

Abstract
We have studied the antagonist action of prazosin and KMD-3213 in a constitutively active mutant of the human alpha-1a adrenoceptor in which Ala271 was substituted to Thr and was expressed in CHO cells. Inverse agonism was characterized by up-regulation of receptor density, a decrease in basal GTPγS binding, and a reduction in basal inositol-1,4,5-trisphosphate (IP3) level.
According to the above criteria, prazosin acted as an inverse agonist, whilst KMD-3213 behaved as a neutral antagonist.
Compared with the wild-type receptor, mutant receptor exhibited single affinity sites for [3H]-prazosin, [3H]-KMD and the non-radioactive ligands tested, and displayed significantly higher affinities for several agonists but not for the two antagonists.
Administration of KMD-3213 to prazosin-treated CHO cells expressing the mutant receptor reversed the inverse agonism of prazosin resulting in rapid increases in cellular IP3, in intracellular [Ca2+] and in the rate of extracellular acidification.
These results indicated that a neutral antagonist can reverse the action of an inverse agonist at the receptor site. The distinct properties of inverse agonist and neutral antagonist in affecting receptor function may be important for the clinical use of such antagonists.
Keywords: Alpha-1 adrenoceptor, mutant, constitutive activity, inverse agonist, neutral antagonist
Introduction
It has been shown that many G protein-coupled receptors (GPCRs) may exist in a spontaneously active form in the absence of an agonist (Costa et al., 1992; Kenakin, 1996; Lefkowitz et al., 1993; Mewes et al., 1993; Milano et al., 1994). This phenomenon has been termed ligand-independent or constitutive receptor activation and has been most readily observed in cell lines in which receptors are overexpressed or mutated (Barker et al., 1994; Bond et al., 1995; Chidiac et al., 1994; Newman-Tancredi et al., 1997). This situation may be explained by a two-state receptor model in which GPCRs exist in an equilibrium between two conformations, an active form (R*) and an inactive form (R) (Lefkowitz et al., 1993; Samama et al., 1993). Based on this model, agonists shifts the equilibrium to the active conformation (R*), thereby increasing receptor activity, whereas some antagonists, called inverse agonists lead preferentially to the inactive conformational state (R), resulting in a reduction in basal receptor activity. In addition, there is another type of antagonist, a neutral antagonist, which is believed to bind equally to both R* and R without affecting the equilibrium. Thus, such neutral antagonist can inhibit the effects of both agonists (reducing the receptor signal) and inverse agonists (thereby increasing receptor signalling in the concurrent presence of an inverse agonist). In constitutively active receptor systems, treatment with an inverse agonist has been forced to increase receptor density, to alter basal second messenger levels (Lee et al., 1997; MacEwan & Milligan, 1996; Smit et al., 1996) and to reduce G protein activity (Newman-Tancredi et al., 1997; Shryock et al., 1998).
The alpha-1 adrenoceptor (AR) family comprises at least three cloned subtypes, alpha-1a, -1b and -1d ARs (Minneman et al., 1994; Muramatsu et al., 1998). These subtypes have distinctive pharmacological characteristics with subtype-selective ligands (Graham et al., 1996). It has been previously reported that a series of antagonists can act as inverse agonists for the alpha-1a AR and alpha-1b AR subtypes (Hwa & Perez, 1996; Lee et al., 1997). A recent study has reported that Recordati compounds, such as REC 15/3039, REC 15/2739 and REC 15/3011, act as neutral antagonists at the alpha-1a AR but as inverse agonists at the alpha-1b AR (Rossier et al., 1999). However, further studies are warranted to examine the interactions of neutral antagonist and inverse agonist with the alpha-1 AR subtypes.
Here, we have constructed a constitutively active mutant of the alpha-1a AR, by changing Ala271 to Thr in the third intracellular loop, and have examined the action of prazosin and KMD-3213 in terms of either inverse agonist or neutral antagonist activities. We also measured the binding affinities of a number of ligands for the wild-type and mutant receptors, employing [3H]-prazosin and [3H]-KMD3213 as radioligands, and have analysed the results based on a two-state receptor model. We have further investigated the interactions of prazosin and KMD-3213 in regulating the signalling properties of the mutant receptor system.
