

Abstract
Nociceptin, the endogenous ORL1 receptor agonist inhibited the motor response to electrical-field stimulation in the rat anococcygeus muscle. This effect was characterized using the peptide ligands acetyl-Arg-Tyr-Tyr-Arg-Trp-Lys-NH2 (Ac-RYYRWK-NH2), acetyl-Arg-Tyr-Tyr-Arg-Ile-Lys-NH2 (Ac-RYYRIK-NH2) and [Phe1ψ(CH2-NH)Gly2]nociceptin(1-13)NH2 ([F/G]NC(1-13)NH2), and the non-selective opioid antagonist naloxone benzoylhydrazone (NalBzOH).
Nociceptin produced a concentration-dependent inhibition of the adrenergic motor response to electrical-field stimulation (EC50 19 nM, pEC50 7.7±0.1, n=8), but the response to exogenous noradrenaline (0.2–1 μM) was unaffected. The inhibitory nerve response was not affected by up to 1 μM nociceptin.
After inhibition of nitric oxide synthase (Nω-nitro-L-arginine 100 μM), and in the presence of peptidase inhibitors, nociceptin produced full inhibition of the pure adrenergic motor response (EC50 4 nM; pEC50 8.4±0.1, Emax 98.3±1.2%, n=12). Ac-RYYRWK-NH2 was a potent partial-agonist (pEC50 9.0±0.1, Emax 66.4±5.2; n=11) but the efficacy of Ac-RYYRIK-NH2 (pEC50 8.0±0.2, Emax 36.7±3.5; n=12) was lower and the peptide could be tested as an antagonist (pA2 9.01).
[F/G]NC(1-13)NH2 and NalBzOH had little or no efficacy and were competitive antagonists with pKB values of 7.4 (95% c.l. 7.1, 7.7) and 6.9 (95% c.l. 6.7, 7.1) respectively. Both increased the response to field stimulation at high concentrations, suggesting the release of an endogenous agonist for the ORL1 receptor during stimulation.
Rat nocistatin did not affect the response to electrical-field stimulation, nor did it modify the inhibitory action of nociceptin.
Our findings suggest there is a significant endowment of ORL1 receptors on sympathetic terminals of the rat anococcygeus, where nociceptin mediates a powerful inhibitory effect on adrenergic neuromuscular transmission.
Keywords: Nociceptin, ORL1 receptor, anococcygeus
Introduction
Nociceptin (also known as orphanin FQ) is a heptadecapeptide (H-Phe-Gly -Gly-Phe-Thr-Gly-Ala -Arg-Lys-Ser-Ala-Arg-Lys-Leu-Ala-Asn-Gln-OH) recently isolated from the brain, and is the endogenous agonist for the orphan opioid-receptor-like (ORL1) receptor (Meunier et al., 1995; Reinscheid et al., 1995). Despite its similarity to the endogenous opioid peptides such as dynorphin A, nociceptin has no appreciable activity at μ-, δ- or κ-receptors. Likewise, although there is a high degree of structural homology between ORL1 and the ‘classical' opioid receptors, none of the known agonists or antagonists for those receptors interact with high affinity at the ORL1 receptor (see Henderson & McKnight, 1997).
Since the ORL1 receptor and nociceptin were discovered, only very few synthetic ligands with significant affinity and selectivity for the receptor have been identified. Screening of combinatorial libraries was used to identify hexapeptides with activity at the ORL1 receptor; these have been shown to be partial agonists with varying affinity, potency or efficacy depending on the test (Dooley et al., 1997; Nicholson et al., 1997; Mason & McKnight, 1998; Nicholson & McKnight, 1998; Berger et al., 1999a,1999b). The synthetic peptide [Phe1ψ(CH2-NH)Gly2]nociceptin(1-13)NH2 ([F/G]NC(1-13)NH2), which is an analogue of nociceptin(1-13) made to be resistant to aminopeptidase action, was initially shown to be an antagonist for the ORL1 receptor (Guerrini et al., 1998). More recent studies report varying degrees of efficacy with this peptide analogue, particularly in the whole animal or after transfection of the ORL1 receptor in host CHO cells (Butour et al., 1998; Calò et al., 1998; Carpenter & Dickenson, 1998; Grisel et al., 1998; Mason & McKnight, 1998; Nicholson & McKnight, 1998; Xu et al., 1998; Chiou, 1999; Okawa et al., 1999). To date, no selective antagonists have been identified for the ORL1 receptor.
The presence of opioid receptors in the peripheral nervous system has been exploited to provide functional in vitro models of opioid action, using preparations of the isolated ileum of the guinea-pig, or of the vas deferens from various species (see Corbett et al., 1993). Recently the presence of the ORL1 receptor on sympathetic nerves in the vas deferens (rat and rabbit) and in myenteric neurones (guinea-pig) has been demonstrated functionally from the powerful opioid-like inhibitory effect on neuromuscular transmission of nociceptin (Nicholson et al., 1996; 1998; Zhang et al., 1997). The peptide also inhibits the response of the mouse vas deferens to field stimulation, though less potently (Berzetei-Gurske et al., 1996; Calò et al., 1996).
Here we report the discovery of a powerful inhibitory effect of nociceptin, pointing to the probable presence of the ORL1 receptor at another peripheral site: on sympathetic motor nerves in the anococcygeus muscle of the rat. Exceptionally the investment with the ORL1 receptor in the anococcygeus occurs in the absence of μ-, κ- or δ-opioid receptors, offering the potential of this tissue as a pharmacological model for the ORL1 receptor, free from the possible complication of interference by activity at ‘conventional' opioid receptors. This work has been the subject of a preliminary communication (Ho et al., 1999).
Methods
Animals were housed and cared for in accordance with the stipulations of the Animals (Scientific Procedures) Act 1986.
