

Abstract
We investigated the effects of chronic pravastatin treatment on the impaired endothelium-dependent relaxation seen in aortae from established streptozotocin (STZ)-induced diabetic rats. Starting at 6 weeks of diabetes, pravastatin (10 mg kg−1) was administered to STZ-induced diabetic rats for 4 weeks.
The increased total cholesterol and low-density lipoprotein (LDL) cholesterol levels seen in STZ-induced diabetic rats were not restored to normal by pravastatin. Aortae from pravastatin-treated diabetic rats did not show an impaired endothelium-dependent relaxation to acetylcholine. The expression of the mRNA for endothelial nitric oxide synthase was unaffected by diabetes or pravastatin.
The enhanced level of malondialdehyde (MDA)-modified LDL seen in STZ-induced diabetic rats was normalized by pravastatin treatment. The resistance of LDL to oxidation was assessed by measuring the amount of MDA or conjugated dienes generated by incubation with copper ions. LDL isolated from diabetic rats, but not those from pravastatin-treated diabetics, showed enhanced the susceptibility to oxidation, but incubation in vitro with pravastatin had no effect on LDL oxidation.
Following incubation of control aortae for 6 h with LDL (0.1 mg protein ml−1) isolated from diabetic rats, the endothelium-dependent relaxation to acetylcholine or A23187 was impaired, but LDL isolated from control or pravastatin-treated rats had no such effect. This inhibitory effect of diabetic LDL was prevented by superoxide dismutase (SOD), a superoxide scavenger.
These results suggest that pravastatin preserves endothelial function in aortae from STZ-induced diabetic rats without lowering plasma cholesterol, and its effect may be due to decreased LDL oxidation.
Keywords: Pravastatin, diabetes, endothelium, LDL, oxidized LDL, relaxation, streptozotocin
Introduction
The relaxation responses of aortic strips to endothelium-dependent agents are weaker in streptozotocin (STZ)-induced diabetic rats than in normal rats (Oyama et al., 1986; Kamata et al., 1989; 1996; Poston & Taylor, 1995; Kamata & Kobayashi, 1996; Pieper, 1998). Oxidized LDL (ox-LDL) is thought to promote atherogenesis by a multitude of different mechanisms. Indeed, ox-LDL acts more potently than native LDL in markedly inhibiting the endothelium-dependent relaxation of the rabbit aorta (Kugiyama et al., 1990) and pig coronary artery (Simon et al., 1990). Thus, the circulating modified-LDL in diabetes may be responsible for the impaired endothelium-dependent relaxation. However, few studies of endothelial dysfunction in diabetes have directly assessed the relationship between circulating modified LDL and endothelium-dependent relaxation.
Pravastatin, an 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, has been shown to reduce adverse cardiovascular events in patients suffering from coronary artery disease with or without hypercholesterolemia (Sacks et al., 1996). Furthermore, It has been shown that HMG-CoA reductase inhibitors improve the defective endothelium-dependent vasodilation of atherosclerotic vessels in humans or animals by lowering plasma cholesterol (Kroon, et al., 1993; John et al., 1998). Although the mechanism by which HMG-CoA reductase inhibitors preserve vascular function is primarily attributed to their inhibition of hepatic HMG-CoA reductase and the subsequent lowering of plasma cholesterol levels, some recent studies have questioned the exclusive involvement of the decrease in plasma LDL levels in the effects of HMG-CoA reductase inhibitors (Hussein et al., 1997; O'Driscoll et al., 1997; Essig et al., 1998; Laufs et al., 1998). Thus, it remains unclear how an early improvement in endothelial function might occur during treatment with an HMG-CoA reductase inhibitor.
The aims of the present study were (1) to investigate the influence of chronic pravastatin treatment on the impairment of endothelium-dependent relaxation seen in aortae from rats with established STZ-induced diabetes and (2) to evaluate the effects of diabetes-modified LDL on the endothelium-dependent relaxation of the rat aorta.
Methods
Animals and experimental design
Male Wistar rats, 8 weeks old and 220–300 g in weight, received a single injection via the tail vein of STZ 75 mg kg−1 dissolved in a citrate buffer. Age-matched control rats were injected with the buffer alone. Food and water were given ad libitum. Starting 6 weeks after the STZ injection, pravastatin (10 mg kg−1, p.o., daily for 4 weeks) was administered to STZ-induced diabetic rats. In preliminary experiments, the endothelium-dependent relaxation was restored by this dose of pravastatin. Thus, we used this dose of pravastatin in the present experiment. As a control, STZ-induced diabetic rats received saline.
Measurement of plasma glucose, cholesterol, HDL, triglycerides, and phospholipid
Ten weeks after the STZ injection, glucose, total cholesterol, free cholesterol, triglycerides, and phospholipid were determined using a commercially available enzyme kit (Wako Chemical Company, Osaka, Japan). Cholesterol ester mass was calculated as (total cholesterol–free cholesterol)×1.68 (Tsai et al., 1992).
Preparation of VLDL and LDL
Lipoproteins (VLDL, d=0.96–1.019; LDL, d=1.019–1.063) were isolated by density gradient ultracentrifugation (Havel et al., 1955) from pooled plasma (EDTA, 1 mM) from age-matched controls, diabetics, or pravastatin-treated diabetic rats. The lipoproteins were extensively dialysed against phosphate-buffered saline (PBS).
Measurement of MDA levels
The oxidation state of rat total plasma and VLDL and LDL samples was analysed after the incubation period by means of the thiobarbituric acid-reactive substances (TBARS) assay, which is used to measure malondialdehyde (MDA) equivalents (Yagi, 1976).
