–5) seizure 28 min after drug injection (Table 1), and was therefore not used for ADT determination. Increase of dosage to 20 mg kg−1 induced severe respiratory depression, so that only two rats were treated with this dose. One of these rats showed loss of righting reflexes. ADT was unchanged in one rat, but increased in the other. Further increase in dosage to 30 mg kg−1 led to generalized (stage 4) seizures in both rats treated with this dose (Table 1), so that ADT was not determined. Furthermore, these rats showed respiratory depression, ataxia, catalepsy, extended hind legs and loss of righting reflexes. One rat died 12 h following drug injection. In nonkindled rats, no seizures were observed after 20 or 30 mg kg−1 of the (+)-enantiomer, but rats displayed severe respiratory depression.
Figure 4.
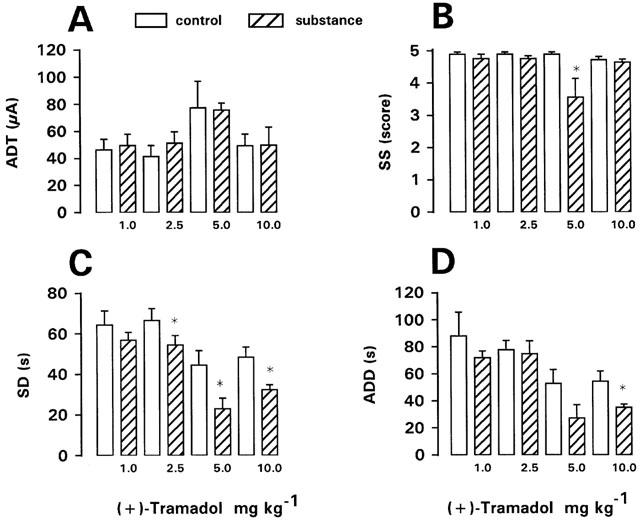
Effect of (+)-tramadol on afterdischarge threshold (ADT) and seizure severity (SS), seizure duration (SD), and afterdischarge duration (ADD) recorded at ADT in fully kindled rats. Doses in mg kg−1 i.p. are indicated below each bar. Data from predrug control trials are shown as open bars. Data are means±s.e.mean of groups of 7–8 rats per dose. Significance of difference between a drug trial and the individual predrug control experiment in the same group of rats is indicated by asterisk (P<0.05).
(+)-Desmethyltramadol (M1 metabolite)
The (+)-enantiomer of the M1 metabolite was tested at doses of 1, 2.5, 5, 10 and 30 mg kg−1 in kindled rats. At 1 or 2.5 mg kg−1, no significant effects on ADT or seizure parameters recorded at ADT were determined (Figure 5). Straub tail and moderate sedation were observed after 2.5 mg kg−1. Increase of dosage to 5 mg kg−1 led to severe respiratory depression, so that only two rats were tested with this dose. One of these rats exhibited myoclonic twitches before ADT determination (Table 1). ADT was not affected (not illustrated). At 10 and 30 mg kg−1 (tested in two rats each), severe respiratory depression and catalepsy with muscle rigidity, but no seizures were observed. ADT was not affected at 10, but markedly increased at 30 mg kg−1 (not illustrated). One of the rats treated with 30 mg kg−1 died within 24 h after drug injection. In nonkindled rats, severe respiratory depression was observed following 10 or 30 mg kg−1 of the metabolite (Table 1).
Figure 5.
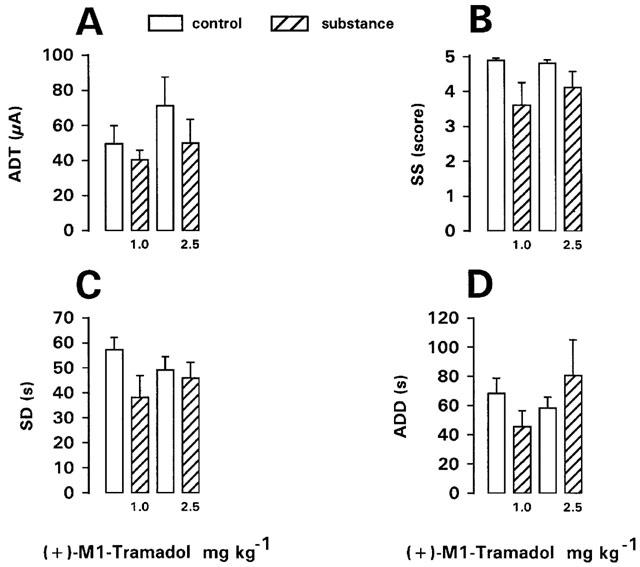
Effect of (+)-desmethyltramadol, i.e., the active M1 metabolite of tramadol on afterdischarge threshold (ADT) and seizure severity (SS), seizure duration (SD), and afterdischarge duration (ADD) recorded at ADT in fully kindled rats. Doses in mg kg−1 i.p. are indicated below each bar. Data from predrug control trials are shown as open bars. Data are means±s.e.mean of groups of 8–9 rats per dose. Significance of difference between a drug trial and the individual predrug control experiment in the same group of rats is indicated by asterisk (P<0.05).
