

Abstract
We have investigated the pharmacological properties of LY344545, a structurally related epimer of the broad spectrum competitive metabotropic glutamate receptor antagonist, LY341495. We have found that LY344545 also antagonizes competitively nearly all mGlu receptor subtypes, but with a wide spectrum of activity. The order of potency for the human receptor isoforms was mGlu5a (IC50 of 5.5±0.6 μM)>mGlu2=mGlu3>mGlu1α=mGlu7>mGlu6=mGlu8. No significant mGlu4 receptor antagonist activity was detected at the highest concentration used (100 μM). 100 μM LY344545 displaced 50±5% of [3H]-CGP39653 binding, but less than 30% of [3H]-kainate or [3H]-AMPA in radioligand binding assays.
LY344545 antagonized L-glutamate stimulated Ca2+ release in CHO cells transfected with mGlu receptors in a concentration dependent manner with a 10 fold higher affinity for the rat mGlu5a receptor (Ki=2.1±0.6 μM) compared to the rat mGlu1α receptor (Ki=20.5±2.1 μM). 50 μM (1S, 3R)-ACPD-induced Ca2+ rises in hippocampal CA1 neurones were also antagonized (IC50=6.8±0.7 μM).
LY344545 antagonized 10 μM (S)-3,5-DHPG-induced potentiation of NMDA depolarizations in CA1 neurones (EC50=10.6±1.0 μM). At higher concentrations (⩾100 μM), LY344545 was an NMDA receptor antagonist.
LY344545 also blocked the induction, but not the expression, of LTP at CA3 to CA1 synapses with an IC50>300 μM. This effect is consistent with its weak activity at NMDA receptors.
These results demonstrate that the binding of ligands to mGlu receptor subtypes is critically dependent on the spatial orientation of the same molecular substituents within a given chemical pharmacophore. The identification of LY344545 as the first competitive antagonist to show selectivity towards mGlu5 receptors supports the potential to design more selective and potent competitive antagonists of this receptor.
These results further indicate that mGlu receptor-mediated potentiation of NMDA responses is not essential for the induction of LTP.
Keywords: Metabotropic glutamate receptors, LTP, mGluR, excitatory amino acid
Introduction
L-Glutamate, the principal excitatory neurotransmitter in the brain, activates a family of G-protein linked receptors, termed metabotropic glutamate (mGlu) receptors, of which eight subtypes have been cloned (Nakanishi et al., 1994; Conn & Pin, 1997). These receptors are subdivided into three groups (Group I, mGlu1 and mGlu5; Group II, mGlu2 and mGlu3; Group III, mGlu4, mGlu6, mGlu7 and mGlu8) based on sequence homology, pharmacology and transduction mechanisms of recombinant receptors expressed in cell lines. However, identification of roles for these receptors in physiological and pathological processes has, until recently, been hindered by the lack of potent and selective antagonists (Schoepp et al., 1999). As part of a program to develop potent and subtype selective mGlu receptor antagonists we have described the actions of LY341495 (Fitzjohn et al., 1998), which is a highly potent mGlu receptor antagonist (Kingston et al., 1998). This compound can be used in nanomolar concentrations to selectively antagonise group II mGlu receptors (Kingston et al., 1998), but inhibits all mGlu receptor subtypes at high concentrations and can thus be seen as a broad spectrum mGlu receptor antagonist (Fitzjohn et al., 1998). Here we report the activity of a structurally related epimer of LY341495 (Figure 1). We have characterized this compound (LY344545; Ornstein et al., 1998b) using clonal cell lines expressing individual mGlu receptor subtypes as well as rat hippocampal slices. We show that LY344545 has a similar potency to LY341495 at mGlu5 receptors, but for all other mGlu receptors the compound exhibits significantly weaker antagonist activity. Moreover, unlike LY341495, the compound displayed weak NMDA receptor antagonist activity. These data underline the exquisite molecular and stereoselective ligand binding requirements of individual mGlu receptor subtypes.
Figure 1.
