

Abstract
Tumour necrosis factor-α (TNF-α) and interleukin 1β (IL-1β) have been implicated in the pathogenesis of asthma. The p38 kinase inhibitor, SB 203580 inhibits TNF-α and IL-1β production in vitro and in vivo. In this study the effect of SB 203580 on allergen-induced airway TNF-α production and inflammatory cell recruitment was investigated in sensitized Brown Norway rats. The allergen-induced increase in bronchoalveolar lavage (BAL) TNF-α was inhibited by SB 203580 at every dose tested (10–100 mg kg−1, p.o.). In contrast, neither ovalbumin-induced eosinophilia or neutrophilia were inhibited by SB 203580 (10–100 mg kg−1, p.o.). In conclusion, SB 203580 inhibits BAL TNF-α production by 95% without inhibiting either antigen-induced airway eosinophilia or neutrophilia. This data suggests that either the residual TNF-α is sufficent to drive allergen-induced inflammatory cell recruitment into the lung or that TNF-α is not involved in allergen-induced inflammatory cell recruitment.
Keywords: p38 Kinase, allergic airway inflammation, tumour necrosis factor-α
Introduction
Asthma is an inflammatory disease of the airways which involves many different inflammatory cell types (e.g. eosinophils) and the release of a large variety of inflammatory mediators including cytokines. There is both clinical and pre-clinical evidence to suggest that cytokines such as TNF-α and IL-1β are involved in the pathogenesis of asthma (Shah et al., 1995; Okada et al., 1995). BAL TNF-α and IL-1β levels are elevated in symptomatic asthmatics and administration of TNF-α to normal subjects mimics some of the clinical features of asthma such as bronchial hyperreactivity and airway inflammation (Broide et al., 1992; Thomas et al., 1995). Furthermore, inhibition of TNF-α, using the TNF fusion protein R045-2081 or soluble TNF-α receptors, prevents the development of allergen-induced airway eosinophilia and hyperreactivity in rats, guinea-pigs and mice (Renzetti et al., 1996; Lukacs et al., 1995). Also, IL-1 receptor antagonists suppress bronchial hyperreactivity, the late bronchoconstrictor response and eosinophil recruitment in allergen challenged guinea-pigs (Watson et al., 1993; Okada et al., 1995).
The p38 kinases are members of the mitogen activated protein (MAP) kinases which are important signal transducers. They modulate the production of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, IL-8) and other mediators such as nitric oxide (Lee & Young 1996; Matsumoto et al., 1998; Badger et al., 1998). Recently, a series of pyridinyl-imidazole compounds, including SB 203580 and SB 220025, have been shown to selectively inhibit p38 kinases and the in vitro production of IL-1β and TNF-α (Lee et al., 1994). SB 203580 inhibits lipopolysaccharide (LPS) induced plasma TNF production, endotoxic shock and displays anti-arthritic activity in mice and rats (Badger et al., 1996). SB 220025 is reported to inhibit angiogenesis and to prevent the progression of an established arthritic response in mice (Jackson et al., 1998). These findings suggest that p38 kinases are involved in several inflammatory processes and are potential therapeutic targets for the treatment of asthma.
The p38 kinases are present in the lungs of humans and rats (Wang et al., 1997; Birrell et al., 1998). The modulation of TNF-α production has been used as an indicator of p38 kinase inhibition in vivo (Badger et al., 1996). We have demonstrated that SB 203580 does not inhibit ovalbumin-induced airway eosinophilia in the mouse (Escott et al., 1998). However, inhibition of airway TNF-α was not demonstrated in this study. Therefore, the aim of this study was to investigate the effect of SB 203580 on both airway TNF-α production and inflammatory cell number following aerosolized ovalbumin challenge in sensitized Brown Norway rats.
Methods
Female Brown Norway rats (180–200 g) were obtained from Harlan, U.K. and allowed free access to food and water. All procedures were carried out according to strict U.K. Home Office Guidelines (Animals Scientific Procedures Act 1986). In this study, the doses, route (p.o.) and times of administration of the p38 kinase inhibitor, SB 203580 were chosen based on those used in the published literature which inhibited acute inflammatory responses such as serum LPS induced TNF-α release and chronic inflammatory diseases such as collagen or adjuvant-induced arthritis in rats and mice (Badger et al., 1996).