Oxidation of LDL
The lipoproteins were diluted with EDTA-free PBS to a final concentration of 0.2 mg of protein ml−1 and then incubated with 10 μM CuSO4 at 37°C for 4 or 24 h. The oxidative state of the LDL was then assayed by means of a TBARS assay. In addition, the formation of conjugated dienes during LDL (25 μg protein ml−1) oxidation was assessed by monitoring the change in 234 nM absorbance at 15 min intervals for 300 min (using a spectrophotometer). In an in vitro experiment, plasma LDL from STZ-induced diabetic rats were incubated with pravastatin (0.1 or 0.001 mg ml−1) for 0.5 h before administration of CuSO4.
Measurement of isometric force
Rats were anaesthetized with diethyl ether and killed by decapitation 10 weeks after treatment with STZ or buffer. A section of the thoracic aorta from between the aortic arch and the diaphragm was then removed and placed in oxygenated, modified Krebs-Henseleit solution (KHS). The solution consisted of (mM): NaCl 118.0, KCl 4.7, NaHCO325.0, CaCl2 1.8, NaH2PO4 1.2, MgSO4 1.2, dextrose 11.0. The aorta was cleaned of loosely adhering fat and connective tissue and cut into helical strips 3 mm in width and 20 mm in length. The tissue was placed in a well-oxygenated (95% O2, 5% CO2) bath of 10 ml KHS at 37°C with one end connected to a tissue holder and the other to a force-displacement transducer (Nihon Kohden, TB-611T). The tissue was equilibrated for 60 min under a resting tension of 1.0 g (determined to be optimum in preliminary experiments). During this period, the KHS in the tissue bath was replaced every 20 min. After equilibration, each aortic strip was contracted with 10−7 M noradrenaline (NA). The relaxation response to ACh was expressed as a percentage of the contractile force induced by 10−7 NA. For the relaxation studies, the aortic strips, which were weighed at the end of each experiment, were precontracted with an equieffective concentration of NA (5×10−8∼3×10−7 M). This concentration produced 75–85% of the maximal response, each strip developing a tension of approximately 95 mg mg−1 tissue whether it was from an age-matched control or a diabetic rat. When the NA-induced contraction reached a plateau level, ACh (10−9∼10−5 M), sodium nitroprusside (SNP) (10−9∼10−5 M) or A23187 (10−8∼10−5 M) was added in a cumulative manner. When the effects of each of the LDLs and superoxide dismutase (SOD) on the response to ACh, SNP, or A23187 were to be examined in control aortae, the LDL or SOD was added to the bath either 30 min or 6 h before the administration of NA.
Measurement of the expression of the mRNA for endothelial nitric oxide synthase
Oligonucleotides
Rat endothelial nitric oxide synthase oligonucleotides (ON) were used with primers, as described previously (Seki et al., 1996). The respective Gen Bank data library accession numbers and the amplification of the PCR product in the coding sequence are given in brackets: rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (X02231, position 492–799, amplification of a 308 bp) ON 1 : 5′-TCCCTCAAGATTGTCAGCAA-3′, ON 2 : 5′-AGATCCACAACGGATACATT-3′: rat endothelial nitric oxide synthase (eNOS)(RNU02534, the amplification of a 693 bp) ON 3 : 5′-TCCAGTAACACAGACAGTGCA-3′, ON 4 : 5′-CAGGAAGTAAGTGAGAGC-3′.
RNA isolation and RT–PCR
RNA was isolated by the guanidinium method (Chomczynski & Sacchi, 1987). Rat aortae were carefully isolated and cleaned of adhering parenchyma and connective tissue. The aortae were homogenized in RNA buffer, and the RNA was quantified by ultraviolet absorbance spectrophotometry. For the RT–PCR analysis, first-strand cDNA was synthesized from total RNA using Oligo (dT)12–18 and a cDNA Synthesis Kit (Life Sciences). Twenty or twenty-eight PCR cycles (94°C for 1 min, 62°C for 1 min, 72°C for 1 min) were performed with half of the reverse transcription (RT) mixture. The obtained PCR products were analysed on ethidium bromide-stained agarose (1.5%) gels. The eNOS and GAPDH products were quantified by scanning densitometry. GAPDH levels were determined to control for rates of transcription of RNA and amplification of cDNA. All eNOS data (eNOS density/GAPDH density) were normalized with respect to GAPDH, which was measured using the same technique and samples, except that only 20 cycles of amplification were used, this number being in the linear portion of the amplification curve for GAPDH.
Drugs
Streptozotocin, (−)norepinephrine hydrochloride, superoxide dismutase (bovine erythrocytes) and sodium nitroprusside were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.), and acetylcholine chloride from Daiichi Pharmaceuticals (Tokyo, Japan). Pravastatin was purchased from Sankyo Pharmaceuticals (Tokyo, Japan). All drugs were dissolved in saline, except where otherwise noted.
Statistical analysis
The contractile force developed by aortic strips from control and diabetic rats is expressed in mg tension mg−1 tissue. Data are expressed as the mean±s.e.mean. In some experiments, statistical differences were determined by Dunnett's test for multiple comparisons after a one-way analysis of variance, a probability level of P<0.05 being regarded as significant. Statistical comparisons between concentration-response curves were determined by a two-way ANOVA with Bonferroni's correction performed post-hoc to correct for multiple comparisons. A two-tailed value of P<0.05 was considered significant.
Results
Plasma glucose, cholesterol, and triglyceride levels
As indicated in Table 1, plasma glucose levels were significantly elevated in STZ-induced diabetics by comparison with controls. This raised level was not affected by the chronic administration of pravastatin. Plasma total cholesterol, triglyceride, HDL, and LDL cholesterol levels were all significantly raised in STZ-induced diabetic rats, and while pravastatin treatment did not significantly change the raised total cholesterol, triglyceride, or LDL levels, it did reduce the HDL cholesterol levels. The doses of pravastatin used in our experiments were such that plasma cholesterol and LDL levels in pravastatin-treated diabetic rats were similar to those seen in diabetic rats. The various parameters was not different between controls and pravastatin-treated control of rats.