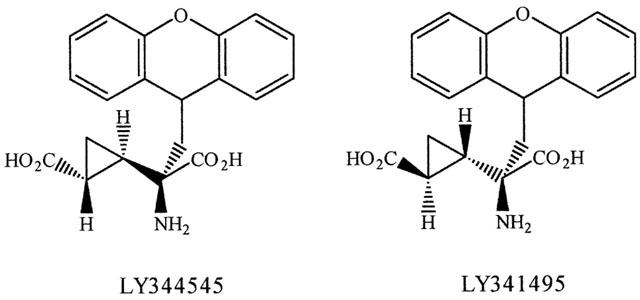
Structure of LY344545 and LY341495.
Methods
Materials
L-Quisqualate, (1S,3R)-aminocyclopentanedicarboxylic acid (ACPD), (S)-3,5-dihydroxyphenylglycine ((S)-DHPG) and L-2-amino-4-phosphobutanoate (L-AP4) were obtained from Tocris Cookson (U.K. and U.S.A.). [3H]-myo-inositol was purchased from Amersham International (U.K.) specific activity 10–20 Ci mmol−1. LY344545 was synthesized as described by Ornstein et al, (1998b). [3H]-CGP 39653 ([3H]-D,L-(E)-2-amino-4-propyl-5-phosphono-3-pentenoic acid), [3H]-AMPA ([3H]-α-amino-3-hydroxy-5-methyl-4-isoxazole propionate) and [3H]-kainate were obtained from NEN Dupont. Cell culture materials: Dulbecco's modified Eagle's medium (DMEM), antibiotics, glutamine, pyruvate, HEPES buffer, geneticin G418 were from Life Technologies; hygromycin B was obtained from Boehringer Mannheim (Lewes, U.K.); heat inactivated dialysed foetal calf serum from Life Technologies (Paisley, U.K., RGT cells) or Sigma (Poole, U.K., CHO cells) and cell culture plates from Becton Dickinson.
Cell culture
RGT cell lines stably expressing human mGlu1α, mGlu2, mGlu3, mGlu4, mGlu5a, mGlu7 and mGlu8 receptors were derived as previously described (Schoepp et al, 1997). Cells were cultured in DMEM supplemented with 5% foetal bovine serum, (in mM) glutamine 1, sodium pyruvate 1, HEPES 10, 50 μg ml−1 G418 and 0.2 mg ml−1 hygromycin B. CHO cells stably expressing rat mGlu1α and mGlu5a receptors were kindly provided by Professor St Nakanishi. Cells were cultured in DMEM (4.5 gl−1 D-glucose) supplemented with 10% dialysed foetal bovine serum (10,000 Mr cut-off, Sigma), 2 mM glutamine and 1% L-proline. All cells were treated with 1 mg l−1 penicillin and 100 u l−1 streptomycin and were maintained at 37°C in a 5% CO2 humidified atmosphere.
Phosphoinositide hydrolysis
Cells were seeded into 24-well plates at 2.5×105 cells per well in medium containing no added glutamine and cultured overnight. After 24 h, cells were labelled with [3H]-inositol (4 μCi ml−1) for a further 20 h. Cells were then washed in assay medium containing HEPES (10 mM), inositol (10 mM) and LiCl (10 mM). Antagonists were added to the cell cultures 20 min prior to the addition of quisqualate and then further incubated in the presence of agonist for 60 min. The reaction was terminated by replacing the medium with acetone : methanol (1 : 1) and the cultures incubated on ice for a further 20 min. Separation of the [3H]-inositol phosphates was carried out as previously described (Kingston et al, 1995).