Experimental procedures
Single anococcygeus muscles from mature male rats (200–250 g, Lister Hooded or Wistar) were mounted under a resting tension of around 10 mN in 3 ml silanized glass organ baths containing modified Krebs' solution of varying composition. In the preliminary experiments the Krebs' solution comprised: (mM) NaCl 118, KCl 4.74, CaCl2 2.5, KH2PO4 1.19, MgSO4.7H2O 1.2, NaHCO3 25, glucose 11, pH 7.4 when gassed with 95% O2-5% CO2 at 37°C.
Electrical-field stimulation of intramural nerves in the anococcygeus was with trains of biphasic pulses (30 Hz for preliminary experiments on the motor response, 10 Hz for the pure motor response, 10 or 20 Hz for the inhibitory response) of supramaximal voltage (∼70 mA) delivered from a Grass S88 stimulator (Grass Instruments, Quincy, MA, U.S.A.) through platinum electrodes at the top and bottom of the organ baths. Isometric responses were recorded using Grass FT03C force-displacement transducers.
In the few experiments where the response to stimulation of the non-adrenergic-non-cholinergic (NANC) inhibitory nerves was studied, the Krebs' solution contained guanethidine sulphate at 30 μM. In experiments on the pure motor adrenergic response, the effects of concomitant activation of the inhibitory nerves were removed by the addition of 100 μM Nω-nitro-L-arginine (NOARG). Generally in this case the Krebs' solution was modified by the reduction of the concentrations of the salts of calcium and magnesium by half (without compensation for the small change in osmolarity), and by the addition of the peptidase inhibitors amastatin, bestatin, phosphoramidon and captopril (all at 30 μM; Nicholson et al., 1998). The bulk of the work was under these conditions, examining the effects of test substances on the pure motor response to field stimulation with trains of 5×0.5 ms pulses at 10 Hz every 30 s. Agents producing an inhibition of the motor response were added cumulatively to the organ bath, generally with a period of around 2 min between additions, using a 30 min period of washout and recovery between agonist concentration-response schedules. For experiments involving potential antagonists, a 30 min preincubation time was generally allowed before the addition of agonist.
Drugs and chemicals
The following compounds were used: nonadrenaline bitartrate, guanethidine sulphate, [D-Ala2,Me-Phe4,Gly-ol5)enkephalin, [D-Pen2,D-Pen5]enkephalin and Nω-nitro-L-arginine (Sigma Chemical Co., St. Louis, MO, U.S.A.); naloxone benzoylhydrazone (RBI, Natick, MA, U.S.A.); nociceptin (Bachem, U.K.); Ac-RYYRWK-NH2 and Ac-RYYRIK-NH2 (Albachem, Edinburgh, U.K.); U-69,593 (Upjohn Labs., MI, U.S.A.); [Phe1ψ(CH2-NH)Gly2]nociceptin(1-13)NH2 (Tocris Cookson Ltd., Bristol, U.K.); rat nocistatin (‘nocistatin-35', prepro-nociceptin(98-132), Phoenix Pharmaceuticals Inc., CA, U.S.A.). The peptidase inhibitors were amastatin (Peptide Institute, Osaka, or Sigma), bestatin (Peninsula, U.K., or Sigma), captopril (Sigma), and phosphoramidon (Peptide Institute).
All stocks of peptides were made up in 0.5 M acetic acid in 50% (w v−1) aqueous methanol containing 0.1% (w v−1) bovine serum albumin (Fraction V, Sigma) to reduce adsorptive losses. Stock solutions of the other agents were in aqueous solvents.
Statistical analysis
Responses to agonists were measured as the percentage inhibition of the stimulation-evoked motor response of the rat anococcygeus muscle, against the level of the stable baseline response before addition of agonist. Pooled data for responses to the one concentration are expressed as mean±s.e.mean. Potencies of agonists were compared in terms of values for EC50 expressed as the geometric mean, or as the arithmetic mean of pEC50±s.e.mean. Values for EC50 and Emax the maximum effect, were obtained by fitting (GraphPad Prism 3.0) the log(concentration)-response curves to the standard four-parameter logistic equation, usually without any constraint:
Where E is the measured response, Emin and Emax are the minimum and maximum asymptotes, EC50 is obtained from the ‘location parameter' of the curve, x is the concentration and n is the slope factor of the curve (‘Hill slope').
Antagonist affinity was measured by Schild regression of the line:
with the concentration ratio CR, obtained from the difference between the value for EC50 for nociceptin in the presence of antagonist B and the control value. At least three observations for concentration ratio from three or more concentrations of antagonist were used. When the slope of the Schild regression m was not significantly different from one, the regression was constrained to unit slope and the value for −logKB was expressed as pKB with 95% confidence limits (c.l.), otherwise this value was expressed as pA2.
Results
Characterization of the actions of nociceptin in the anococcygeus
In preliminary experiments, electrical-field stimulation of the anococcygeus muscle with 30 Hz trains produced reproducible motor responses, where the contraction (usually between 15 and 20 mN) matched that obtained by the addition of exogenous noradrenaline 0.2–1 μM. From four experiments the mean EC50 for noradrenaline was 2.6 μM (pEC50 5.59±0.09). The response to electrical-field stimulation was abolished by the addition of either tetrodotoxin (1 μM) or phentolamine (100 μM), confirming that the contraction was mediated by release of noradrenaline from intramural nerves (Gillespie, 1972).