Discussion
High doses of morphine and other opioid analgesics can induce naloxone-insensitive electrographic epileptiform patterns and behavioural convulsions in a variety of species, including humans (Frenk, 1983; Reisine & Pasternak, 1996). In contrast, administration of opioid analgesics at lower doses within the analgesic range have been reported to induce anticonvulsant effects (Frenk, 1983; Czuczwar & Frey, 1986; Schwark et al., 1986). However, depending on affinity and intrinsic efficacy at multiple opioid receptors, the seizure model, or alterations in brain function, such low doses of opioid analgesics may be proconvulsant, too (Frenk, 1983). For instance, fentanyl and alfentanyl have been reported to enhance focal seizure activity in patients with temporal lobe epilepsy (Bowdle, 1998). In line with this clinical finding, small doses of fentanyl decreased the focal seizure threshold (ADT) in the amygdala kindling model of temporal lobe epilepsy in rats (Schwark et al., 1986). Epilepsy may also increase the sensitivity to proconvulsant action of morphine; e.g., extremely low doses of morphine elicited seizures in genetically epilepsy-prone rats (Reigel et al., 1988) and amygdala kindled rats showed a heightened sensitivity to morphine's convulsive effects which could be blocked by naloxone (Mansour & Valenstein, 1984). Indeed, increased opioid receptor binding has been described in patients with temporal lobe epilepsy and different models of epilepsy, including amygdala kindling (Lee et al., 1986; Frost et al., 1988; Fisher & Frost, 1991; Engel & Rocha, 1992; Rocha et al., 1993), which may explain the increased susceptibility to proconvulsant effects of opioid analgesics.
In the present experiments with (±)-tramadol, myoclonic seizures and generalized convulsions were induced by administration of 30 mg kg1 in kindled but not in nonkindled rats, which seem to substantiate that seizure proneness increases the sensitivity to opioid receptor activation. Although (±)-tramadol is only a weak mu receptor agonist (Ki 8.3 μM vs 0.0021 μM for morphine; Frink et al., 1996; see Table 2), the (+)-enantiomer of the M1 metabolite, which is rapidly formed in vivo (see Figure 1), is much more potent (Frink et al., 1996; Lai et al., 1996). It has therefore been suggested that (+)-M1 might be a major source of the opioid component of tramadol-induced analgesia (Lai et al., 1996). Compared to affinity for mu receptors, affinities of (±)-tramadol or its enantiomers to delta and kappa opioid receptors are much lower (Table 2). Nevertheless, in view of previous findings indicating an important role of the delta receptor in proconvulsant effects of opioids (Frenk, 1983; Dua et al., 1985; Reisine & Pasternak, 1996), this receptor type may be involved in the convulsant activity seen after high doses of tramadol, too. In this respect, it also important to note that the (+)-M1 metabolite of tramadol has an about 150 times higher potency at the delta receptor (Ki 0.69 μM) than the parent compound (Table 2).
Table 2.
Inhibition of opioid binding or monoamine uptake by tramadol, its enantiomers and the M1 metabolite
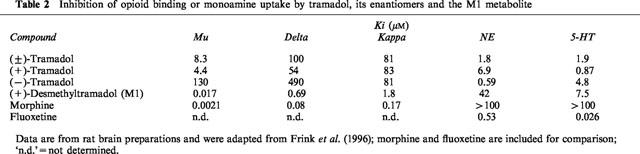
In rodent antinociception tests, (±)-tramadol exerts antinociceptive effects with ED50s ranging between 3 and 20 mg kg−1 after s.c. or i.p. administration (Friderichs et al., 1978; Raffa et al., 1993). Evaluation of the enantiomers of tramadol in such tests has shown complementary and synergistic antinociceptive interaction between (+)- and (−)-tramadol (Raffa et al., 1993). (+)-Tramadol is 30 times as potent as (−)-tramadol at the mu site and five times more potent to inhibit uptake of 5-HT, whereas the (−)-enantiomer is about 10 times more potent than the (+)-enantiomer in blocking NE uptake (see Table 2). Both the mu agonism and the 5-HT and NE uptake inhibition are thought to contribute to the antinociception produced by (±)-tramadol in vivo (Lee et al., 1993; Raffa et al., 1993; Dayer et al., 1994; Lai et al., 1996; Bamigbade & Langford, 1998).