Cyclic AMP
Cyclic AMP assays were carried out for cells expressing mGlu2, mGlu3, mGlu4, mGlu7 and mGlu8 receptors. Cells were washed in Dulbecco's phosphate buffered saline (PBS) plus 3 mM glucose and 500 μM isobutyl-methylxanthine (IBMX), and pre-incubated for 30 min at 37°C. Each well was then washed and antagonist was added for 15 min, followed by agonist and forskolin (15 μM final concentration, 0.5 ml final volume per well). Cells were incubated for 20 min at 37°C and then terminated by adding 6 mM EDTA solution (0.75 ml) to each well and placing the plate in a boiling water bath. An Amersham [3H]-cyclic AMP SPA kit was used to determine concentrations of cyclic AMP. Protein content in each well was determined using the modified Bradford-Pierce assay (Pierce Chemicals, U.S.A.).
iGlu binding assays
Measurement of radioligand binding to ionotropic glutamate receptors was carried out using binding of [3H]-AMPA ((±)-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate, 5 nM) with washed crude membranes derived from male Sprague Dawley rats (150–175 g), and [3H]-kainate (5 nM) or [3H]-CGP 39653 (10 nM) binding with synaptosomal membranes of rat forebrain (Kingston et al., 1998). Data were analysed with non-linear regression analysis of displacement data transformed to the Hill equation to give the Ki values for each ligand.
Ca2+ imaging
Ca2+ imaging experiments with cultured cells were performed and the fluorescence intensity of up to 10 individual cells per experiment determined using NIH-Image as previously described (Doherty et al., 1999). Responses were normalized to baseline and the initial peak response taken as a measure of the intracellular Ca2+ concentration. Ca2+ responses in rat hippocampal CA1 neurones were measured in slices (200 μm thick), perfused with Kreb's buffer comprising (in mM) NaCl, 124, KCl, 3, NaHCO3, 26, NaH2PO4, 1.25, CaCl2, 2, MgSO4, 1, D-glucose 10 bubbled with O2/ CO2 95 : 5%. In addition, picrotoxin (50 μM), L-689,560 (5 μM) and NBQX (10 μM) were present throughout the experiments to block GABAA, NMDA and AMPA receptors, respectively. Cells were patched blind and then filled with fluo-3 free acid as previously described (Frenguelli et al., 1993) and held at −60 mV throughout the experiment. Cells were imaged using a Bio-Rad MRC 1024 laser scanning confocal microscope equipped with an argon ion laser and using 488 nm excitation/515 nm bp emission filters. All agonists and antagonists were added by bath perfusion. Agonist was added at 15 min intervals; antagonists were preincubated for 7 min. Mean pixel intensity was measured using Bio-Rad LaserSharp software.
Electrophysiology
Depolarizations induced by NMDA (10–30 μM) in rat hippocampal CA1 neurones were measured by the grease-gap technique as previously described (Fitzjohn et al., 1996; Doherty et al., 1997). Slices were taken from 8–10 week old rats and NMDA was applied in 2 ml aliquots by bath perfusion.
Extracellular microelectrode recordings using two inputs were performed on hippocampal slices as previously described (Fitzjohn et al., 1998). Stimuli to the Schaffer collateral-commissural pathway were delivered alternately to each of two pathways every 15 s (30 s interval for each input). Individual responses were displayed and the average field EPSP slopes of four successive responses were plotted on-line using the LTP program (Anderson & Collingridge, 1997; available from http://www.ltp-program.com). LTP was induced by delivering 100 shocks at 100 Hz at test intensity.
Results
Clonal cell lines
The pharmacological profile of LY344545 for all human mGlu receptor subtypes expressed in the CNS is presented in Table 1. LY344545 was slightly more potent on mGlu5 (IC50 ∼ 5 μM) than mGlu2/3 (IC50 ∼ 10 μM). It had intermediate activity on mGlu1,6,7 and 8(IC50 ∼ 40–65 μM) but had no significant antagonist activity on mGlu4 receptors at the highest concentration used (100 μM). 100 μM LY344545 displaced 50±5% of [3H]-CGP 39653 binding to NMDA receptors and less than 30% radiolabelled ligands for AMPA and kainate receptors in radioligand binding assays (Table 2).
Table 1.
Pharmacology profile of LY344545 on metabotropic glutamate receptor subtypes

Table 2.