The motor response to electrical field stimulation was unaffected by up to 10 μM of the selective agonists [D-Ala2, Me-Phe4,Gly-ol5]enkephalin,[D-Pen2,D-Pen5]enkephalin or U-69,593 (not shown), confirming the absence of μ-, δ- and κ-receptors respectively. The addition of nociceptin however, produced a concentration-related inhibition with a threshold around 1 nM and a maximum effect of near complete abolition of the response at 1–3 μM (EC50 19 nM; pEC50 7.72±0.13, n=18), without affecting the response to the matching concentration of exogenous noradrenaline (not shown). The inhibitory effect of nociceptin was quick to develop, with a stable effect being achieved within a few minutes, and was rapidly reversed on washout to produce full recovery of the tissue.
In high tone after guanethidine 30 μM the relaxations produced by activation of the NANC inhibitory nerves (stimulation with 10 or 20 Hz trains for 2 or 1 s respectively) were seen. The inhibitory nitrergic NANC response (Liu et al., 1991) was blocked by 1 μM tetrodotoxin or 100 μM Nω-nitro-L-arginine (NOARG) but was unaffected by up to 1 μM nociceptin (not shown).
With the influence of the inhibitory nerves removed in the presence of 100 μM NOARG, the strength of the pure motor adrenergic response to electrical field stimulation was roughly doubled, but the maximum inhibition achieved by the addition of nociceptin was reduced by half. When the experiment on the pure motor response was repeated in Krebs' solution with reduced concentrations of Ca2+ (1.25 mM) and Mg2+ (0.6 mM), and the peptidase inhibitors added (Nicholson et al., 1998), the efficacy of nociceptin was restored and the potency was increased almost 5 fold (EC50 4 nM; pEC50 8.4±0.1, Emax 98.3±1.2%, n=12, Figure 1).
Figure 1.
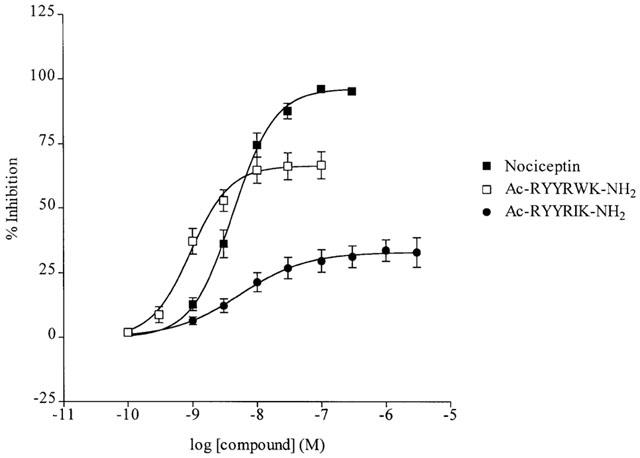
Inhibition of stimulation-evoked contractions in the rat anococcygeus muscle by the ORL1 agonists nociceptin, Ac-RYYRWK-NH2 and Ac-RYYRIK-NH2. Each point is the mean±s.e.mean of 11 or 12 observations.
A reduction in the concentration of Ca2+ ions to the physiologically more relevant level, or even lower, has commonly been employed to increase the potency or efficacy of opioid agonists in isolated organs (see e.g. Dougall & Leff, 1987). In such cases restoration of the contractile response can be achieved by a corresponding reduction of the concentration of Mg2+ ions in the buffer, or as in the classical example of the mouse vas deferens (Hughes et al., 1975) by omitting Mg2+ all together. The addition of peptidase inhibitors had little effect on the potency and efficacy of nociceptin in the anococcygeus using the modified Krebs' solution (peptidase inhibitors absent, EC50 8 nM; pEC50 8.1±0.1, Emax 97.1±1.3%, n=9), however it was routinely done so that a direct comparison with our data from the vas deferens could be made, and to prevent breakdown of the other more labile peptide ligands. All subsequent experiments were with the low-calcium Krebs' solution containing peptidase inhibitors and NOARG.
The actions of the hexapeptides
As we had seen in the vas deferens (Nicholson et al., 1997) the effect of nociceptin on the anococcygeus was reproduced by the hexapeptide Ac-RYYRWK-NH2 with higher potency (pEC50 9.0±0.1), though reduced efficacy (Emax 66.4±5.2%, n=11, Figure 1). With the related peptide Ac-RYYRIK-NH2 there was a reduction both in potency (pEC50 8.0±0.2) and efficacy (Emax 36.7±3.5%, n=12, Figure 1).
The low efficacy of Ac-RYYRIK-NH2 permitted an attempt at its use as an antagonist of the response to the full agonist nociceptin. In the presence of Ac-RYYRIK-NH2 1 nM to 1 μM there was a concentration-dependent rightward shift of the nociceptin response curve indicative of surmountable antagonism (Figure 2). A value for pA2 for the interaction between Ac-RYYRIK-NH2 and nociceptin of 9.01 was obtained from the Schild regression on the results of these experiments, suggesting high affinity for Ac-RYYRIK-NH2, but the slope was too low (0.6) to permit derivation of pKB (Figure 2).
Figure 2.
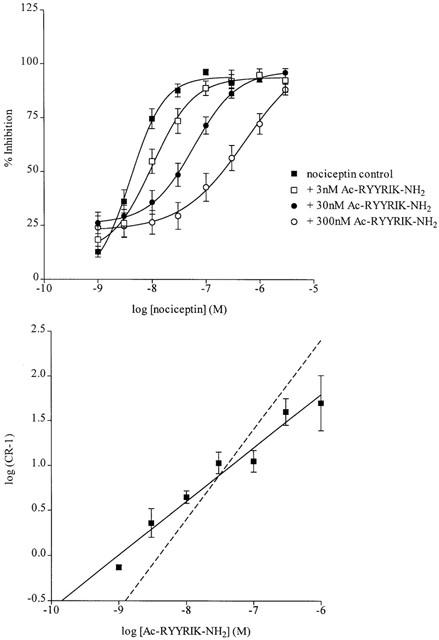
Antagonism by the partial agonist Ac-RYYRIK-NH2 (1 nM–1 μM) of the effect of nociceptin in the rat anococcygeus. Top: concentration-response curves to nociceptin in untreated preparations, or after incubation with Ac-RYYRIK-NH2 at 3, 30, or 300 nM. Each point is the mean±s.e.mean of at least four observations. Data for the experiments with 1, 10 and 100 nM and 1 μM Ac-RYYRIK-NH2 have been omitted for the sake of clarity. Bottom: Schild regression for the interaction between Ac-RYYRIK-NH2 and nociceptin, showing the line of best fit (log{CR−1}=0.6{log[Ac-RYYRIK-NH2]}+9.01), and the theoretical line of unit slope (dashed line).