Respiratory depression is the most severe opioid adverse effect and is thought to be primarily mediated via mu receptors (Bowdle, 1998). The differences in mu potencies between tramadol, its enantiomers and the (+)-M1 metabolite ((+)-M1 > > (+) - tramadol >( ±)- tramadol > > (−) - tramadol; Raffa et al., 1993; Frink et al., 1996; Lai et al., 1996; see Table 2) therefore explain the present finding that respiratory depression was observed at relatively low doses of the (+)-M1 metabolite (5 mg kg−1) and (+)-tramadol (20 mg kg−1), but not the racemate and the (−)-enantiomer. Respiratory depression limited the dose range of (+)-tramadol and the (+)-M1 metabolite which could be tested for proconvulsant activity. Both compounds induced seizures in kindled rats at doses below those of (±)-tramadol, which could indicate that the mu receptor was involved in this convulsant activity. However, previous experiments in nonkindled rats and mice have shown that the seizures occurring after high doses of (±)-tramadol are not blocked by high doses (10 mg kg−1) of naloxone (Friderichs et al., unpublished data), which is similar to the naloxone-insensitive convulsions observed after high doses of morphine and other opioid analgesics (Frenk, 1983; Reisine & Pasternak, 1996), and argues against a critical role of the mu-receptor in tramadol-induced seizures. Furthermore, (−)-tramadol, which is about 15 times less potent at mu receptors in rat brain preparations than the racemate (Table 2), exhibited a similar convulsant potency in kindled rats than the (±)-racemate in the present experiments. In this respect it is important to note that the type of seizures differed. At 30 mg kg−1, (±)-tramadol predominantly induced generalized convulsive seizures, whereas (−)-tramadol induced myoclonic (nonconvulsive) seizures. It has been shown previously in rats that classical opioid analgesics such as morphine produce generalized convulsive seizures while delta agonists produce nonconvulsive seizure activity (Snead & Bearden, 1982). This could indicate that a delta receptor agonistic effect was involved in the present observations with the (−)-enantiomer of tramadol which, however, is unlikely because of the weak potency of (−)-tramadol at this opioid receptor subtype (Raffa et al., 1993; Lai et al., 1996).
In lower (analgesic range) doses (c.f., Raffa et al., 1993), (±)-tramadol and its (−)-enantiomer increased the focal seizure threshold (ADT) in kindled rats, while the (+)-enantiomer and the (+)-M1 metabolite did not exert such an anticonvulsant effect. Instead, (+)-tramadol was relatively potent to decrease seizure and afterdischarge duration, indicating that this drug inhibited seizure propagation from the focus. This observation suggests that different mechanisms underlie the anticonvulsant properties of the two tramadol enantiomers. Furthermore, the differences in anticonvulsant activity between (±)-tramadol, its enantiomers, and the (+)-M1 metabolite seem to indicate that these effects were not primarily mediated via mu receptors. However, previous experiments with opioid analgesics also resulted in differences between different mu agonists in the kindling model (Albertson et al., 1984; Schwark et al., 1986). For instance, analgesic doses of morphine increased ADT but exerted no significant effects on SS, SD, or ADD, while fentanyl did not increase ADT but significantly decreased SS, SD, and ADD (Schwark et al., 1986). With respect to tramadol and its enantiomers, in addition to mu receptor activation the inhibitory effects on monoamine uptake could be involved in the anticonvulsant activity observed at low doses of these compounds in the kindling model. It is well established that drugs enhancing serotonergic or noradrenergic activity exert anticonvulsant activity in different seizure models (Kilian & Frey, 1973; Peterson & Albertson, 1982; Przegalinski, 1985; Corcoran & Weiss, 1990). Antidepressant drugs, which block the uptake of NE, 5-HT, or both biogenic amines, were consistently shown to exert anticonvulsant activity after acute administration in fully kindled animals (Stach et al., 1980; Knobloch et al., 1982; Clifford et al., 1985; Minabe et al., 1987; Yacobi & Burnham, 1991). However, clinically many antidepressant drugs are thought to have the potential to provoke seizures, particularly in patients with a preexisting lowered seizure threshold (Trimble, 1984; Lipka & Lathers, 1987; Brodie, 1992). Correlations between the incidence of antidepressant-induced seizures and doses or plasma concentrations strongly indicate that this apparent paradox between preclinical and clinical findings is related to the fact that an increased risk for seizures in patients almost exclusively occurs after high doses or elevated plasma concentrations of antidepressant agents (Preskorn & Fast, 1992; Dailey & Naritoku, 1996; Alldredge, 1999). Thus, the antidepressant drugs are like several antiepileptic drugs that can both prevent and cause seizures (Dailey & Naritoku, 1996). It is therefore possible that tramadol's inhibitory effect on 5-HT and NE uptake is involved both in its anticonvulsant and convulsant activity. In this respect, it is interesting to note that concomitant treatment with antidepressant medications appears to increase the risk of tramadol-related seizures (Kahn et al., 1997).