Binding of LY344545 to ionotropic glutamate receptors

With respect to its activity at group II mGlu receptors, LY344545 is much less potent than other known compounds, such as LY341495 (Kingston et al., 1998). However, an interesting property of LY344545 is its activity at mGlu5 receptors. In contrast to all known competitive group I mGlu receptor antagonists, this compound shows a degree of selectivity for mGlu5 versus mGlu1 receptors. Therefore we explored further the selectivity of LY344545 activity in cell lines expressing the equivalent rat mGlu receptor clones (Figure 2). LY344545 showed a concentration-dependent antagonism of Ca2+ release evoked by glutamate (100 μM and 10 μM) in CHO cells expressing rat mGlu5 receptors. Consistent with the human receptor data, LY344545 showed less antagonist activity of rat mGlu1 in parallel assays. Approximate Ki values of 2.1±0.6 μM and 20.5±2.1 μM were calculated for the antagonism of L-glutamate by LY344545 using rat mGlu5 and mGlu1 receptor-expressing cells, respectively whereas EC50 values for glutamate evoked Ca2+ release in these two cell lines were similar, (10.4±2.4 μM, n=6 and 7.6±1.6 μM, n=4, respectively) as reported previously (Doherty et al., 1997, 1999). These results confirm that LY344545 shows a differential affinity for mGlu5 receptors over mGlu1 receptors.
Figure 2.
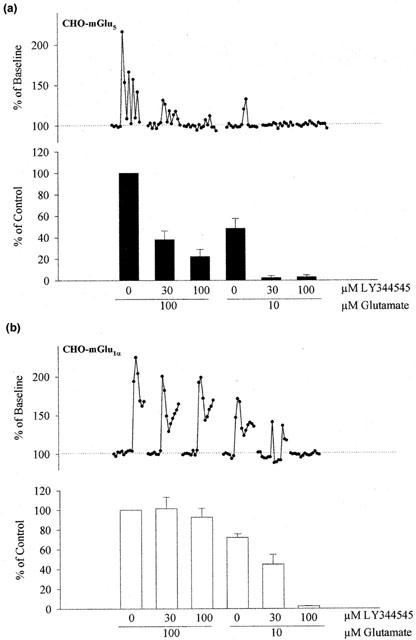
LY344545 is an antagonist of rat mGlu5a receptors. The effects of LY344545 on the release of Ca2+ from internal stores evoked by activation of the rat mGlu5a (a) and mGlu1α (b) receptors expressed in CHO cells are shown. Traces of fluorescence intensity from an individual cell are shown above pooled data under the same conditions from six or four experiments, respectively. Concentrations of both glutamate and LY344545 used are indicated. Ki values for the inhibition of Ca2+ release were calculated to be 2.1±0.6 μM (n=6) for mGlu5a receptors and 20.5±2.1 μM (n=4) for mGlu1α receptors.
Hippocampal slices
The ability of LY344545 to antagonize Ca2+ release evoked by native group I mGlu receptor activity was investigated in rat hippocampal slices. Ca2+ responses in CA1 neurones following (1S,3R)-ACPD (50 μM) application were measured in the absence and presence of LY344545 (Figure 3). A reversible, concentration-dependent reduction in the release of Ca2+ was seen with co-application of LY344545, with an IC50 of 6.8±0.7 μM (n=3), consistent with antagonism of mGlu5 receptors.
Figure 3.
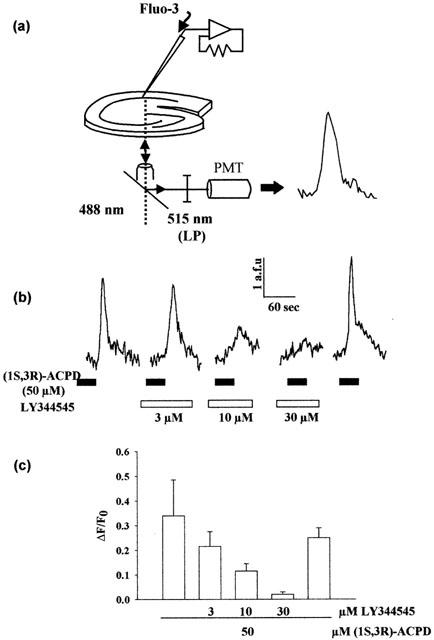
LY344545 blocks (1S,3R)-ACPD evoked Ca2+ release in voltage clamped CA1 hippocampal neurons. Individual hippocampal neurons voltage clamped at −60 mV were filled with the Ca2+ indicator Fluo-3 as described in Methods and imaged by confocal microscopy (a). (b) Traces of fluorescence intensity measured from a single cell. Bath application of (1S,3R)-ACPD caused a transient rise in Ca2+ concentration that was blocked in a dose-dependent and reversible manner. (c) Dose-response profile showing antagonism of (1S,3R)-ACPD induced Ca2+ release by LY344545. The IC50 calculated from this data is 6.8±0.7 μM (n=3).