The actions of the antagonists [F/G]NC(1-13)NH2 and NalBzOH
By itself the synthetic peptide [F/G]NC(1-13)NH2 at concentrations up to 100 nM produced weak inhibitions of the pure motor adrenergic response of the anococcygeus, consistent with the known partial agonist action of this ligand on the ORL1 receptor. At concentrations above 100 nM however, [F/G]NC(1-13)NH2 produced enhancement of the stimulation-evoked response (Figure 3). The non-selective opioid NalBzOH had little overt effect between 10 and 300 nM, but a clear enhancement of the response to field stimulation was obtained with this non-peptide ORL1-receptor antagonist above 1 μM (Figure 3). In tissues not treated with NOARG, [F/G]NC(1-13)NH2 still produced an enhancement of the motor response to electrical field stimulation (not shown).
Figure 3.
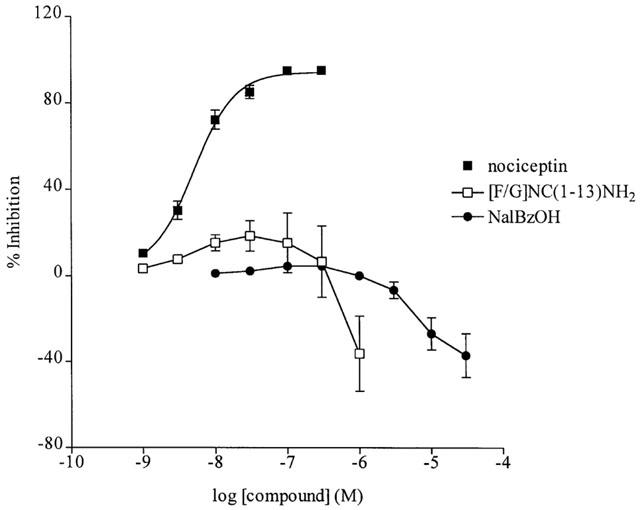
The effect of [F/G]NC(1-13)NH2 and NalBzOH on the adrenergic motor response to electrical field stimulation in the rat anococcygeus muscle. Responses to nociceptin, [F/G]NC(1-13)NH2 and NalBzOH are the mean±s.e.mean of 3–6 observations.
To assess their antagonist activity NalBzOH was added at 1, 3 or 10 μM, and [F/G]NC(1-13)NH2 at 0.1, 0.3 and 1 μM. The enhancement of the motor response by these agents stabilized during the 30 min equilibration period, and the response to addition of nociceptin was measured against this new baseline. Both NalBzOH and [F/G]NC(1-13)NH2 produced parallel rightwards shift of the concentration-response curve to nociceptin, with the agonist still able to produce a near complete inhibition of the response to field stimulation (not shown). The results of the Schild regressions (Figure 4) suggested that both agents were competitive antagonists of modest affinity (NalBzOH; pA2 6.67, slope 1.21; pKB 6.9, 95% c.l. 6.7, 7.1. [F/G]NC(1-13)NH2: pA2 7.24, slope 1.18; pKB 7.4, 95% c.l. 7.1, 7.7).
Figure 4.
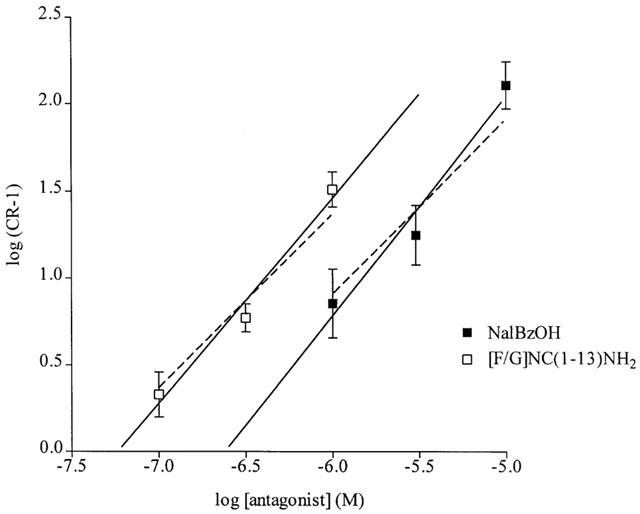
Analysis of the antagonism by NalBzOH and [F/G]NC(1-13)NH2 of the response to nociceptin in the rat anococcygeus. Schild regressions for the interactions between NalBzOH (1, 3 and 10 μM) and nociceptin, showing the line of best fit (log{CR−1}=1.21{log[NalBzOH]}+6.67), or between [F/G]NC(1-13)NH2 (0.1, 0.3 and 1 μM) and nociceptin, showing the line of best fit (log{CR−1}=1.18{log[[F/G]NC(1-13)NH2]}+7.24). The theoretical lines of unit slope are indicated by the dashed lines. Data are mean±s.e.mean of 3–12 observations.