Ovalbumin-induced BAL TNF-α production in sensitized rats
Initially, a pilot study was carried out to identify an appropriate time point to evaluate the effect of SB 203580 on BAL TNF-α production following aerosolized ovalbumin challenge in sensitized Brown Norway rats. Then the effect of SB 203580 on this cytokine response was investigated.
Brown Norway rats were sensitized on days 0, 12 and 21 with ovalbumin (100 μg) administered intraperitoneally (i.p.) with aluminium hydroxide (100 mg) in saline (1 ml). On day 28, rats were challenged with aerosolized ovalbumin (1% ovalbumin in saline for 30 min) in a chamber using a deVilbiss Pulmosonic nebulizer (deVilbiss Healthcare, London, U.K.). BAL was performed at various time points (1–48 h) after ovalbumin challenge by flushing the airways with two aliquots (2×10 ml kg−1) of RPMI 1640 medium for 30 s per aliquot. A total of 5 ml of BAL fluid was collected from each rat and centrifuged at 200×g, 4°C for 10 min. The cell free supernatant was removed, frozen at −20°C and assayed for TNF-α levels using a commercially available rat TNF-α ELISA kit from Genzyme, U.K. (detection limit 12.5 pg ml−1). Figure 1 shows the BAL TNF-α response to aerosolized ovalbumin challenge in sensitized rats. The peak increase in TNF-α level, observed 1 h after aerosolized ovalbumin challenge, was selected to investigate the effect of SB 203580.
Figure 1.

Time course of ovalbumin induced TNF-α production in BAL fluid. BAL TNF-α levels were measured at several time points (1=48 h) after aerosolized ovalbumin challenge (1% for 30 min) in sensitized Brown Norway rats. Group size was 12 with results expressed as mean±s.e.mean. *P<0.05 compared to the unchallenged group.
In order to investigate the effect of SB 203580 on ovalbumin-induced BAL TNF-α, vehicle (1% carboxymethylcellulose in distilled water, 2 ml kg−1) or SB 203580 (10–100 mg kg−1) were administered orally 30 min prior to ovalbumin challenge. One hour after the end of challenge, rats were euthanized with sodium pentobarbitone (200 mg kg−1, i.p.), the trachea was cannulated, BAL performed and BAL TNF-α levels measured as described above.
Ovalbumin-induced airway inflammation in sensitized rats
The time points used to measure inflammatory cell recruitment in the airways of rats were chosen to observe peak airway eosinophilia and were based on previous studies performed and optimized in-house and those seen in the published literature (Underwood et al., 1997). In order to evoke an allergic airway inflammatory response in Brown Norway rats, rats were sensitized and challenged as outlined above. Vehicle (1% carboxy methylcellulose in distilled water, 2 ml kg−1) or SB 203580 (10–100 mg kg−1) were administered orally 30 min before and 4 h after challenge. Twenty-four hours after antigen challenge, rats were euthanized with sodium pentobarbitone (200 mg kg−1, i.p.) and BAL was performed using RPMI containing 10% foetal calf serum (FCS) as outlined above. Immediately after BAL, lungs were removed and the pulmonary vasculature flushed with RPMI 1640 containing 10% FCS. Cell suspensions were recovered by incubating lung tissue (300 mg) with 10 ml collagenase (20 u ml−1 for 2 h, 60 u ml−1 for 1 h). The recovered cells were filtered (mesh size 70 μm), washed three times and resuspended in a final volume of 1 ml RPMI 1640 containing 10% FCS (Underwood et al., 1997).
Total cell counts were obtained in BAL or lung tissue samples by using an automated cell counter (COBAS Argos, Roche U.K.). Cytospins of these samples were prepared by cytocentrifugation and stained with Wright-Giemsa stain. Differential cell counts (neutrophils, eosinophils, monocytes/macrophages and lymphocytes) were obtained using light microscopy and the percentage of each cell population were determined after counting a total of 200 cells per slide.