Table 1.
Levels of various plasma parameters in age-matched controls, STZ-diabeyic rats, and pravastatin-treated rats

Relaxation responses to ACh or SNP in aortae from age-matched controls, STZ-induced diabetic, and pravastatin-treated diabetic rats
The relaxation caused by ACh was significantly weaker in strips from STZ-induced diabetic rats (Figure 1A). Aortic strips from STZ-induced diabetic rats chronically treated with pravastatin relaxed in a normal way to ACh (Figure 1A). The relaxation caused by SNP (10−9∼10−5 M) did not differ significantly among the three groups (Figure 1B).
Figure 1.
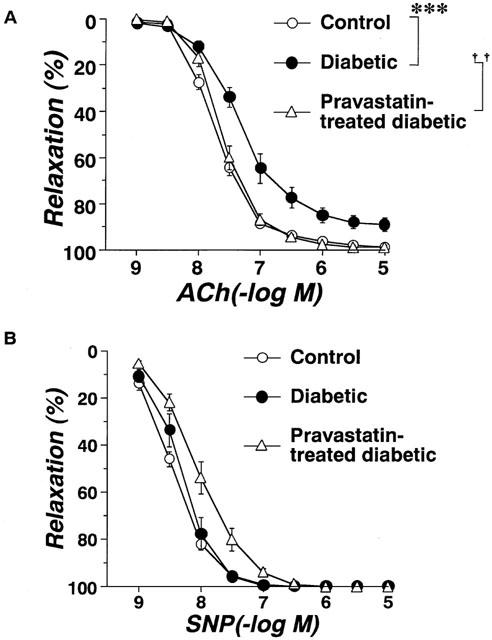
Concentration-response curves for ACh(A)- or SNP(B)-induced relaxation of aortic strips obtained from age-matched controls, untreated diabetic rats, and pravastatin-treated diabetic rats. Ordinate shows relaxation of aortic strips as a percentage of the contraction induced by an equieffective concentration of norepinephrine (5×10−8∼3×10−7 M). Each data point represents mean±s.e.mean of 6–8 experiments; the s.e. is included only when it exceeds the dimension of the symbol used. ***P<0.01, diabetic vs control; ††P<0.01, diabetic vs pravastatin-treated diabetic.
Expression of the mRNA for endothelial nitric oxide synthase
Using RT–PCR on the total RNA isolated from the aortae of age-matched controls, untreated diabetic, and chronic pravastatin-treated diabetic rats, we found the following: (i) the expression of GAPDH mRNA did not differ among the three groups; and (ii) the expression of the mRNA for eNOS did not differ significantly among the aortae from the three groups (Figure 2).
Figure 2.
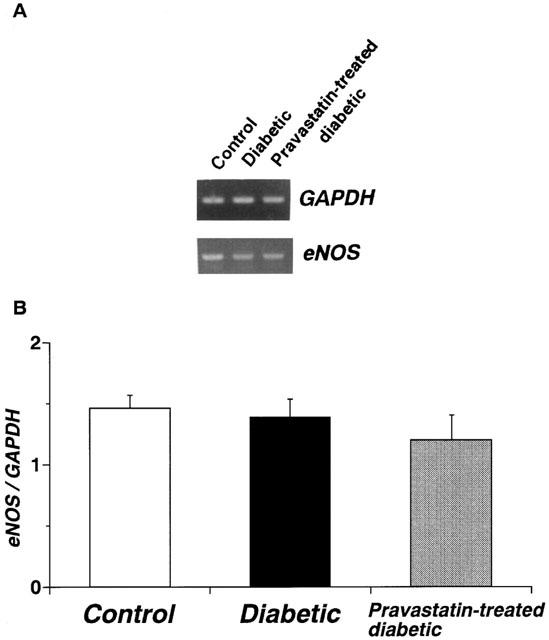
RT–PCR assay of the expression of mRNA for endothelial nitric oxide synthase (eNOS) in control, diabetic, and pravastatin-treated diabetic rat aortae. (A) Expression of mRNA for eNOS as assayed by RT–PCR. (B) Quantitative analysis of expression of mRNA for eNOS by scanning densitometry. Control rats (n=6, open column); STZ-induced diabetic rats (n=6, closed column); pravastatin-treated diabetic rats (n=6, stippled column). Values are mean±s.e.mean of six determinations (eNOS/GAPDH ratios). The RT–PCR assay was performed as described in Methods. Each total RNA preparation (1.0 mg) was reverse-transcribed, and half of the cDNA product was PCR-amplified, using various primers, for 20 or 28 cycles. A portion of the PCR reaction product was electrophoresed on a 1.5% agarose gel containing ethidium bromide.
Measurement of MDA levels
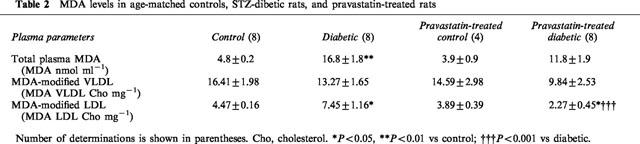
The oxidation state of rat total plasma and VLDL and LDL samples was analysed after the incubation period by means of the TBARS assay, which is used to measure MDA equivalents. As indicated in Table 2, the MDA content of the plasma was significantly higher in the diabetic rats, and it was not changed by chronic treatment with pravastatin. The MDA content of the VLDL fraction of the plasma was not significantly different among the three groups. In contrast, the MDA content of the LDL fraction of the plasma was significantly higher in the diabetic rats, and it was restored to the control level by the chronic administration of pravastatin (Table 2). The oxidation state of rat total plasma and VLDL and LDL samples were not different between controls and pravastatin-treated control of rats.
Table 2.