The action of nocistatin
The addition of rat nocistatin (‘nocistatin-35', rat prepro-nociceptin(98-132)) up to 1 μM produced no remarkable effect by itself on the pure motor response of the anococcygeus to electrical-field stimulation (not shown). Preincubation with nocistatin at 1 μM did not affect the inhibitory action of nociceptin on motor nerve transmission (control: pEC50 8.27±0.04 n=5, treated: pEC50 8.07±0.08 n=5).
Discussion
Like the ‘classical' opioid receptor types, the ORL1 receptor is widely distributed in the central nervous system but is also present at peripheral sites. Transcripts for the ORL1 receptor have been found in the liver, spleen, intestine and vas deferens of the rat (Wang et al., 1994), and in pre- and paravertebral ganglia in the guinea-pig (Kummer & Fischer, 1997). The presence of a functional ORL1 receptor at a number of sites has been suggested by the observation of an inhibitory effect in isolated tissue preparations, usually with nociceptin alone. Thus ORL1 receptors may be present in the guinea-pig in the renal pelvis (Giuliani & Maggi, 1996), the atrium of the heart (Giuliani & Maggi, 1997), and in the trachea (Patel et al., 1997) and bronchus (Fischer et al., 1998). Significantly, in view of their place in the development of our knowledge of the pharmacology of opioids, the most extensive studies have been with preparations of vasa deferentia from the mouse (Calò et al., 1996; Berzetei-Gurske et al., 1996; Bigoni et al., 1999; Zhang et al., 1997), rat and rabbit (Nicholson et al., 1996; 1997; 1998; Nicholson & McKnight, 1998), or the guinea-pig ileum (Salvadori et al., 1997; Zhang et al., 1997; Nicholson et al., 1998).
Our current study on the rat isolated anococcygeus has been as extensive as is practicably possible, in terms of the availability of ligands, and now suggests the presence of the ORL1 receptor in this tissue. We did not measure the release of the motor transmitter directly, but the lack of effect on the response of the anococcygeus to exogenous noradrenaline, points to the powerful inhibitory effect of nociceptin on motor transmission being due to inhibition of release of noradrenaline. The absence of a post-junctional inhibitory effect of nociceptin seems to rule out an indirect effect through modulation of the release of other putative neuropeptides in the anococcygeus, such as vasoactive intestinal polypeptide (Blank et al., 1986) or neuropeptide Y (Vila et al., 1992). The action of those peptides, if it is important, seems predominantly to be postjunctional so the most likely site for the ORL1 receptor is on the post-ganglionic adrenergic nerves. The receptor seems not to be present on the inhibitory nerves.
Our finding that the hexapeptide Ac-RYYRWK-NH2 was a potent partial agonist in the anococcygeus mirrors our original findings with this synthetic peptide in the vas deferens (Nicholson et al., 1997), where we believe the ORL1 receptor is also present on the sympathetic nerves. The assessment of the antagonist activity in the anococcygeus of the non-selective opioid NalBzOH suggested a competitive interaction with nociceptin, and a value for pKB of 6.9 was assigned. This finding is again in accord with our observation in the vas deferens, where the antagonist activity of NalBzOH at the ORL1 receptor was first identified (Nicholson et al., 1996; Dunnill et al., 1996). Likewise the analogue of the N-terminal tridecapeptide of nociceptin [F/G]NC(1-13)NH2 was a competitive antagonist with about 3 fold higher affinity, as in the rat vas deferens (Nicholson & McKnight, 1998). Interestingly both NalBzOH and FG/NC13 are surmountable antagonists of the action of nociceptin to inhibit the evoked release of noradrenaline in mouse cortex in vitro, and the values for pA2 reported in that study (6.6 and 7.2, respectively; Schlicker et al., 1998) are in close agreement with our findings for the ORL1 receptor in the sympathetic nerves.
The hexapeptide Ac-RYYRIK-NH2 was recently reported to be an antagonist at ORL1 sites in rat (Berger et al., 1999a), although it was later reported also to exhibit the properties of an agonist (Berger et al., 1999b). This may point merely to partial agonism with low efficacy, as we found in the present study. The levels of relative efficacy in the anococcygeus for nociceptin, Ac-RYYRWK-NH2 and Ac-RYYRIK-NH2 are the same as we have seen in the frontal cortex of the rat, measuring the stimulation of the binding of GTPγ35S by these agents (Mason & McKnight, 1998; Ho & McKnight, unpublished). The level of investment of the ORL1 receptor in the frontal cortex of the rat is high (the Bmax for the binding of [3H]-nociceptin is around 0.25 pmol mg−1; Mason & McKnight, 1998), but whether the comparability of the findings with the three agonists of different efficacies points also to a high density of the ORL1 receptor in the anococcygeus, has yet to be proven. The low efficacy of Ac-RYYRIK-NH2 allowed its use as an antagonist in the anococcygeus and the findings point to an interaction with high affinity that is surmountable. The low slope of the Schild regression precludes the conclusion that the hexapeptide is acting competitively.
The stimulatory effect of the ORL1 antagonists [F/G]NC(1-13)NH2 and NalBzOH on the response to activation of the motor nerves deserves comment. That the same action was obtained with the two structurally unrelated compounds suggests that the effect may be a consequence of antagonism of the ORL1 receptor, rather than by some other pharmacological effect such as e.g. block of prejunctional α-adrenoceptors. The corollary is, however, that field stimulation in the anococcygeus must activate intramural nerves that store and release nociceptin, or another endogenous agonist for the ORL1 receptor.