With respect to tramadol's anticonvulsant activity seen at low doses in the present experiments in kindled rats, previous studies in other seizure models yielded inconsistent results. In the MES test in mice, (±)-tramadol, 10–40 mg kg−1 orally, exerted no anticonvulsant effects (Osterloh et al., 1978). When seizures were induced by pentylenetetrazol in mice, tramadol, 10–40 mg kg−1, did not affect seizures or mortality (Osterloh et al., 1978). However, in a more recent study on MES in mice, i.p. administration of tramadol resulted in a dose-dependent reduction in the duration of tonic hindlimb extension with an ED50 of 33 mg kg−1 (Manocha et al., 1998). The anticonvulsant effect was antagonized by a selective kappa receptor antagonist and also by high doses of naloxone, whereas low doses of naloxone were ineffective. The authors concluded that the anti-MES effect of tramadol is mediated by kappa receptors (Manocha et al., 1998). In rat brain preparations, (±)-tramadol and its enantiomers inhibit kappa opioid binding with Kis of about 80 μM (Table 2), i.e., with about the same potency, while the respective potency of the (+)-M1 metabolite is much higher (Ki 1.8 μM; Frink et al., 1996). The differences in anticonvulsant activity between (±)-tramadol and its enantiomers and the (+)-M1 metabolite observed in the present study in kindled rats make it unlikely that effects on kappa receptors are critically involved. On the other hand, based on experiments with opioid analgesics in an electroconvulsive model in mice, Frey (1988) has proposed that stimulation of both mu and kappa receptors acts synergistically in evoking anticonvulsant effects, and that these effects may be inhibited or abolished when one of both binding sites is blocked, which could explain the observations of Manocha et al. (1998) with tramadol.
Interestingly, low doses of (±)-tramadol and its (+)-enantiomer markedly increased the duration of postictal behavioural depression in the present experiments in kindled rats. There is evidence that endogenous opioids mediate limbic postictal events, including behavioural suppression and decreased seizure susceptibility (Frenk et al., 1979; Frenk, 1983; Caldecott-Hazard & Engel, 1987; Tortella, 1988; Hong, 1992; Rocha et al., 1997). For instance, the inhibition that one kindled seizure produces upon a subsequent seizure is reduced by naloxone (Frenk, 1983; Tortella, 1988). Various types of seizures, including amygdala kindled seizures, have shown to be followed by a transient increase in release of endogenous opioids, and it has been suggested that endogenous opioids may play an important functional role in the mechanisms of seizure arrest and postseizure refractoriness (Frenk, 1983; Tortella, 1988; Hong, 1992; Rocha et al., 1997). The significant increase in the duration of postictal behavioural depression seen after low doses of tramadol and its (+)-enantiomer in the present study may thus indicate that these drugs interacted in an additive manner with endogenous opioids.
In conclusion, the present data from the kindling model show that tramadol exerts anticonvulsant activity at doses within the analgesic range, but only moderately higher doses induce myoclonic activity and generalized convulsions, which are not seen at these i.p. doses in nonkindled rats. After i.v. administration, clonic or tonic-clonic convulsions have been reported previously in NMRI mice, Sprague-Dawley rats, rabbits, and dogs following doses of tramadol of 10–20 mg kg−1 (Osterloh et al., 1978). After i.p. application of (±)-tramadol in female Wistar rats (as used in the present study), convulsions were observed after doses of 50–60 mg kg−1 (Friderichs, unpublished data). The present results indicate that epileptogenesis as induced by kindling enhances the convulsant potency of tramadol. Convulsant activity was found with both enantiomers of tramadol and the (+)-M1 metabolite, indicating that these compounds interact in a complementary manner to produce seizures. In view of the complex effects of tramadol on opioid receptors and monoamine uptake, it is not possible to conclude which mechanism is primarily responsible for the observed effects in kindled rats.