Methods
Materials
Alpha minimum essential medium, foetal bovine serum, G418 and Lipofectamine were from GIBCO BRL (Grand Island, NY, U.S.A.); phentolamine HCl, prazosin HCl, (−)-noradrenaline HCI, methoxamine HCl, phenylephrine HCl, oxymetazoline HCl, GTPγS and GDP from Sigma (St. Louis, U.S.A.); (−)-(R)-1-(3-hydroxypropyl)-5-[2-[[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl]amino]propyl] indoline-7-carboxamide (KMD-3213) from Kissei Pharmaceutical Co. td. (Matsumoto, Japan); 2-(2,6-dimethoxyphenoxyethyl)aminomethyl-1,4-benzodioxane hydrochloride (WB4101) and 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4,5] decane-7,9-dione dihydrochloride (BMY7378) from Research Biochemicals Inc. (Natick, U.S.A.); alpha-ethyl-3,4,5-trimethoxy-alpha-(3-((2-(2-methoxy-phenoxy)ethyl)-amino)-propyl) benzeneacetonitrile fumarate (HV723) from Hokuriku Seiyaku (Katsuyama, Japan); tamsulosin from Yamanouchi Pharmaceutical Co. Ltd. (Tsukuba, Japan); acetoxymethyl ester of fura2 (fura2-AM) from Dojin (Kumamoto, Japan) [3H]-prazosin (2.59∼3.22 TBq mmol−1), inositol-1,4,5-trisphosphate (IP3) [3H]-radioreceptor assay kit, [3H]-myoinositol (0.37∼0.925 TBq mmol−1) and [35S]-GTPγS (46.2 TBqmmol−1) from Dupont–New England Nuclear Inc. (Boston, U.S.A.); [3H]-KMD-3213 (1.8∼1.9 TBq mmol−1) from Amersham (U.K.).
Construction of wild-type and mutated alpha-1a AR plasmids
The human alpha-1a AR cDNA (Taniguchi et al., 1999) was constructed in an expression vector, pCR3 (Invitrogen). The codon substitution was produced by a modified site-specific PCR-mediated mutagenesis technique (Kunkel, 1985). The sequence of the mutagenic primer was 5′-ACTAAGACGTTGGGCATTGTGG in which the underlined nucleotides indicate the Ala to Thr mutation at residue 271. The PCR products were subcloned into pCR3. The mutation was confirmed by DNA sequencing.
Transfection and cell culture
The wild-type and mutant plasmids were stably transfected into Chinese hamster ovary (CHO) cells using Lipofectamine according to the manufacturer's protocol. The stable cell lines expressing alpha-1a AR were established by G418 selection (400 μg ml−1) and were screened for alpha-1a AR expression by [3H]-prazosin binding. Cells were maintained in Alpha Minimum Essential medium containing 10% foetal bovine serum and 200 μg ml−1 G418 at 37°C in a humidified atmosphere of 5% CO2/95% O2.
Membrane preparations
CHO cells grown to confluence were harvested and homogenized in ice-cold assay buffer (50 mM Tris-HCl, 1 mM EDTA, pH 7.4) using an Ultraturax homogenizer and were centrifuged at 1700×g for 10 min. The supernatant was then centrifuged at 48,000×g for 30 min, and the resulting pellet was resuspended in the appropriate binding buffer (see below). All procedures were performed at 4°C.
Radioligand binding
For binding saturation analysis, CHO cell membranes (20–60 μg of protein) were incubated for 40 min at 30°C with five concentrations of [3H]-prazosin or [3H]-KMD in a total volume of 1 ml. Nonspecific binding was defined as binding in the presence of 1 μM tamsulosin. For binding competition analysis, membranes were incubated with 200 pM [3H]-prazosin or 100 pM for [3H]-KMD and unlabelled drugs (six concentrations in one point per one log unit) for 40 min at 30°C. Reactions were terminated and evaluated as described previously (Taniguchi et al., 1999).