It is not yet known whether nociceptin is present in the anococcygeus, but the above findings point to the possibility, and provided the rationale for the experiment to test for an interaction between nocistatin and nociceptin. Nocistatin, a putative endogenous antagonist of nociceptin (Okuda-Ashitaka et al., 1998), is the name originally given to the heptadecapeptide that occurs between the −Lys109-Arg110-sequence in the prepro-nociceptin peptide from the bull, and the −Lys128-Arg129-cleavage site before the N-terminus of nociceptin. The former -Lys-Arg- motif is absent from the homologous peptides in man, rat and mouse, so the putative additional product of the processing of the nociceptin precursor in those species will be the N-terminally extended 30-, 35- and 41-amino acid peptides (see Okuda-Ashitaka et al., 1998). Although data for the rat peptide are lacking, the human and murine N-terminally extended nocistatin peptides are reported also to act like bovine nocistatin by blocking the allodynia after intrathecal administration of nociceptin (Minami et al., 1998), supporting the view that the activity of the nocistatins resides in the highly conserved C-terminus. We thought it most appropriate to work with the rat nocistatin-35 peptide (rat prepro-nociceptin(98-132)) for our experiments in the anococcygeus, but could detect no effect on the response to nociceptin, suggesting that the interaction between nocistatin and nociceptin may not be a general phenomenon.
Whether the putative nociceptin-ORL1 receptor system subserves a physiological role in controlling adrenergic neuromuscular transmission in the anococcygeus is not clear. The effect is not confined to the rat, as we have seen signs of an analogous inhibitory effect of nociceptin in preliminary experiments on the anococcygeus muscle of the rabbit (Ho et al., unpublished), in spite of the difficulty in obtaining a stable motor response in this tissue (see Gillespie, 1980). The effect may extend to other species, and other organs (albeit related) since nociceptin is reported to produce a powerful effect to promote penile erection in the cat after intracavernosal injection of quite small amounts (Champion et al., 1997).
In summary, we have found a powerful effect of nociceptin to inhibit adrenergic neuromuscular transmission in the isolated anococcygeus muscle of the rat. We investigated the pharmacology of this effect with as much rigour as the paucity of useful ligands permits, and conclude that an ORL1 receptor exists on the sympathetic terminals in this tissue. The pharmacology of this receptor is like that in the sympathetic nerves in the vas deferens, or on central (frontal cortex) neurones. The ORL1 receptor in the anococcygeus occurs without the co-presence of the ‘classical' opioid receptor types, which is rather unusual. This could prove to be a useful feature for the possible future use of this popular isolated-tissue preparation in the characterization of the pharmacology of ligands for the ORL1 receptor.
Abbreviations
- Ac-RYYRIK-NH2
acetyl-Arg-Tyr-Tyr-Arg-Ile-Lys-NH2
- Ac-RYYRWK-NH2
acetyl-Arg-Tyr-Tyr-Arg-Trp-Lys-NH2
- CHO
Chinese hamster ovary
- c.l.
confidence limits
- CR
concentration ratio
- [F/G]NC(1-13)NH2
[Phe1ψ(CH2-NH)Gly2]nociceptin(1-13)NH2
- NalBzOH
naloxone benzoylhydrazone
- NANC
non-adrenergic, non-cholinergic
- NOARG
Nω-nitro-L-arginine
- ORL1
opioid-receptor-like
References
- BERGER H., ALBRECHT E., WALLUKAT G., BIENERT M. Antagonism by acetyl-RYYRIK-NH2 of G protein activation in rat brain preparations and of chronotropic effect on rat cardiomyocytes evoked by nociceptin/orphan FQ. Br. J. Pharmacol. 1990a;126:555–558. doi: 10.1038/sj.bjp.0702353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERGER H., ALBRECHT E., WALLUKAT G., CALO G., BIGONI R., BIENERT M. Acetyl-RYYRIK-NH2 a nociceptin/orphanin FQ (NOC/OFQ) receptor ligand, exhibits properties of an antagonist and agonist as well. Regul. Peptides. 1999b;80:122. [Google Scholar]
- BERZETEI-GURSKE I.P., SCHWARTZ R.W., TOLL L. Determination of activity for nociceptin in the mouse vas deferens. Eur. J. Pharmacol. 1996;302:R1–R2. doi: 10.1016/0014-2999(96)00238-5. [DOI] [PubMed] [Google Scholar]
- BIGONI R., GIULIANI S., CALO' G., RIZZI A., GUERRINI R., SALVADORI S., REGOLI D., MAGGI C.A. Characterization of nociceptin receptors in the periphery: in vitro and in vivo studies. Naunyn Schmiedeberg's Arch. Pharmacol. 1999;359:160–167. doi: 10.1007/pl00005338. [DOI] [PubMed] [Google Scholar]
- BLANK M.A., BROWN J.R., HUNTER J.C., BLOOM S.R., TYERS M.B. Effects of VIP and related peptides and Gila monster venom on genitourinary smooth muscle. Eur. J. Pharmacol. 1986;132:155–161. doi: 10.1016/0014-2999(86)90600-x. [DOI] [PubMed] [Google Scholar]