MDA levels in age-matched controls, STZ-dibetic rats, and pravastatin-treated rats
The oxidative susceptibility of LDL subfractions in vitro experiments
In in vitro experiments, the resistance of LDL to oxidation was assessed by measuring the amount of conjugated dienes or TBARS generated by incubation with copper ions. Three parameters were derived from the kinetics of conjugated diene formation. The lag time was significantly decreased in LDL isolated from diabetic rats relative to control and it was increased by chronic administration of pravastatin (10 mg kg−1, p.o., daily for 4 weeks). However, following incubation in vitro with pravastatin (1.0 μg ml−1), LDL oxidation levels were not different from the levels seen in non-pravastatin-treated STZ-induced diabetic rats (Figure 3A). Neither propagation rates nor maximum conjugated levels differed among the various groups (Figure 3A). The MDA levels of LDL isolated from diabetic rats were higher than those of LDL isolated from control rats at 4 or 24 h of incubation. Chronic administration of pravastatin (10 mg kg−1, p.o., daily for 4 weeks) inhibited these increases in the MDA levels at 4 h of incubation. However, following incubation in vitro with pravastatin (1 μg ml−1–0.1 mg ml−1), LDL oxidation levels were not different from the levels seen in non-pravastatin-treated STZ-induced diabetic rats (Figure 3B). In marked contrast, incubation in vitro with vitamin E (0.1 mg ml−1) inhibited the LDL oxidation (Figure 3B).
Figure 3.
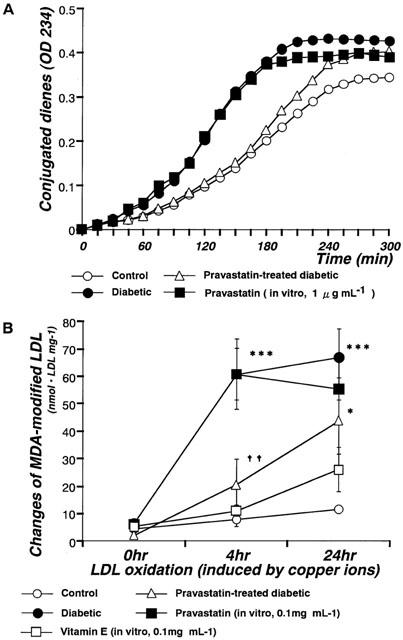
Changes in conjugated dienes (A) or MDA-modified LDL (B) induced by copper ions in control, diabetic, and pravastatin-treated diabetic rats, and by incubation with pravastatin in vitro. (A) Formation of conjugated dienes analysed by continuous monitoring of change of absorbance at 234 nM in samples containing 25 μg protein ml−1. (B) To measure MDA levels, LDL were incubated to a final concentration of 0.2 mg protein ml−1 with 10 μM CuSO4 at 37°C for 0, 4, or 24 h. Each data point represents mean±s.e.mean of 4–8 experiments; the s.e. is included only when it exceeds the dimension of the symbol used. *P<0.05, ***P<0.001, diabetic vs control; ††P<0.01, diabetic vs pravastatin-treated diabetic.
Chemical composition of LDL subfractions
Changes in LDL composition were observed among the groups (Table 3). The phospholipid (mg protein mg−1) content of the LDL fraction of the plasma was significantly higher in the diabetic rats, and it was not changed by the chronic administration of pravastatin. The triglyceride content of the LDL fraction was similar in all groups. The total cholesterol content of the LDL fraction was lower in the diabetic rats, and it was not changed by chronic treatment with pravastatin. However, the free cholesterol/cholesterol ester ratio, which was significantly raised in the diabetics, was significantly reduced by pravastatin, although it was still higher than in the controls.
Table 3.
LDL composition in age-matched controls, STZ-diabetic rats, and pravastatin-treated STZ-diabetic rats
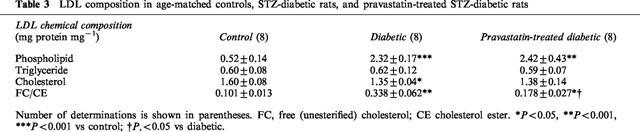
Effects of control-LDL, diabetic-LDL, and SOD on ACh-induced vasorelaxation
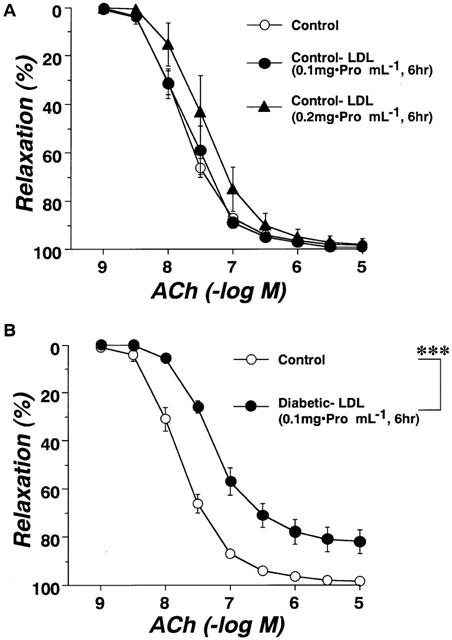
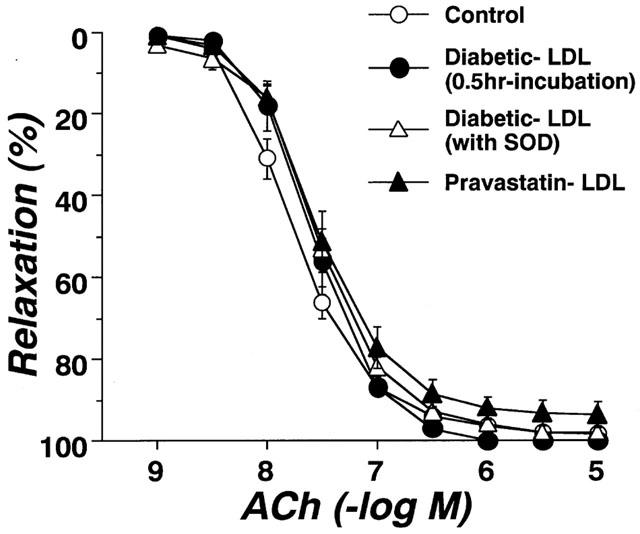
Circulating LDL isolated from control, diabetic, or pravastatin-treated diabetic rats did not affect the resting tonus of rat aortic strips. Following incubation of aortae from control rats for 6 h with LDL (0.1 mg protein ml−1) isolated from control rats, the endothelium-dependent relaxation to ACh was unchanged. Even an increased increasing LDL concentration of 0.2 mg protein ml−1 failed to produce any effect (Figure 4A). Following incubation of aortae from control rats for 6 h with LDL (0.1 mg protein ml−1) isolated from diabetic rats, the endothelium-dependent relaxation showed an impaired response compared to control (Figure 4B). However, LDL (0.1 mg protein ml−1) isolated from rats chronically treated with pravastatin had no such effect (Figure 5). The inhibitory effects of LDL (0.1 mg protein ml−1) isolated from diabetic rats were prevented by SOD (180 ml−1), a scavenger of superoxide anions (Figure 5). Incubation for only 0.5 h with LDL from diabetic rats had no effect on the ACh-induced relaxation of control aortic strips (Figure 5). Following incubation of control aortic strips with LDL from diabetic rats, the A23187-induced relaxation was inhibited (Figure 6A). In contrast, prior incubation with LDL from diabetic rats had no significant effect on SNP-induced relaxation in such strips (Figure 6B).