Acknowledgments
We thank Mrs M. Schröder for technical assistance. The study was supported by grants from Grünenthal (Aachen, Germany).
Abbreviations
- ADD
afterdischarge duration
- ADT
afterdischarge threshold
- EEG
electroencephalogram
- 5-HT
5-hydroxytryptamine
- M1
mono-O-desmethyltramadol
- MES
maximal electroshock seizure test
- NE
norepinephrine
- SD
seizure duration
- SS
seizure severity
References
- ALBERTSON T.E., JOY R.M., STARK L.G. Modification of kindled amygdaloid seizures by opiate agonists and antagonists. J. Pharmacol. Exp. Ther. 1984;228:620–627. [PubMed] [Google Scholar]
- ALLDREDGE B.K. Seizure risk associated with psychotropic drugs: clinical and pharmacokinetic considerations. Neurology. 1999;53 Suppl. 2:S68–S75. [PubMed] [Google Scholar]
- BAMIGBADE T.A., LANGFORD R.M. Tramadol hydrochloride: an overview of current use. Hosp. Med. 1998;59:373–376. [PubMed] [Google Scholar]
- BOWDLE T.A. Adverse effects of opioid agonists and agonists-antagonists in anaesthesia. Drug Saf. 1998;19:173–189. doi: 10.2165/00002018-199819030-00002. [DOI] [PubMed] [Google Scholar]
- BRODIE M.J. Drug interactions in epilepsy. Epilepsia. 1992;33 Suppl. 1:S13–S22. doi: 10.1111/j.1528-1157.1992.tb05896.x. [DOI] [PubMed] [Google Scholar]
- CALDECOTT-HAZARD S., ENGEL J.J. Limbic postictal events: anatomical substrates and opioid receptor involvement. Prog. Neuro-Psych. Biol. Psych. 1987;11:389–418. doi: 10.1016/0278-5846(87)90014-5. [DOI] [PubMed] [Google Scholar]
- CLIFFORD D.B., RUTHERFORD J.L., HICKS F.G., ZORUMSKI C.F. Acute effects of antidepressants on hippocampal seizures. Ann. Neurol. 1985;18:692–697. doi: 10.1002/ana.410180611. [DOI] [PubMed] [Google Scholar]
- CORCORAN M.E., WEISS G.K.Noradrenaline and kindling revisited Kindling 4 1990New York, Plenum Press; 141–156.ed. Wada J.A., pp [Google Scholar]
- CZUCZWAR S.J., FREY H.-H. Effect of morphine and morphine-like analgesics on susceptibility to seizures in mice. Neuropharmacology. 1986;25:465–469. doi: 10.1016/0028-3908(86)90169-3. [DOI] [PubMed] [Google Scholar]
- DAILEY J.W., NARITOKU D.K. Antidepressants and seizures: clinical anecdotes overshadow neuroscience. Biochem. Pharmacol. 1996;52:1323–1329. doi: 10.1016/s0006-2952(96)00509-6. [DOI] [PubMed] [Google Scholar]
- DAYER P., COLLART L., DESMEULES J. The pharmacology of tramadol. Drugs. 1994;47 Suppl. 1:3–7. doi: 10.2165/00003495-199400471-00003. [DOI] [PubMed] [Google Scholar]
- DUA A.K., PINSKY C., LABELLA F.S. Mu- and delta-opioid receptor-mediated epileptoid responses in morphine-dependent and non-dependent rats. Electroencephalogr. Clin. Neuro. 1985;61:569–572. doi: 10.1016/0013-4694(85)90976-9. [DOI] [PubMed] [Google Scholar]
- ENGEL J.J., ROCHA L.L. Interictal behavioral disturbances: a search for molecular substrates. Epilepsy Res. 1992;9 Suppl.:341–349. [PubMed] [Google Scholar]
- FISHER R.S., FROST J.J. Epilepsy J. Nucl. Med. 1991. pp. 651–659. [PubMed]
- FREEMAN F.G., JARVIS M.F. The effect of interstimulation interval on the assessment and stability of kindled seizure threshold. Brain Res. Bull. 1981;7:629–633. doi: 10.1016/0361-9230(81)90109-x. [DOI] [PubMed] [Google Scholar]