Data analysis
Results are expressed as mean±s.e.mean with n=8–12 animals per group. BAL TNF-α levels are expressed as pg ml−1 TNF-α. Inflammatory cell numbers are expressed per ml of BAL fluid or per mg of lung tissue. Inflammatory cell results were analysed using Kruskal–Wallis multiple comparison test. BAL TNF-α was analysed using an ANOVA with a Dunnet's post-hoc test.
Materials
SB 203580 [4-(4-fluorophenyl)-2-(4-methylsulphnyl phenyl)-5-(4-pyridyl)imidazole] was synthesized by Aventis Pharma (Dagenham Research Centre) and all other materials were purchased from Sigma (Poole, U.K.) except for: aluminium hydroxide from Prolabo (Fontenay, France), sodium pentobarbitone (Euthatal) from Rhone Merieux (Harlow, U.K.), RPMI 1640 and foetal calf serum from Gibco (Paisley, U.K.).
Results
Effect of SB 203580 on BAL TNF-α production in Brown Norway rats
Aerosolized ovalbumin challenge produced a time-dependent increase in BAL TNF-α levels and the peak increase was observed 1 h after the end of challenge (Figure 1). Therefore, this time point was selected to investigate the effect of SB 203580. Figure 2 shows that there was a significant increase in BAL TNF-α 1 h after aerosolized ovalbumin challenge and SB 203580 significantly (P<0.05) inhibited BAL TNF-α levels at all of the doses tested (10–100 mg kg−1).
Figure 2.
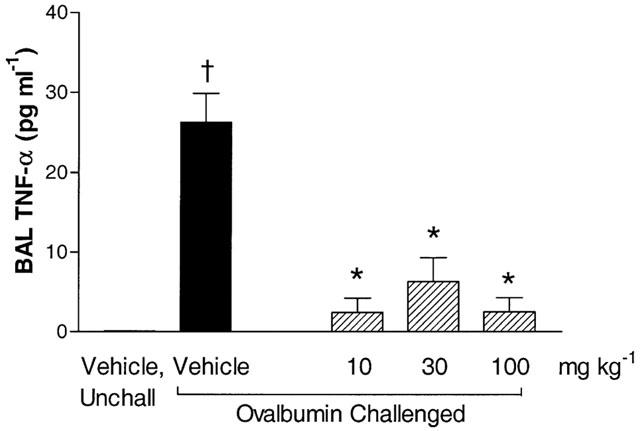
The effect of SB 203580 on BAL TNF-α levels in sensitized Brown Norway rats. SB 203580 (10-100 mg kg−1) or vehicle (2 ml kg−1) were administered orally 30 min prior to challenge and BAL TNF-α production was measured 1 h after aerosolized ovalbumin challenge. Group size was 12 with results expressed as mean±s.e.mean. †P<0.05 compared to unchallenged (unchall) group, pretreated with vehicle. *P<0.05 compared to challenged group, pretreated with vehicle.
Effect of SB 203580 on BAL and lung tissue inflammatory cell recruitment
Aerosolized ovalbumin challenge produced a significant increase in BAL fluid and lung tissue eosinophil numbers at 24 h (Figure 3a and Table 1). A significant increase in BAL fluid, but not lung tissue, neutrophil numbers was observed after ovalbumin challenge (Figure 3b and Table 1). No changes in lung tissue monocyte/macrophage cell numbers were observed following ovalbumin challenge (Table 1).
Figure 3.
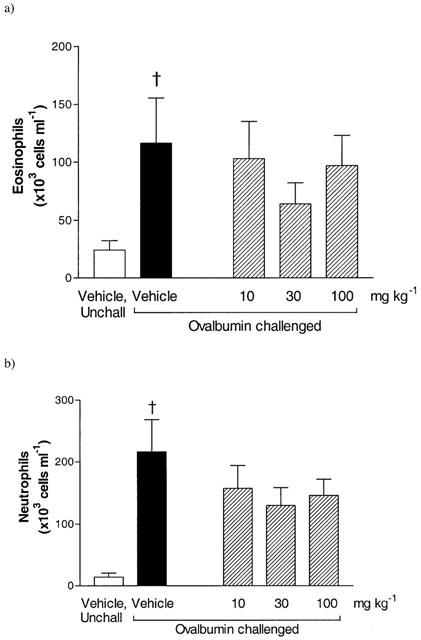
Effect of SB 203580 (10–100 mg kg−1, p.o.) on BAL eosinophils (a) and neutrophils (b) 24 h after aerosolized ovalbumin challenge (1% for 30 min) in sensitized Brown Norway rats. Group size was 10 with results expressed as mean±s.e.mean. †P<0.05 compared to unchallenged (unchall) group, pretreated with vehicle.