- BUTOUR J.L., MOISAND C., MOLLEREAU C., MEUNIER J.C. [Phe1ψ(CH2-NH)Gly2]nociceptin-(1-13)-NH2 is an agonist of the nociceptin (ORL1) receptor. Eur. J. Pharmacol. 1998;349:R5–R6. doi: 10.1016/s0014-2999(98)00273-8. [DOI] [PubMed] [Google Scholar]
- CALÒ G., RIZZI A., BOGONI G., NEUGEBAUER V., SALVADORI S., GUERRINI R., BIANCHI C., REGOLI D. The mouse vas deferens: a pharmacological preparation sensitive to nociceptin. Eur. J. Pharmacol. 1996;311:R3–R5. doi: 10.1016/0014-2999(96)00563-8. [DOI] [PubMed] [Google Scholar]
- CALÒ G., RIZZI A., MARZOLA G., GUERRINI R., SALVADORI S., BEANI L., REGOLI D., BIANCHI C. Pharmacological characterization of the nociceptin receptor mediating hyperalgesia in the mouse tail withdrawal assay. Br. J. Pharmacol. 1998;125:373–378. doi: 10.1038/sj.bjp.0702087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARPENTER K.J., DICKENSON A.H. Evidence that [Phe1ψ(CH2-NH)Gly2]nociceptin-(1-13)-NH2, a peripheral ORL-1 receptor antagonist, acts as an agonist in the rat spinal cord. Br. J. Pharmacol. 1998;125:949–951. doi: 10.1038/sj.bjp.0702188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAMPION H.C., WANG R., HELLSTROM W.J., KADOWITZ P.J. Nociceptin, an endogenous ligand for the ORL1 receptor, has potent erectile activity in the cat. Am. J. Physiol. 1997;273:214–219. doi: 10.1152/ajpendo.1997.273.1.E214. [DOI] [PubMed] [Google Scholar]
- CHIOU L.C. [Phe1ψ(CH2-NH)Gly2]nociceptin-(1-13)-NH2 activation of an inward rectifier as a partial agonist of ORL1 receptors in rat periaqueductal gray. Br. J. Pharmacol. 1999;128:103–107. doi: 10.1038/sj.bjp.0702746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORBETT A.D., PATERSON S.J., KOSTERLITZ H.W.Selectivity of ligands for opioid receptors Opioids I: Handbook of Experimental Pharmacology 1993104/1Berlin: Springer-Verlag; 645–679.ed. Hertz, A [Google Scholar]
- DOOLEY C.T., SPAETH C.G., BERZETEI-GURSKE I.P., CRAYMER K., ADAPA I.D., BRANDT S.R., HOUGHTEN R.A., TOLL L. Binding and in vitro activities of peptides with high affinity for the nociceptin/orphanin FQ receptor, ORL1. J. Pharmacol. Exp. Ther. 1997;283:735–741. [PubMed] [Google Scholar]
- DOUGALL I.G., LEFF P. Pharmacological analysis of the calcium-dependence of μ-receptor agonism. Br. J. Pharmacol. 1987;92:723–731. doi: 10.1111/j.1476-5381.1987.tb11376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNNIL R.J., KAKIZAWA K., MCKNIGHT A.T., HENDERSON G. Characterisation of the actions of naloxone benzoylhydrazone at mu-opioid, kappa-opioid and ORL1 receptors in isolated tissues from rat and guinea-pig. Br. J. Pharmacol. 1996;119:275P. [Google Scholar]
- FISCHER A., FORSSMANN W.G., UNDEM B.J. Nociceptin-induced inhibition of tachykinergic neurotransmission in guinea pig bronchus. J. Pharmacol. Exp. Ther. 1998;285:902–907. [PubMed] [Google Scholar]
- GILLESPIE J.S. The rat anococcygeus muscle and its response to nerve stimulation and to some drugs. Br. J. Pharmacol. 1972;45:404–416. doi: 10.1111/j.1476-5381.1972.tb08097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILLESPIE J.S. The physiology and pharmacology of the anococcygeus muscle. Trends Pharmacol. Sci. 1980;1:453–457. [Google Scholar]
- GIULIANI S., MAGGI C.A. Inhibition of tachykinin release from peripheral endings of sensory nerves by nociceptin, a novel opioid peptide. Br. J. Pharmacol. 1996;118:1567–1569. doi: 10.1111/j.1476-5381.1996.tb15576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIULIANI S., MAGGI C.A. Prejunctional modulation by nociceptin of nerve-mediated ionotropic responses in guinea-pig left atrium. Eur. J. Pharmacol. 1997;332:231–236. doi: 10.1016/s0014-2999(97)01076-5. [DOI] [PubMed] [Google Scholar]
- GRISEL J.E., FARRIER D.E., WILSON S.G., MOGIL J.S. [Phe1ψ(CH2-NH)Gly2]nociceptin-(1-13)-NH2 acts as an agonist of the orphanin FQ/nociceptin receptor in vivo. Eur. J. Pharmacol. 1998;357:R1–R3. doi: 10.1016/s0014-2999(98)00567-6. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO G., RIZZI A., BIGONI R., BIANCHI C., SALVADORI S., REGOLI D. A new selective antagonist of the nociceptin receptor. Br. J. Pharmacol. 1998;123:163–165. doi: 10.1038/sj.bjp.0701640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENDERSON G., MCKNIGHT A.T. The orphan opioid receptor and its endogenous ligand–nociceptin/orphanin FQ. Trends Pharmacol. Sci. 1997;18:293–300. [PubMed] [Google Scholar]
- HO M., WATSON A., CORBETT A.D., MCKNIGHT A.T. Characterisation of the ORL1 receptor on adrenergic nerves in the anococcygeus. Br. J. Pharmacol. 1999;128:84P. doi: 10.1038/sj.bjp.0703583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES J., KOSTERLITZ H.W., LESLIE F.M. Effect of morphine on adrenergic transmission in the mouse vas deferens. Assessment of agonist and antagonist potencies of narcotic analgesics. Br. J. Pharmacol. 1975;53:371–381. doi: 10.1111/j.1476-5381.1975.tb07373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUMMER W., FISCHER A. Nociceptin and its receptor in guinea-pig sympathetic ganglia. Neurosci. Lett. 1997;234:35–38. doi: 10.1016/s0304-3940(97)00662-9. [DOI] [PubMed] [Google Scholar]