Figure 4.
Effects of control-LDL (A; 0.1 mg or 0.2 mg protein ml−1) or diabetic-LDL (B; 0.1 mg protein ml−1) on concentration-response curve for acetylcholine-induced relaxation of aortic strips from control rats. Aortic strips were incubated with an LDL preparation for 6 h. The LDL were taken from age-matched controls or untreated diabetic rats. Each data represents mean±s.e.mean of 4–8 experiments; the s.e. is included only when it exceeds the dimension of the symbol used. ***P<0.001, control vs diabetic LDL.
Figure 5.
Effects of incubation for 0.5 h with diabetic-LDL, or for 6 h with either diabetic-LDL plus SOD or pravastatin-LDL on concentration-response curves for acetylcholine-induced relaxation in aortic strips from control rats. The LDL were taken from untreated diabetic or pravastatin-treated diabetic rats. Each data point represents mean±s.e.mean of six experiments; the s.e. is included only when it exceeds the dimension of the symbol used.
Figure 6.
Effects of diabetic-LDL (0.1 mg protein ml−1) on concentration-response curves for the A23187 (A) and SNP (B) induced relaxation of aortic strips from control rats. Aortic strips were incubated with LDL preparation for 6 h. Each data point represents mean±s.e.mean of four experiments; the s.e. is included only when it exceeds the dimension of the symbol used. **P<0.01, conrol vs diabetic LDL.
Discussion
We found in the present study that chronic administration of pravastatin improves endothelial dysfunction in the diabetic aorta without alterating the expression of eNOS mRNA, suggesting that the mechanism underlying the altered endothelial function seen in diabetics or in pravastatin-treated diabetics does not dependent on a change in eNOS expression.
It has been reported that acute administration of L-arginine or tetrahydrobiopterin (BH4), as a co-factor for NO, normalizes the defective cyclic GMP production and relaxation to ACh seen in diabetes (Pieper, 1998). Recently, it has been suggested that depletion of BH4 reduces NO release by stimulating eNOS-dependent O2− generation in the endothelium (Cosentino et al., 1998). Thus, the impaired vasorelaxation seen in the diabetic state may be, at least in part, due to the formation of O2− from NO synthase.
Elevations in the plasma levels of oxidized LDL and MDA-modified LDL are thought to be important steps both in the alteration of a variety of endothelial functions and in the initiation of atherosclerosis (Tamal et al., 1997; Holvoet et al., 1998). In fact, some studies have shown that ox-LDL impairs endothelium-dependent relaxation by inhibiting the synthesis of NO and by enhancing its inactivation (Kugiyama et al., 1990; Simon et al., 1990). In the present study, the plasma levels of MDA-modified LDL and total MDA were higher in diabetic rats, but MDA-modified VLDL was not affected. Furthermore, both the raised MDA-modified LDL and the impaired endothelial function were normalized by the chronic administration of pravastatin. The above results suggest that the endothelial dysfunction seen in diabetes may be due to an accumulation of MDA-modified LDL in the plasma.
Although HDL are known to exert an antiatherogenic effect by promoting reverse cholesterol transport, some studies have suggested that when HDL are oxidized, the presence of oxidized HDL may be related to coronary artery spasm and impaired endothelium-dependent vasoreactivity (Chin et al., 1992; Ohmura et al., 1999). Furthermore, in insulin-dependent diabetics, a lower antioxidant content in HDL has been reported (Maxwell et al., 1997). In the present study, chronic administration of pravastatin reduced HDL levels in the plasma and prevented the impairment of endothelial function otherwise seen in diabetics. Although it seems reasonable to speculate, on the basis of the above data, that diabetic HDL may have a reduced antiatherogenic effect and may inhibit endothelial function, further investigation is clearly required on this point.