(S)-DHPG has previously been shown to potentiate NMDA-induced depolarizations in hippocampal CA1 neurones (Fitzjohn et al., 1996). The potentiation of NMDA (10–30 μM) responses by 10 μM DHPG was inhibited by co-application of LY344545 (Figure 4). This effect was concentration-dependent, with an IC50 of ∼10 μM and maximal inhibition occurring at 100 μM. LY344545 also inhibited NMDA-induced depolarizations in these neurones, but with lower affinity than its effects on mGlu5 receptors. Thus 300 μM LY344545 reduced the NMDA-induced depolarization by ∼60% of control (Figure 4). These results demonstrate that this compound not only possesses mGlu5 receptor antagonist activity but, at 30 fold higher concentrations, NMDA receptor antagonism.
Figure 4.
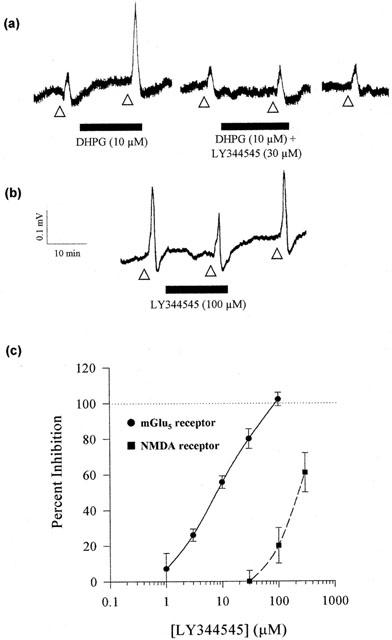
Differential antagonism by LY344545 of 3,5-DHPG induced potentiation of NMDA responses and NMDA responses per se. (a) Gease-gap recordings from the CA1 region of the hippocampus showing inhibition of 3,5-DHPG mediated potentiation of sub-maximal NMDA receptor activation by 30 μM LY344545 (application of NMDA is indicated by the open triangles). (b) Recordings from a single experiment showing that LY344545 inhibits NMDA induced responses in a reversible manner at concentrations above those required to block 3,5-DHPG mediated potentiation. (c) Dose-response profiles for LY344545 antagonism of 3,5-DHPG mediated potentiation of NMDA receptor activation (closed circles, EC50=10.6±1.0 μM) and NMDA induced responses (closed squares). Each point represents data from 3–7 cells.
The effects of LY344545 on the induction of LTP in hippocampal CA1 neurons were also investigated (Figure 5). Application of up to 1 mM LY344545 after the tetanus had no significant effect on the level of pre-established LTP. However, LY344545 prevented the induction of LTP in a concentration–dependent manner. Application of 300 μM LY3445345 prior to tetanus reduced the level of LTP by approximately 40±13 % (n=3) while 1 mM LY344545 completely abolished the induction of LTP (n=4). 100 μM LY344545 had a small but statistically insignificant effect on the induction of LTP (20±15 % inhibition, n=9, P>0.05). The concentration dependency of inhibition of LTP induction mirrors the weak antagonist activity of this compound at NMDA receptors.
Figure 5.