Table 1.
Effect of SB 203580 on lung tissue inflammatory cell number

Oral administration of SB 203580 (10–100 mg kg−1) had no significant effect on airway eosinophilia in comparison to the vehicle treated, challenged group (Figure 3a and Table 1). No reduction in BAL neutrophilia was observed after administration of SB 203580. (Figure 3b). In the lung tissue the basal numbers of neutrophils were not altered by pretreatment with SB 203580 (Table 1). The basal number of BAL monocytes/macrophages were unaffected by SB 203580 (unchallenged, vehicle 443.8±36.2: challenged, vehicle 414.1±29.7: challenged, SB 203580 100 mg kg−1 381.5±41.8×103 cells ml−1). However, SB 203580 produced a dose-related increase in lung tissue monocyte/macrophage cell numbers (Table 1).
Discussion
Previous in vivo studies have demonstrated anti-inflammatory activities following oral administration of the p38 kinase inhibitor, SB 203580. SB 203580 inhibits systemic TNF-α production induced by LPS and also displays anti-arthritic activity in both rats and mice (Badger et al., 1996). However, there are no published reports to date which investigate the effect of SB 203580 on TNF-α production and inflammatory cell recruitment in an in vivo rat model of allergic lung inflammation.
The inhibition of airway TNF-α production by SB 203580 in allergen challenged, sensitized Brown Norway rats suggests a role for p38 kinase in allergen induced airway TNF-α production. Furthermore, these results confirm previous findings that SB 203580 can modulate TNF-α production in vivo. Similar doses of SB 203580 have been shown to inhibit LPS-induced TNF-α release in the BAL fluid of mice and the plasma of both rats and mice in vivo (Escott et al., 1998; Birrell et al., 1998; Badger et al., 1996). This data demonstrates that both rat and mouse p38 isoforms are sensitive to inhibition by SB 203580.
The inability of SB 203580 to inhibit allergen-induced airway eosinophilia was unexpected since the inhibition of TNF-α activity is reported to reduce eosinophil recruitment in murine and rat airways evoked by allergen (Lukacs et al., 1995; Renzetti et al., 1996). These published reports used the soluble TNF receptor (sTNFr-Fc) or the TNF-α fusion protein (Ro 45-2081) to block TNF-α activity. In mice sensitized to SEA, treatment with the sTNFr-Fc only partially inhibits BAL eosinophilia and neutrophilia even though this treatment effectively neutralizes TNF-α in vivo (Lukacs et al., 1995). Similarly, Ro 45-2081 only partially inhibits ovalbumin-induced airway eosinophilia in guinea-pigs and rats (Renzetti et al., 1996). In our study, it is possible that the low levels of TNF-α observed after treatment with SB 203580 may be sufficient to sustain an inflammatory response, and that an inhibitory effect on airway inflammation may only be observed when TNF-α is inhibited by over 95%. It is possible that the residual TNF-α which is not inhibited by SB 203580 may be controlled by an alternative signal transduction pathways. For example, both p38 and extracellular signal regulated kinase (ERK) are involved in macrophage LPS-induced TNF-α production in vitro (Ajizian et al., 1999).