Oxidative modification of LDL is thought to be a key early event in atherogenesis, and thus intervention to reduce LDL oxidation is considered to be antiatherogenic. In our study, we demonstrated that LDL isolated from diabetic rats had an enhanced susceptibility to oxidation (compared to LDL from control rats). The mechanism by which diabetic LDL acquire this increased susceptibility is completely unknown. Previous studies suggest that chemical modification of LDL (affecting, for example, glycation (Bowie et al., 1993; Galle et al., 1998), antioxidant content (Esterbauer et al., 1990), lipid and fatty acid composition (Thomas et al., 1994), and particle size of LDL (de Graaf et al., 1991)) can increase their susceptibility to oxidation. In our study, phospholipid/protein and free cholesterol/esterified cholesterol ratios were significantly higher for diabetic LDL than for the controls. Previously, the same modification of composition has been shown in lesioned LDL isolated from the vascular tissue of atherosclerotic mice and humans, together with an enhanced susceptibility to oxidation (Aviram et al., 1995). Thus, an increase in modified-LDL in the plasma may underlie the enhanced susceptibility to oxidation seen in diabetes. In diabetes or hypercholesterolemia, the attenuation of endothelial-dependent relaxation has been shown to be related to high levels of LDL; however, endothelial dysfunction can occur even under normocholesterolemic conditions with normal LDL levels (Cohen, 1993). In this study, we have directly shown that incubation of control aortic strips with circulating LDL (0.1 mg protein ml−1, 6 h) isolated from diabetics can impair the endothelium-dependent relaxation more powerfully than incubation with LDL from control rats. Oxidized LDL are more potent than native LDL at inhibiting endothelium-dependent relaxation, and we found that LDL isolated from diabetic rats had an enhanced susceptibility to oxidation. Thus, a possible mechanism for the attenuation of endothelium-dependent relaxation seen in diabetes may involve oxidation of LDL by the prolonged formation of superoxide anions, since use of a shorter incubation period (0.5 h rather than 6 h) or the presence of SOD prevented the induction of endothelial dysfunction by diabetic-LDL. At present, however, it is unclear how LDL are oxidized on the cells of the arterial wall. A recent study on isolated arteries indicated that the induction of endothelial dysfunction by native-LDL was time-dependent, since the inhibitory effect was observed only after a long period of incubation (Abebe & Mustafa, 1997; Hein & Kuo, 1998). Thus, it seems likely that a time-dependent oxidation of LDL is involved in the initiation of vascular impairment in diabetes. In several models of LDL oxidation, SOD has been shown to be protective, suggesting that the superoxide anion might be involved in the LDL oxidation process (Steinbrecher, 1988; Kawamura et al., 1994; Fang et al., 1998). On the other hand, modification of LDL by endothelial cells has been shown to involve lipid peroxidation in LDL phospholipids (Steinbrecher et al., 1984). Oxidation of LDL also results in the extensive conversion of phosphatidylcholine (PC) to lysophosphatidylcholine (LPC) in phospholipids. Several earlier studies confirmed that the endothelium-dependent relaxation of aortic strips induced by ACh could be attenuated by pretreatment with LPC (Kugiyama et al., 1992). In our study, the phospholipid/protein ratio in diabetic LDL was 4 fold higher than in the controls. On this basis, the release of LPC from diabetic LDL would be expected to play an important role in altering endothelial function. In fact, Galle et al. (1998) have suggested that oxidized glycated-LDL, (glycated by high glucose and oxidized by copper ions), impair endothelial function more powerfully than oxidized LDL, and that the former occur as a result of an increased LPC ratio. To clarify the potential antioxidant role of pravastatin, we examined the changes in LDL composition. As shown in Table 3, the free cholesterol cholesterol ester ratio, which was significantly raised in the diabetics, was significantly reduced by pravastatin, although it was still higher than in the controls. Although this decomposition of LDL by chronic pravastatin may be responsible for the antioxidant role of pravastatin, further investigation is required on this point.
In the present study, a prior incubation with LDL from diabetic rats inhibited the A23187-induced relaxation of control aortic strips, indicating that LDL from diabetic rats produce their inhibitory effect on vascular function at a point(s) beyond the receptor level. Circulating LDL isolated from diabetic rats did not affect the resting tonus of rat aortic strips, suggesting that the diabetic LDL may only affect the stimulated NO pathway.
In conclusion, the present results provide direct evidence that circulating modified-LDL isolated from diabetic rats can cause a dysfunction of the aortic endothelium. A likely mechanism for the attenuation of the endothelial-dependent relaxation seen in the diabetic aorta involves increased LDL oxidation and altered LDL composition. Furthermore, we suggest that pravastatin may help prevent endothelial dysfunction in large arteries by a mechanism involving not only its hypocholesterolemic effect, but also its inhibitory effect on the increased LDL oxidation that occurs in diabetes. At present, we cannot say whether similar processes affect resistance arteries.
Acknowledgments
This study was supported in part by the Ministry of Education, Science, Sports and Culture, Japan.