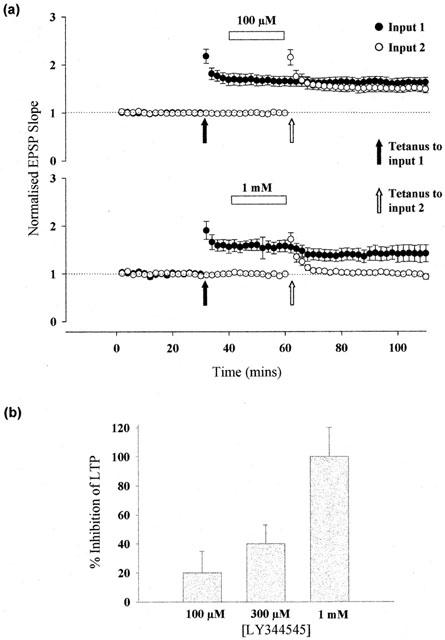
LY344545 blocks the induction of LTP. (a) Pooled data from up to nine separate two-input experiments. In input 1, a tetanus was applied prior to application of the indicated concentration of LY344545, whilst in input 2 a tetanus was applied following 20-min incubation, immediately before washout of antagonist. (b) Histogram showing the dose-dependent percentage inhibition of LTP by LY344545 measured 30 min following the tetanus.
Discussion
A large number of compounds have been described that are antagonists at one or more mGlu receptor subtypes (Schoepp et al., 1999). This study describes the pharmacology of a novel mGlu receptor antagonist, LY344545. The compound is a structurally related epimer of a previously described mGlu receptor antagonist LY341495 (Ornstein et al., 1998a,1998b). LY341495 has a high potency for mGlu2 and mGlu3 receptor subtypes but is active on all other subtypes (Table 1). By altering the stereoisomeric configuration from S,S,S (LY341495) to the S,R,R (LY344545) form, there was a thousand fold loss in potency for group II and III mGlu receptors. There was also a large loss in potency at group III mGlu receptors. However, for group I mGlu receptors, there was only a small loss of activity at mGlu1 receptors (circa 6 fold) and essentially no change in activity at mGlu5 receptors. As a result, LY344545 is weakly selective for mGlu5 receptors. Indeed, it is the first competitive antagonist to show selectivity towards mGlu5 receptors.
These results underline two important points with respect to the development of mGlu receptor antagonists: (1) that ligand binding to mGlu receptor subtypes is critically dependent on the spatial orientation of the same molecular substituents within a given chemical pharmacophore; and (2) that the mode of interaction of the same ligands is not equivalent across the different mGlu subtypes. Recently, potent and selective mGlu5 antagonists have been described (Varney et al., 1999; Gasparini et al., 1999). These compounds are non-competitive in their binding mode and show therapeutic efficacy in models of pain (Walker et al., 1999). The development of both competitive and non-competitive ligands of mGlu receptors will be useful for the treatment of neurological and psychiatric conditions in which glutamatergic transmission contributes to pathology. The pharmacological profile of this type of compound which has both mGlu5 and NMDA receptor antagonist activity could find utility as a neuroprotective or analgesic agent under conditions where both receptors are highly expressed, for example in the CA1 region of the hippocampus, and where their overactivation mediates a neuropathological response.
We have been interested in studying the actions of LY344545 in the CA1 region of the hippocampus, given that mGlu5 receptors are the principal postsynaptically expressed mGlu receptor in this region (Luján et al., 1996). We had suggested previously that the ability of mGlu receptor activation to acutely potentiate NMDA responses was due to activation of mGlu5 receptors on the basis of the activity of CHPG, a weak but selective mGlu5 receptor agonist (Doherty et al., 1997). The potency of LY344545 in antagonising the NMDA potentiating effect of the group I specific agonist DHPG confirms the involvement of mGlu5 receptors in this effect.
It has been suggested that the ability of mGlu receptors to potentiate NMDA responses underlies the involvement of mGlu receptors in the induction of LTP at CA1 synapses (Ben-Ari et al., 1992). If this was the case, then antagonism of mGlu5 receptors should block the induction of LTP. In contrast, we found no significant effect of LY344545 on the induction of LTP at a concentration (100 μM) that eliminated DHPG potentiation of NMDA responses. However, the ability of LY344545 at higher concentrations than 100 μM to inhibit the induction of LTP paralleled its weak activity as an NMDA receptor antagonist. The finding that LTP of AMPA receptor-mediated synaptic transmission is readily obtained following antagonism of mGlu5 receptors confirms the conclusions made using an mGlu5 receptor knockout (Jia et al., 1998) and the broad-spectrum mGlu receptor antagonist LY341495 (Fitzjohn et al., 1998). These findings do not, however, preclude a role for mGlu5 receptors, through potentiation of NMDA receptor-mediated responses or other mechanisms, in synaptic plasticity under different experimental conditions.