- FRENK H. Pro- and anticonvulsant actions of morphine and the endogenous opioids: Involvement and interactions of multiple opiate and non-opiate systems. Brain Res. Rev. 1983;6:197–210. doi: 10.1016/0165-0173(83)90039-5. [DOI] [PubMed] [Google Scholar]
- FRENK H., ENGEL J.J., ACKERMANN R.F., SHAVIT Y., LIEBESKIND J.C. Endogenous opioids may mediate post-ictal behavioral depression in amygdaloid-kindled rats. Brain Res. 1979;167:435–440. doi: 10.1016/0006-8993(79)90842-4. [DOI] [PubMed] [Google Scholar]
- FREY H.-H. Effects of μ- and kappa-opioid agonists on the electroconvulsive seizure threshold in mice and antagonism by naloxone and MR 2266. Pharmacol. Toxicol. 1988;62:150–154. doi: 10.1111/j.1600-0773.1988.tb01863.x. [DOI] [PubMed] [Google Scholar]
- FRIDERICHS E., FELGENHAUER F., JONGSCHAAP P., OSTERLOH G. Pharmakologische Untersuchungen zur Analgesie, Abhngigkeits-und Toleranzentwicklung von Tramadol, einem stark wirksamen Analgetikum. Arzneim.-Forsch. (Drug Res.) 1978;28:122–134. [PubMed] [Google Scholar]
- FRINK M.C., HENNIES H.-H., ENGLBERGER W., HAURAND M., WILFFERT B. Influence of tramadol on neurotransmitter systems of the rat brain. Arzneim.-Forsch. (Drug Res.) 1996;46:1029–1036. [PubMed] [Google Scholar]
- FROST J.J., MAYBERG H.S., FISHER R.S., DOUGLASS K.H., DANNALS R.F., LINKS J.M., WILSON A.A., RAVERT H.T., ROSENBAUM A.E., SNYDER S.H. Mu-opiate receptors measured by positron emission tomography are increased in temporal lobe epilepsy. Ann. Neurol. 1988;23:231–237. doi: 10.1002/ana.410230304. [DOI] [PubMed] [Google Scholar]
- GIBSON T.P. Pharmacokinetics, efficacy, and safety of analgesia with a focus on tramadol HCl. Am. J. Med. 1996;101:47S–53S. doi: 10.1016/s0002-9343(96)00138-6. [DOI] [PubMed] [Google Scholar]
- HÖNACK D., LÖSCHER W. Kindling increases the sensitivity of rats to adverse effects of certain antiepileptic drugs. Epilepsia. 1995;36:763–771. doi: 10.1111/j.1528-1157.1995.tb01613.x. [DOI] [PubMed] [Google Scholar]
- HONG J.S. Hippocampal opioid peptides and seizures. Epilepsy Res. 1992;7 Suppl.:187–195. [PubMed] [Google Scholar]
- JICK H., DERBY L.E., VASILAKIS C., FIFE D. The risk of seizures associated with tramadol. Pharmacotherapy. 1998;18:607–611. [PubMed] [Google Scholar]
- KHAN L.H., ALDERFER R.J., GRAHAM D.J. Seizures reported with tramadol. J. Am. Med. Assoc. 1997;278:1661. [PubMed] [Google Scholar]
- KILIAN M., FREY H.-H. Central monoamines and convulsive thresholds in mice and rats. Neuropharmacology. 1973;12:681–692. doi: 10.1016/0028-3908(73)90121-4. [DOI] [PubMed] [Google Scholar]
- KNOBLOCH L.C., GOLDSTEIN J.M., MALICK J.B. Effects of acute and subacute antidepressant treatment on kindled seizures in rats. Pharmacol. Biochem. Behav. 1982;17:461–465. doi: 10.1016/0091-3057(82)90305-7. [DOI] [PubMed] [Google Scholar]
- LAI J., MA S.-W., PORECCA F., RAFFA R.B. Tramadol, M1 metabolite and enantiomer affinities for cloned human opioid receptors expressed in transfected HN9.10 neuroblastoma cells. Eur. J. Pharmacol. 1996;316:369–372. doi: 10.1016/s0014-2999(96)00770-4. [DOI] [PubMed] [Google Scholar]
- LEE C.R., MCTAVISH D., SORKIN E.M. Tramadol. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in acute and chronic pain states. Drugs. 1993;46:313–340. doi: 10.2165/00003495-199346020-00008. [DOI] [PubMed] [Google Scholar]