Abbreviations
- ACh
acetylcholine
- eNOS
endothelial nitric oxide synthase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HDL
high-density lipoprotein
- HMG-CoA
3-hydroxy-3-methylglutary coenzyme A
- KHS
Krebs-Henseleit solution
- LDL
low-density lipoprotein
- LPC
lysophosphatidylcholine
- MDA
malondialdehyde
- NA
noradrenaline
- ON
oligonucleotides
- ox-LDL
oxidized LDL
- PBS
phosphate-buffered saline
- PC
phosphatidylcholine
- SNP
sodium nitroprusside
- SOD
superoxide dismutase
- STZ
streptozotocin
- TBARS
thiobarbituric acid-reactive substances
- VLDL
very low-density lipoprotein
References
- ABEBE W., MUSTAFA S.J. Effect of low density lipoprotein on adenosine receptor-mediated coronary vasorelaxation in vitro. J. Pharmacol. Exp. Ther. 1997;282:851–857. [PubMed] [Google Scholar]
- AVIRAM M., MAOR I., KEIDAR S., HAYEK T., OIKNINE J., BAR-EL Y., ADLER Z., KERTZMAN V., MILO S. Lesioned low density lipoprotein in atherosclerotic apolipoprotein E-deficient transgenic mice and in humans is oxidized and aggregated. Biochem. Biophys. Res. Commun. 1995;216:501–513. doi: 10.1006/bbrc.1995.2651. [DOI] [PubMed] [Google Scholar]
- BOWIE A., OWENS D., COLLINS P., JOHNSON A., TOMKIN G. Glycosylated LDL is more sensitive to oxidation: implications for the diabetic patient. Atherosclerosis. 1993;102:63–67. doi: 10.1016/0021-9150(93)90084-8. [DOI] [PubMed] [Google Scholar]
- CHIN J.H., AZHAR S., HOFFMAN B.B. Inactivation of endothelial derived relaxing factor by oxidized lipoproteins. J. Clin. Invest. 1992;89:10–18. doi: 10.1172/JCI115549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- COHEN R.A. Dysfunction of vascular endothelium in diabetes mellitus. Circulation. 1993;87 suppl 5:V67. [Google Scholar]
- COSENTINO F., PATTON S., D'USCIO L.V., WERNER E.R., WERNER-FELMAYER G., MOREAU P., MALINSKI T., LÜSCHER T.F. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J. Clin. Invest. 1998;101:1530–1537. doi: 10.1172/JCI650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE GRAAF J., HAK-LEMMERS H.L.M., HECTORS M.P.C., DEMACKER P.N.M., HENDRIKS J.C.M., STALENHOEF A.F.H. Enhanced susceptibility to in vitro oxidation of the dense low density lipoprotein subfraction in healthy subjects. Arterioscler. Thromb. 1991;11:298–306. doi: 10.1161/01.atv.11.2.298. [DOI] [PubMed] [Google Scholar]
- ESSIG M., NGUYEN G., PRIÉ D., ESCOUBET B., SRAER J.D., FRIEDLANDER G. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors increase fibrinolytic activity in rat aortic endothelial cells: Role of geranylgeranylation and Rho proteins. Circ. Res. 1998;83:683–690. doi: 10.1161/01.res.83.7.683. [DOI] [PubMed] [Google Scholar]
- ESTERBAUER H., DIEBER-ROTHENEDER M., WAEG G., STRIEGL G., JÜRGENS G. Biochemical, structural, and functional properties of oxidized low-density lipoprotein. Chem. Res. Toxicol. 1990;3:77–92. doi: 10.1021/tx00014a001. [DOI] [PubMed] [Google Scholar]
- FANG X., WEINTRAUB N.L., RIOS C.D., CHAPPELL D.A., ZWACKA R.M., ENGELHARDT J.F., OBERLEY L.W., YAN T., HEISTAD D.D., SPECTOR A.A. Overexpression of human superoxide dismutase inhibits oxidation of low-density lipoprotein by endothelial cells. Circ. Res. 1998;82:1289–1297. doi: 10.1161/01.res.82.12.1289. [DOI] [PubMed] [Google Scholar]
- GALLE J., SCHNEIDER R., WINNER B., LEHMANN-BODEN C., SCHINZEL R., MÜNCH G., CONZELMANN E., WANNER C. Glyc-oxidized LDL impair endothelial function more potently than oxidized LDL: role of enhanced oxidative stress. Atherosclerosis. 1998;138:65–77. doi: 10.1016/s0021-9150(98)00005-7. [DOI] [PubMed] [Google Scholar]
- HAVEL R.J., EDER H.A., BRAGSON J.H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in humans serum. J. Clin. Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEIN T.W., KUO L. LDLs impair vasomotor function of the coronary microcirculation: Role of superoxide anions. Circ. Res. 1998;83:404–414. doi: 10.1161/01.res.83.4.404. [DOI] [PubMed] [Google Scholar]
- HOLVOET P., VANHAECKE J., JANSSENS S., DE WERF F.V., COLLEN D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487–1494. doi: 10.1161/01.cir.98.15.1487. [DOI] [PubMed] [Google Scholar]
- HUSSEIN O., SCHLEZINGER S., ROSENBLAT M., KEIDAR S., AVIRAM M. Reduced susceptibility of low density lipoprotein (LDL) to lipid peroxidation after fluvastatin therapy is associated with the hypocholesterolemic effect of the drug and its binding to the LDL. Atherosclerosis. 1997;128:11–18. doi: 10.1016/s0021-9150(96)05972-2. [DOI] [PubMed] [Google Scholar]
- JOHN S., SCHLAICH M., LANGENFELD M., WEIHPRECHT H., SCHMITZ G., WEIDINGER G., SCHMIEDER R.E. Increased bioavailability of nitric oxide after lipid-lowering therapy in hypercholesterolemic patients: A randomized, placebo-controlled, double-blind study. Circulation. 1998;98:211–216. doi: 10.1161/01.cir.98.3.211. [DOI] [PubMed] [Google Scholar]