Acknowledgments
This work was supported by the European Economic Community (BioTech PL96-0049 and BioMed BMH4-CT96-0228), The Wellcome Trust and the MRC. The authors would like to acknowledge the technical assistance of Brian Arnold, Tom Bleisch, David Calligaro, Bryan Johnson, Rosemarie Tomlinson and Becky Wright in undertaking this study.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- LTP
long-term potentiation
- mGlu receptor
metabotropic glutamate receptor
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- (S)-3,5-DHPG
(S)-3,5-dihydroxyphenylglycine
- (1S, 3R)-ACPD
(1S, 3R)-aminocyclopentane-1,3-dicarboxylic acid
References
- ANDERSON W.W., COLLINGRIDGE G.L. A data acquisition program for the on-line analysis of Long Term Potentiation and Long Term Depression. Soc. Neurosci. Abst. 1997;23:665. doi: 10.1016/s0165-0270(01)00374-0. [DOI] [PubMed] [Google Scholar]
- BEN-ARI Y., ANIKSZTEIJN L., BREGESTOVSKI P. Protein kinase C modulation of NMDA currents: an important link for LTP induction. Trends in Neurosci. 1992;15:333–339. doi: 10.1016/0166-2236(92)90049-e. [DOI] [PubMed] [Google Scholar]
- CONN P.J., PIN J.P. Pharmacology and functions of metabotropic glutamate receptors. Ann. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- DOHERTY A.J., COLLINGRIDGE G.L., JANE D.E. Antagonist activity of α-substituted 4-carboxyphenylglycine analogues at group I metabotropic glutamate receptors expressed in CHO cells. Br. J. Pharmacol. 1999;126:205–210. doi: 10.1038/sj.bjp.0702297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOHERTY A.J., PALMER M.J., HENLEY J.M., COLLINGRIDGE G.L., JANE D.E. (RS)-2-Chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5, but not mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology. 1997;36:265–267. doi: 10.1016/s0028-3908(97)00001-4. [DOI] [PubMed] [Google Scholar]
- FITZJOHN S.M., BORTOLOTTO Z.A., PALMER M.J., DOHERTY A.J., ORNSTEIN P.L., SCHOEPP D.D., KINGSTON A.E., LODGE D., COLLINGRIDGE G.L. The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology. 1998;37:1445–1458. doi: 10.1016/s0028-3908(98)00145-2. [DOI] [PubMed] [Google Scholar]
- FITZJOHN S.M., IRVING A.J., PALMER M.J., HARVEY J., LODGE D., COLLINGRIDGE G.L. Activation of group I mGluRs potentiates NMDA responses in rat hippocampal slices. Neurosci. Lett. 1996;203:211–213. doi: 10.1016/0304-3940(96)12301-6. [DOI] [PubMed] [Google Scholar]
- FRENGUELLI B.G., POTIER B., SLATER N.T., ALFORD S., COLLINGRIDGE G.L. Metabotropic glutamate receptors and calcium signalling in dendrites of hippocampal CA1 neurones. Neuropharmacology. 1993;32:1229–1237. doi: 10.1016/0028-3908(93)90017-w. [DOI] [PubMed] [Google Scholar]
- GASPARINI F., LINGENHÖHL K., STOEHR N., FLOR P.J., HEINRICH M., VRANESIC I., BIOLLAZ M., ALLEIER H., HECKENDORN R., URWYLER S., VARNEY M.A., JOHNSON E.C., HESS S.D., RAO S.P., SACAAN A.I., SANTORI E.M., VELIÇELEBI G., KUHN R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1504. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- JIA Z., LU Y., HENDERSON J., TAVERNA F., ROMANO C., ABRAMOW-NEWERLY W., WOJTOWICZ J.M., RODER J. Selective abolition of the NMDA component of long-term potentiation in mice lacking mGluR5. Learning & Memory. 1998;5:331–343. [PMC free article] [PubMed] [Google Scholar]