- LIU X.R., GILLESPIE J.S., GIBSON I.F., MARTIN W. Effects of NG-substituted analogues of L-arginine on NANC relaxation of the rat anococcygeus and bovine retractor penis muscles and the bovine penile artery. Br. J. Pharmacol. 1991;104:53–58. doi: 10.1111/j.1476-5381.1991.tb12384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASON S.L., MCKNIGHT A.T. Pharmacological characterisation of the novel synthetic hexapeptide Ac-Arg-Tyr-Tyr-Arg-Trp-Lys-NH2 at the rat brain ORL1 receptor using [3H]nociceptin and [35S]GTPγS binding assays. Br. J. Pharmacol. 1998;124:13P. [Google Scholar]
- MEUNIER J.C., MOLLEREAU C., TOLL L., SUAUDEAU C., MOISAND C., ALVINERIE P., BUTOUR J.L., GUILLEMOT J.C., FERRARA P., MONSARRAT B., MAZARGUILL H., VASSART G., PARMENTIER M., COSTENTIN J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- MINAMI T., OKUDA-ASHITAKA E., NISHIUCHI Y., KIMURA T., TACHIBANA S., MORI H., ITO S. Anti-nociceptive responses produced by human putative counterpart of nocistatin. Br. J. Pharmacol. 1998;124:1016–1018. doi: 10.1038/sj.bjp.0701995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLSON J.R., MCKNIGHT A.T. The effect of the agonist Ac-RYYRWK-NH2 and the antagonist [Phe1ψ(CH2-NH)Gly2]nociceptin(1-13)NH2 at the ORL1 receptor of central and peripheral sites. Br. J. Pharmacol. 1998;125:5P. [Google Scholar]
- NICHOLSON J.R., PATERSON S.J., MCKNIGHT A.T. Characterisation of the response in the rat vas deferens to the ORL-1 agonist nociceptin. Br. J. Pharmacol. 1996;119:36P. [Google Scholar]
- NICHOLSON J.R., PATERSON S.J., MCKNIGHT A.T. Effect of nociceptin, nociceptin analogues and a novel hexapeptide agonist at the ORL1 receptor in central and peripheral sites. Br. J. Pharmacol. 1997;122:28P. [Google Scholar]
- NICHOLSON J.R., PATERSON S.J., MENZIES J.R., CORBETT A.D., MCKNIGHT A.T. Pharmacological studies on the “orphan” opioid receptor in central and peripheral sites. Can. J. Physiol. Pharmacol. 1998;76:304–313. [PubMed] [Google Scholar]
- OKAWA H., NICOL B., BIGONI R., HIRST R.A., CALO' G., GUERRINI R., ROWBOTHAN D.J., SMART D., MCKNIGHT A.T., LAMBERT D.G. Comparison of the effects of [Phe1ψ(CH2-NH)Gly2]Nociceptin(1-13)NH2 in rat brain, rat vas deferens and CHO cells expressing recombinant human nociceptin receptors. Br. J. Pharmacol. 1999;127:123–130. doi: 10.1038/sj.bjp.0702539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKUDA-ASHITAKA E., MINAMI T., TACHIBANA S., YOSHIHARA Y., NISHIUCHI Y., KIMURA T., ITO S. Nocistatin, a peptide that blocks nociceptin action in pain transmission. Nature. 1998;392:286–289. doi: 10.1038/32660. [DOI] [PubMed] [Google Scholar]
- PATEL H.J., GIEMBYCZ M.A., SPICUZZA L., BARNES P.J., BELVISI M.G. Naloxone-insensitive inhibition of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by the novel opioid, nociceptin. Br. J. Pharmacol. 1997;120:735–736. doi: 10.1038/sj.bjp.0701013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REINSCHEID R.K., NOTHACKER H.P., BOURSON A., ARDATI A., HENNINGSEN R.A., BUNZOW J.R., GRANDY D.K., LANGEN H., MONSMA F.J., JR, CIVELLI O. Orphanin FQ: a neuropeptide that activates an opioid like G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- SALVADORI S., PICONE D., TANCREDI T., GUERRINI R., SPADACCINI R., LAZARUS L.H., REGOLI D., TEMUSSI P.A. Solution conformation of nociceptin. Biochem. Biophys. Res. Commun. 1997;233:640–643. doi: 10.1006/bbrc.1997.6285. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., WERTHWEIN S., KATHMANN M., BAUER U. Nociceptin inhibits noradrenaline release in the mouse brain cortex via presynaptic ORL1 receptors. Naunyn Schmiedeberg's Arch. Pharmacol. 1998;358:418–422. doi: 10.1007/pl00005273. [DOI] [PubMed] [Google Scholar]
- VILA E., TABERNERO A., FERNANDES F., SALAICES M. Effect of neuropeptide Y on adrenergic and non-adrenergic, non-cholinergic responses in the rat anococcygeus muscle. Br. J. Pharmacol. 1992;107:66–72. doi: 10.1111/j.1476-5381.1992.tb14464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J.B., JOHNSON P.S., IMAI Y., PERSICO A.M., OZENBERGER B.A., EPPLER C.M., UHL G.R. cDNA cloning of an orphan opiate receptor gene family member and its splice variant. FEBS Lett. 1994;348:75–79. doi: 10.1016/0014-5793(94)00557-5. [DOI] [PubMed] [Google Scholar]
- XU I.S., WIESENFELD-HALLIN Z., XU X.J. [Phe1ψ(CH2-NH)Glu2]nociceptin-(1-13)-NH2, A proposed antagonist of the nociceptin receptor, is a potent and stable agonist in the rat spinal cord. Neurosci. Lett. 1998;249:127–130. doi: 10.1016/s0304-3940(98)00411-x. [DOI] [PubMed] [Google Scholar]
- ZHANG G., MURRAY T.F., GRANDY D.K. Orphanin FQ has an inhibitory effect on the guinea pig ileum and the mouse vas deferens. Brain Res. 1997;772:102–106. doi: 10.1016/s0006-8993(97)00858-5. [DOI] [PubMed] [Google Scholar]