In mice, SB 203580 inhibited BAL neutrophilia at the highest dose tested (100 mg kg−1) suggesting that p38 kinases may be involved in neutrophil recruitment into the airway lumen in vivo (Escott et al., 1998). In fact, SB 203580 prevents the release of the neutrophil chemoattractant, IL-8 from cytokine-stimulated human bronchial epithelial cells in vitro (Matsumoto et al., 1998). Furthermore, SB 203580 (100 mg kg−1) has been shown to inhibit airway neutrophilia in BAL fluid, 4 h after aerosolized LPS in rats (Birrell et al., 1998). However, as the inhibitory effect was not dose-dependent and was only observed at the highest dose of SB 203580 (100 mg kg−1) a non-specific effect, not related to p38 kinase inhibition, cannot be ruled out. The fact that airway neutrophilia was unaffected by SB 203580 in this study in rats provides further evidence against a role for p38 in allergen-induced neutrophilia. SB 203580 produced a dose-dependent rise in rat lung tissue monocyte/macrophage cell numbers. The reason for this increase is unclear, as most published reports have shown an inhibition of macrophage function with SB 203580 treatment in vitro. For example, SB 203580 inhibits LPS-induced TNF-α and IL-1β production in human monocytes in vitro (Lee et al., 1994).
In summary, inhibition of p38 kinase with SB 203580 reduces airway ovalbumin-induced TNF-α production suggesting a role for p38 kinase in allergen induced TNF-α production in rats. However, this study does not support a role for p38 kinase in the development of ovalbumin-induced eosinophilia in rat airways. The development of small molecules which inhibit TNF-α production by >95% may help to further elucidate the role of TNF-α in allergen induced airway inflammatory cell recruitment.
Acknowledgments
Many thanks to Ian McLay and John Souness for their technical and intellectual contribution to this work. We would also like to acknowledge the technical assistance of Sarah McMillan, Kerryn McCluskie, Jonathon Hunt, Rebecca Reeves, Colleen Taylor and Paul Woodman.
Abbreviations
- ANOVA
analysis of variance
- BAL
bronchoalveolar lavage
- FCS
foetal calf serum
- LPS
lipopolysaccharide
- MAP
mitogen acitivated protein
- TNF-α
tumour necrosis factor-α
References
- AJIZIAN S.J., ENGLISH B.K., MEALS E.A. Specific inhibitors of p38 and extracellular signal-regulated kinase and mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-gamma. J. Infect. Dis. 1999;179:939–944. doi: 10.1086/314659. [DOI] [PubMed] [Google Scholar]
- BADGER A.M., BRADBEER J.N., VOTTA B., LEE J.C., ADAMS J.L., GRISWOLD D.E. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxic shock and immune function. J. Pharmacol. Exp. Ther. 1996;279:1453–1461. [PubMed] [Google Scholar]
- BADGER A.M., COOK M.N., LARK M.W., NEWMAN-TARR T.M., SWIFT B.A., NELSON A.H., BARONE F.C., KUMAR S. SB 203580 inhibits p38 mitogen-activated protein kinase, nitric oxide production and inducible nitric oxide synthase in bovine cartilage-derived chondrocytes. J. Immunol. 1998;161:467–473. [PubMed] [Google Scholar]
- BIRRELL M., HADDAD E.-B., MCCLUSKIE K., HELE D., PHIPPS S., WEBBER S.E., FOSTER M., BELVISI M.G. Effect of the p38 kinase inhibitor, SB 203580, in a model of airway inflammation. Br. J. Pharmacol. 1998;125:89P. [Google Scholar]
- BROIDE D.H., LOTZ M., CUOMO A.J., COBURN D.A., FEDERMAN E.C., WASSERMAN S.I. Cytokines in symptomatic asthma airways. J. Allergy Clin. Immunol. 1992;89:958–967. doi: 10.1016/0091-6749(92)90218-q. [DOI] [PubMed] [Google Scholar]
- ESCOTT K.J., BIRRELL M.A., MCCLUSKIE K., BELVISI M.G., MCMILLAN S.J., WEBBER S.E., SARGENT C.A. Effect of SB 203580 on murine airway inflammation. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:R329. [Google Scholar]