- KAMATA K., KOBAYASHI T. Changes in superoxide dismutase mRNA expression by streptozotocin-induced diabetes. Br. J. Pharmacol. 1996;119:583–589. doi: 10.1111/j.1476-5381.1996.tb15712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAMATA K., MIYATA N., KASUYA Y. Impairment of endothelium-dependent relaxation and changes in levels of cyclic GMP in aorta from streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1989;97:614–618. doi: 10.1111/j.1476-5381.1989.tb11993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAMATA K., SUGIURA M., KOJIMA S., KASUYA Y. Preservation of endothelium-dependent relaxation in cholesterol-fed and streptozotocin-induced diabetic mice by the chronic administration of cholestyramine. Br. J. Pharmacol. 1996;118:385–391. doi: 10.1111/j.1476-5381.1996.tb15414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWAMURA M., HEINECKE J.W., CHAIT A. Pathophysiological concentrations of glucose promote oxidative modification of low density lipoprotein by a superoxide-dependent pathway. J. Clin. Invest. 1994;94:771–778. doi: 10.1172/JCI117396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KROON A.A., STALENHOEL A.F.H., BUIKEMA H., DEMACKER P.N.M., DE WILDE P.C.M., LEIJTEN P.A., VAN GILST W.H. The effect of cholesterol reduction on the endothelial function and progression of atherosclerosis in WHHL rabbits. Atherosclerosis. 1993;103:221–230. doi: 10.1016/0021-9150(93)90265-v. [DOI] [PubMed] [Google Scholar]
- KUGIYAMA K., KERNS S.A., MORRISETT J.D., ROBERTS R., HENRY P.D. Impairment of endothelium-dependent arterial relaxation by lysolethicin in modified low-density lipoprotein. Nature. 1990;344:160–162. doi: 10.1038/344160a0. [DOI] [PubMed] [Google Scholar]
- KUGIYAMA K., OHGUSHI M., SUGIYAMA S., MUROHARA T., FUKUNAGA K., MIYAMOTO E., YASUE H. Lysophosphatidylcholine inhibits surface receptor-mediated intracellular signals in endothelial cells by a pathway involving protein kinase C activation. Circ. Res. 1992;71:1422–1428. doi: 10.1161/01.res.71.6.1422. [DOI] [PubMed] [Google Scholar]
- LAUFS U., FATA V.L., PLUTZKY J., LIAO J.K. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- MAXWELL S., HOLM G., BONDJERS G., WIKLUND O. Comparison of antioxidant activity in lipoprotein fractions from insulin-dependent diabetics and healthy controls. Atherosclerosis. 1997;129:89–96. doi: 10.1016/s0021-9150(96)06033-9. [DOI] [PubMed] [Google Scholar]
- O'DRISCOLL G., GREEN D., TAYLOR R.R. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation. 1997;95:1126–1131. doi: 10.1161/01.cir.95.5.1126. [DOI] [PubMed] [Google Scholar]
- OHMURA H., WATANABE Y., HATSUMI C., SATO H., DAIDA H., MOKUNO H., YAMAGUCHI H. Possible role of high susceptibility of high-density lipoprotein to lipid peroxidative modification and oxidized high-density lipoprotein in genesis of coronary artery spasm. Atherosclerosis. 1999;142:179–184. doi: 10.1016/s0021-9150(98)00235-4. [DOI] [PubMed] [Google Scholar]
- OYAMA Y., KAWASAKI H., HATTORI Y., KANNO M. Attenuation of endothelium-dependent relaxation in aorta from diabetic rats. Eur. J. Pharmacol. 1986;132:75–78. doi: 10.1016/0014-2999(86)90013-0. [DOI] [PubMed] [Google Scholar]
- PIEPER G.M. Review of alterations in endothelial nitric oxide production in diabetes: protective role of arginine on endothelial dysfunction. Hypertension. 1998;31:1047–1060. doi: 10.1161/01.hyp.31.5.1047. [DOI] [PubMed] [Google Scholar]
- POSTON L., TAYLOR P.D. Endothelium-mediated vascular function in insulin-dependent diabetes mellitus. Clin. Sci. 1995;88:245–255. doi: 10.1042/cs0880245. [DOI] [PubMed] [Google Scholar]
- SACKS F.M., PFEFFER M.A., MOYE L.A., ROULEAU J.L., RUTHERFORD J.D., COLE T.G., BROWN L, , WARNICA J.W., ARNOLD J.M., WUN C.C., DAVIS B.R., BRAUNWALD E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators. N. Engl. J. Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- SEKI T., HAGIWARA H., NARUSE K., KADOWAKI M., KASHIWAGI M., DEMURA H., HIROSE S., NARUSE M. In site identification of messenger RNA of endothelial type nitric oxide synthase in rat cardiac myocytes. Biochem. Biophys. Res. Commun. 1996;218:601–605. doi: 10.1006/bbrc.1996.0106. [DOI] [PubMed] [Google Scholar]
- SIMON B.C., CUNNINGHAM L.D., COHEN R.A. Oxidized low density lipoproteins cause contraction and inhibit endothelium-dependent relaxation in the pig coronary artery. J. Clin. Invest. 1990;86:75–79. doi: 10.1172/JCI114718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEINBRECHER U.P. Role of superoxide in endothelial-cell modification of low-density lipoproteins. Biochim. Biophys. Acta. 1988;959:20–30. doi: 10.1016/0005-2760(88)90145-2. [DOI] [PubMed] [Google Scholar]
- STEINBRECHER U.P., PARTHASARATHY S., LEAKE D.S., WITZTUM J.L., STEINBERG D. Modification of low density lipoproteins by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc. Natl. Acad. Sci. U.S.A. 1984;81:3883–3887. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAMAI O., MATSUOKA H., ITABE H., WADA Y., KOHNO K., IMAIZUMI T. Single LDL apheresis improves endothelium-dependent vasodilatation in hypercholesterolemic humans. Circulation. 1997;95:76–82. doi: 10.1161/01.cir.95.1.76. [DOI] [PubMed] [Google Scholar]
- THOMAS M.J., THORNBURG T., MANNING J., HOOPER K., RUDEL L.L. Fatty acid composition of low-density lipoprotein influences its susceptibility to autoxidation. Biochemistry. 1994;33:1828–1834. doi: 10.1021/bi00173a028. [DOI] [PubMed] [Google Scholar]
- TSAI M.Y., YUAN J., HUNNINGHAKE D.B. Effect of gernfibrozil on composition of lipoprotein and distribution of LDL subspecies. Atherosclerosis. 1992;95:35–42. doi: 10.1016/0021-9150(92)90173-e. [DOI] [PubMed] [Google Scholar]
- YAGI K. A simple fluorometric assay for lipoperoxide in blood plasma. Biochem. Med. 1976;15:212–216. doi: 10.1016/0006-2944(76)90049-1. [DOI] [PubMed] [Google Scholar]