- KINGSTON A.E., BURNETT J.P., MAYNE N.G., LODGE D. Pharmacological analysis of 4-carboxyphenylglycine derivatives: comparison of effects on mGluR1a and mGluR5a subtypes. Neuropharmacology. 1995;34:887–894. doi: 10.1016/0028-3908(95)00069-i. [DOI] [PubMed] [Google Scholar]
- KINGSTON A.E., ORNSTEIN P.L., WRIGHT R.A., JOHNSON B.G., MAYNE N.G., BURNETT J.P., BELAGAJE R., WU S., SCHOEPP D.D. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology. 1998;37:1–12. doi: 10.1016/s0028-3908(97)00191-3. [DOI] [PubMed] [Google Scholar]
- LUJÁN R., NUSSER Z., ROBERTS J.D.B., SHIGEMOTO R., SOMOGYI P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur. J. Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- NAKANISHI S., MASU M., BESSHO Y., NAKAJIMA Y., HAYASHI Y., SHIGEMOTO R. Molecular diversity of glutamate receptors and their physiological functions. EXS. 1994;71:71–80. doi: 10.1007/978-3-0348-7330-7_8. [DOI] [PubMed] [Google Scholar]
- ORNSTEIN P.L., BLEISCH T.J., ARNOLD M.B., WRIGHT R.A., JOHNSON B.G., SCHOEPP D.D. 2-Substituted 2SR-2-amino-2-(1SR,2SR-2-carboxycyclopropan-1-yl)glycines as potent and selective antagonists of group 2 metabotropic glutamate receptors: Part 1 Effects of alkyl, arylalkyl and diarylalkyl substitution. J. Med Chem. 1998a;41:346–357. doi: 10.1021/jm970497w. [DOI] [PubMed] [Google Scholar]
- ORNSTEIN P.L., BLEISCH T.J., ARNOLD M.B., WRIGHT R.A., JOHNSON B.G., TIZZANO J.P., HELTON D.R., KALLMAN M.J., SCHOEPP D.D. 2-Substituted 2SR-2amino-2-(1SR,2SR-2-carboxycyclo-propan-1-yl)glycines as potent and selective antagonists of group 2 metabotropic glutamate receptors: Part 2: Effects of aromatic substitution: pharmacological characterisation and bioavailability. J. Med Chem. 1998b;41:358–378. doi: 10.1021/jm970498o. [DOI] [PubMed] [Google Scholar]
- SCHOEPP D.D., JANE D.E., MONN J.A. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- SCHOEPP D.D., JOHNSON B.G., WRIGHT R.A., SALHOFF C.R., MAYNE N.G., WU S., COCKERHAM S.L., BURNETT J.P., BELEGAJE R., BLEAKMAN D., MONN J.A. LY354740 is a potent and highly selective group II metabotropic glutamate receptor agonist in cells expressing human glutamate receptors. Neuropharmacology. 1997;36:1–11. doi: 10.1016/s0028-3908(96)00160-8. [DOI] [PubMed] [Google Scholar]
- VARNEY M.A., COSFORD N.D.P., JACHEC C., RAO S.P., SACAAN A., LIN F.-F., BLEICHER L., SANTORI E.M., FLOR P.J., ALLGEIER H., GASPARINI F., KUHN R., HESS S.D., VELICELEBI G., JOHNSON E.C. SIB-1757 and SIB-1893: Selective, non-competitive antagonists of metabotropic glutamate receptor type 5. J. Pharm. Exp. Therap. 1999;290:170–181. [PubMed] [Google Scholar]
- WALKER K., BOWES M., GASPARINI F., KUHN R., URBAN L. The novel, selective mGlu5 receptor antagonist, 2-methyl-6-(phenylethynyl)-pyridine, reverses mechanical hyperalgesia in rat models of inflammatory pain. Neuropharmacology. 1999;38:A46. [Google Scholar]