- LEE R.J., MCCABE R.T., WAMSLEY J.K., OLSEN R.W., LOMAX P. Opioid receptor alterations in a genetic model of generalized epilepsy. Brain Res. 1986;380:76–82. doi: 10.1016/0006-8993(86)91431-9. [DOI] [PubMed] [Google Scholar]
- LEWIS K.S., HAN N.H. Tramadol: a new centrally acting analgesic. Am. J. Health Syst. Pharm. 1997;54:643–652. doi: 10.1093/ajhp/54.6.643. [DOI] [PubMed] [Google Scholar]
- LIPKA L.J., LATHERS C.M. Psychoactive agents, seizure production, and sudden death in epilepsy. J. Clin. Pharmacol. 1987;27:169–183. doi: 10.1002/j.1552-4604.1987.tb02180.x. [DOI] [PubMed] [Google Scholar]
- LÖSCHER W. Pharmacology of glutamate receptor antagonists in the kindling model of epilepsy. Prog. Neurobiol. 1998;54:721–741. doi: 10.1016/s0301-0082(97)00092-0. [DOI] [PubMed] [Google Scholar]
- LÖSCHER W., HÖNACK D. Responses to NMDA receptor antagonists altered by epileptogenesis. Trends Pharmacol. Sci. 1991;12:52. doi: 10.1016/0165-6147(91)90496-f. [DOI] [PubMed] [Google Scholar]
- LÖSCHER W., HÖNACK D. The behavioural effects of MK-801 in rats: involvement of dopaminergic, serotonergic and noradrenergic systems. Eur. J. Pharmacol. 1992;215:199–208. doi: 10.1016/0014-2999(92)90029-4. [DOI] [PubMed] [Google Scholar]
- LÖSCHER W., HÖNACK D. Anticonvulsant and antiepileptogenic effect of L-deprenyl (selegiline) in the kindling model of epilepsy. J. Pharmacol. Exp. Ther. 1995;274:307–314. [PubMed] [Google Scholar]
- MANOCHA A., SHARMA K.K., MEDIRATTA P.K. Tramadol, a centrally acting opioid: anticonvulsant effect against maximal electroshock seizure in mice. Ind. J. Physiol. Pharmacol. 1998;42:407–411. [PubMed] [Google Scholar]
- MANSOUR A., VALENSTEIN E.S. Morphine responsiveness and seizure proneness. Experimental Neurology. 1984;85:346–357. doi: 10.1016/0014-4886(84)90145-6. [DOI] [PubMed] [Google Scholar]
- MATTHIESEN T., WOHRMANN T., COOGAN T.P., URAGG H. The experimental toxicology of tramadol: an overview. Toxicol. Lett. 1998;95:63–71. doi: 10.1016/s0378-4274(98)00023-x. [DOI] [PubMed] [Google Scholar]
- MINABE Y., TANII Y., KURACHI M. Acute effect of some psychotropic drugs on low-frequency amygdaloid kindled seizures. Biol. Psychiatr. 1987;22:1444–1450. doi: 10.1016/0006-3223(87)90102-8. [DOI] [PubMed] [Google Scholar]
- OSTERLOH G., FRIDERICHS E., FELGENHAUER F., GÜNZLER W.A., HENMI Z., KITANO T., NAKAMURA M., HAYASHI H., ISHII I. Allgemeine pharmakologische Untersuchungen mit Tramadol, einem stark wirkenden Analgetikum. Arzneim.-Forsch. (Drug Res.) 1978;28:135–151. [PubMed] [Google Scholar]
- PETERSON S.L., ALBERTSON T.E. Neurotransmitter and neuromodulator function in the kindled seizure and state. Progr. Neurobiol. 1982;19:237–270. doi: 10.1016/0301-0082(82)90008-9. [DOI] [PubMed] [Google Scholar]
- PRESKORN S.H., FAST G.A. Tricyclic antidepressant-induced seizures and plasma drug concentration. J. Clin. Psychiatry. 1992;53:160–162. [PubMed] [Google Scholar]
- PRZEGALINSKI E.Monoamines and the pathophysiology of seizure disorders Antiepileptic Drugs 1985Berlin, Springer; 101–137.eds Frey, H.-H. & Janz, D. pp [Google Scholar]
- RACINE R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroenceph. Clin. Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- RAFFA R.B., FRIDERICHS E., REIMANN W., SHANK R.P., CODD E.E., VAUGHT J.L., JACOBY H.I., SELVE N. Complementary and synergistic antinociceptive interaction between the enantiomers of tramadol. J. Pharmacol. Exp. Ther. 1993;267:331–340. [PubMed] [Google Scholar]