- JACKSON J.R., BOLOGNESE B., HILLEGASS L., KASSIS S., ADAMS J., GRISWOLD D.E., WINKLER J.D. Pharmacological effects of SB 220025, a selective inhibitor of p38 mitogen-activated protein kinase, in angiogenesis and chronic inflammatory disease models. J. Pharmacol. Exp. Ther. 1998;284:687–692. [PubMed] [Google Scholar]
- LEE J.C., LAYDON J.T., MCDONNELL P.C., GALLAGHER T.F., KUMAR S., GREEN D., MCNULTY D., BLUMENTHAL M.J., HEYS J.R., LANDVATTER S.W., STRICKLER J.E., MCLAUGHLIN M.M., SIEMENS I.R., FISHER S.M., LIVI G.P., WHITE J.R., ADAMS J.L., YOUNG P.R. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- LEE J.C., YOUNG P.R. Role of CSBP/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J. Leukocyte. Biol. 1996;59:152–157. doi: 10.1002/jlb.59.2.152. [DOI] [PubMed] [Google Scholar]
- LUKACS N.W., STRIETER R.M., CHENSUE S.W., WIDMER M., KUNKEL S.L. TNF-α mediates recruitment of neutrophils and eosinophils during airway inflammation. J. Immunol. 1995;154:5411–5417. [PubMed] [Google Scholar]
- MATSUMOTO K., HASHIMOTO S., GON Y., NAKAYAMA T., HORIE T. Proinflammatory cytokine-induced and chemical mediator-induced IL-8 expression in human bronchial epithelial cells through p38 mitogen-activated protein kinase-dependent pathway. J. Allergy Clin. Immunol. 1998;101:825–831. doi: 10.1016/S0091-6749(98)70311-2. [DOI] [PubMed] [Google Scholar]
- OKADA S., INOUE H., YAMAUCHI K., LIJIMA H., OHKAWARA Y., TAKISHIMA T., SHIRATO K. Potential role of interleukin-1 in allergen-induced late asthmatic reactions in guinea pigs: Suppressive effect of interleukin-1 receptor antagonist on late asthmatic reaction. J. Allergy Clin. Immunol. 1995;95:1236–1245. doi: 10.1016/s0091-6749(95)70081-1. [DOI] [PubMed] [Google Scholar]
- RENZETTI L.M., PACIOREK P.M., TANNU S.A., RINALDI N.C., TOCKER J.E., WASSERMAN M.A., GATER P.R. Pharmacological evidence for tumor necrosis factor as a mediator of allergic inflammation in the airways. J. Pharmacol. Exp. Ther. 1996;278:847–853. [PubMed] [Google Scholar]
- SHAH A., CHURCH M.K., HOLGATE S.T. Tumour necrosis factor alpha: a potential mediator of asthma. Clin. Exp. Allergy. 1995;25:1038–1044. doi: 10.1111/j.1365-2222.1995.tb03249.x. [DOI] [PubMed] [Google Scholar]
- THOMAS P.S., YATES D.H., BARNES P.J. Tumor necrosis factor-alpha increases airway responsiveness and sputum neutrophilia in normal human subjects. Am. J. Respir. Crit. Care Med. 1995;152:76–80. doi: 10.1164/ajrccm.152.1.7599866. [DOI] [PubMed] [Google Scholar]
- UNDERWOOD S.L.U., RAEBURN D., LAWRENCE C., FOSTER M., WEBBER S., KARLSSON J.-A. RPR 106541, a novel, airways-selective glucocorticoid: effects against antigen-induced CD4+ T lymphocyte accumulation and cytokine gene expression in the Brown Norway rat lung. Br. J. Pharmacol. 1997;122:439–446. doi: 10.1038/sj.bjp.0701398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG X.S., DIENER K., MANTHEY C.L., WANG S.-W., ROSENZWEIG B., BRAY J., DELANEY J., COLE C.N., CHAN-HUI P.-Y., MANTLO N., LICHENSTEIN H.S., ZUKOWSKI M., YAO Z. Molecular cloning and characterization of a novel p38 mitogen-activated protein kinase. J. Biol. Chem. 1997;272:23668–23674. doi: 10.1074/jbc.272.38.23668. [DOI] [PubMed] [Google Scholar]
- WATSON M.L., SMITH D., BOURNE A.D., THOMPSON R.C., WESTWICK J. Cytokines contribute to airway dysfunction in antigen-challenged guinea pigs: Inhibition of airway hyperreactivity, pulmonary eosinophil accumulation and tumor necrosis factor generation by pretreatment with an interleukin-1 receptor antagonist. Am. J. Respir. Crit. Care Med. 1993;8:365–369. doi: 10.1165/ajrcmb/8.4.365. [DOI] [PubMed] [Google Scholar]