- RAFFA R.B., NAYAK R.K., LIAO S., MINN F.L. The mechanism(s) of action and pharmacokinetics of tramadol hydrochloride. Rev. Contemp. Pharmacother. 1995;6:485–497. [Google Scholar]
- REIGEL C.E., JOBE P.C., DAILEY J.W., STEWARD J.J. Responsiveness of genetically epilepsy-prone rats to intracerebroventricular morphine-induced convulsions. Life Sci. 1988;42:1743–1749. doi: 10.1016/0024-3205(88)90040-9. [DOI] [PubMed] [Google Scholar]
- REISINE T., PASTERNAK G.Opioid analgesics and antagonists Goodman & Gilman's The pharmacological basis of therapeutics, 9th edn. 1996New York, McGraw-Hill; 521–556.eds Hardman, J.G. Limbird, L.E. Molinoff, P.B. Ruddon, R.W. & Goodman Gilman, A. pp [Google Scholar]
- ROCHA L., ACKERMANN R.F., NASSIR Y., CHUGANI H.T., ENGEL J.J. Characterization of mu opioid receptor binding during amygdala kindling in rats and effects of chronic naloxone pretreatment: an autoradiographic study. Epilepsy Research. 1993;14:195–208. doi: 10.1016/0920-1211(93)90044-8. [DOI] [PubMed] [Google Scholar]
- ROCHA L.L., EVANS C.J., MAIDMENT N.T. Amygdala kindling modifies extracellular opioid peptide content in rat hippocampus measured by microdialysis. J. Neurochem. 1997;68:616–624. doi: 10.1046/j.1471-4159.1997.68020616.x. [DOI] [PubMed] [Google Scholar]
- RUNDFELDT C., HÖNACK D., LÖSCHER W. Phenytoin potently increases the threshold for focal seizures in amygdala-kindled rats. Neuropharmacology. 1990;29:845–851. doi: 10.1016/0028-3908(90)90159-o. [DOI] [PubMed] [Google Scholar]
- SCHWARK W.S., FREY H.-H., CZUCZWAR S.J. Effect of opiates on the parameters of seizures in rats with full amygdaloid-kindled convulsions. Neuropharmacology. 1986;25:839–844. doi: 10.1016/0028-3908(86)90008-0. [DOI] [PubMed] [Google Scholar]
- SNEAD O.C., BEARDEN L.J. The epileptogenic spectrum of opiate agonists. Neuropharmacology. 1982;21:1137–1144. doi: 10.1016/0028-3908(82)90171-x. [DOI] [PubMed] [Google Scholar]
- STACH R., LAZAROVA M.B., KACZ D. The effects of antidepressant drugs on the seizures kindled from the rabbit amygdala. Pol. J. Pharmacol. Pharm. 1980;32:505–512. [PubMed] [Google Scholar]
- TORTELLA F.C. Endogenous opioid peptides and epilepsy: quieting the seizing brain. Trends Pharmacol. Sci. 1988;9:366–372. doi: 10.1016/0165-6147(88)90256-8. [DOI] [PubMed] [Google Scholar]
- TRIMBLE M.R. Epilepsy, antidepressants, and the role of nomifensine. J. Clin. Psychiatry. 1984;45:39–42. [PubMed] [Google Scholar]
- WAHNSCHAFFE U., LÖSCHER W. Lack of changes in seizure susceptibility during the estrous cycle in kindled rats. Epilepsy Research. 1992;13:199–204. doi: 10.1016/0920-1211(92)90053-v. [DOI] [PubMed] [Google Scholar]
- WLAZ P., LÖSCHER W.Evaluation of associated behavioral and cognitive deficits in anticonvulsant drug testing Neuropharmacology methods in epilepsy research 1998Boca Raton, CRC Press; 172–192.eds Peterson, S.L. & Albertson, T.E. pp [Google Scholar]
- YACOBI R., BURNHAM W.M. The effect of tricyclic antidepressants on cortex- and amygdala-kindled seizures in the rat. Can. J. Neurol. Sci. 1991;18:132–136. doi: 10.1017/s0317167100031589. [DOI] [PubMed] [Google Scholar]