To determine the GTPγS binding activity, membranes (30–80 μg protein) were preincubated with antagonists for 30 min at 22°C in 600 μl of 50 mM Tris-HCl, pH 7.4, containing (in mM): EGTA 0.2, MgCl2 5 and NaCl 100, GDP 10 μM and dithiothreitol 0.1. [35S]-GTPγS was then added to the reaction mixture to make up 150 pM [35S]-GTPγS in 800 μl final volume and incubated further for 60 min at 22°C. Nonspecific binding was defined as binding in the presence of 10 μM GTPγS in the incubation. Assays were terminated and evaluated as described above.
Assay for inositol phosphates accumulation
CHO cells expressing mutant alpha-1a AR, grown in a 12-well plate (4×105 cells/well), were labelled with [3H]-myoinositol at 185 kBq ml−1 (5 μCi ml−1) and treated with various drugs at 100 nM for 16 h. Then, cells were washed and incubated with Krebs–HEPES buffer ((in mM): NaCl 110, KCl 4.5, CaCl2 1.3, KH2PO4 1.2, MgSOK4 1.2, NaHCO3 25, glucose 11.7, HEPES 5, pH 7.4) with 10 mM LiCl in the presence or absence of drugs for 2 h. The reaction was stopped by the addition of ice-cold 3% (v v−1) perchloric acid. The sample was neutralized with 200 mM KOH/50 mM Tris and was centrifuged. The resulting supernatant was applied to 1 ml of packed AG1-X8 (100–200 mesh, chloride form, Bio-Rad). Total inositol phosphates were eluted with 1 ml of 1 N HCl and were estimated in a liquid scintillation counter.
IP3 measurement
Cells were seeded in 24-well plates (2–4×105 cells per well) 48 h before experiments and were washed once with the serum-free medium and incubated in the serum-free medium for 30 min at 37°C. Then, cells were challenged by drugs for the appropriate time, and were lysed with 0.4 ml of ice-cold 0.4 M perchloric acid. The lysate was neutralized with 2 M KOH, 0.1 M HEPES and IP3 content was assayed using a [3H]-radioreceptor assay kit (NEN) according to the manufacturer's specifications. No LiCl was used in this assay.
Estimation of intracellular [Ca2+]
Cells on a coverslip were loaded with 3 μM fura2-AM for 30 min, rinsed and mounted in a quartz cuvette of CAF-110 Ion Analyzer (Nihon Bunko, Tokyo, Japan). Ratiometric fluorescence (excitation 340/380 nm, emission 500 nm) measurement were continuously taken during experiments.
Measurement of extracellular acidification rate
Extracellular acidification rate was measured in a microphysiometer as described previously (Taniguchi et al., 1999).
Data analysis
Nonlinear regression analysis of saturation, competition binding assay and dose-response data was performed using Prism (GraphPAD Software, San Diego, CA, U.S.A.). Data are presented as the mean±standard error mean (s.e.mean). Differences between groups were analysed using a one-way ANOVA, to obtain a P value.
Results
Up-regulation of the alpha-1a ARs and reduction of inositol phosphates
As shown in Table 1, we employed two CHO clones which stably expressed the wild-type and mutant alpha-1a AR, respectively, at comparable receptor densities of about 1100 fmol mg−1 of membrane protein. As shown in Figure 1A, sustained treatment of the CHO clone expressing the mutant alpha-1a AR with a variety of antagonists (100 nM) for 72 h resulted in an up-regulation of the receptor density caused by all antagonists except KMD-3213. In contrast, the equivalent treatment of the CHO clone expressing the wild-type alpha-1a AR failed to produce any significant alteration in receptor levels. The degree of up-regulation varied amongst the antagonists: 3–4 fold for prazosin, BMY7378 and WB4101, 2 fold or less for phentolamine and HV723, and no increase in the case of KMD-3213 compared with the densities of the mutant receptor without antagonist treatment. We next tested these antagonists for their ability to reduce intracellular inositol phosphates in the CHO cells expressing the mutant alpha-1a AR. As shown in Figure 1B, treatment with a line of antagonists lead to various reduction of inositol phosphates level; prazosin induced the strongest reduction (40%), and KMD-3213 and BMY7378 exhibited small reduction (less than 10%). Since the concentrations of antagonist in the experiments were fixed at 100 nM, the variation of up-regulation and inositol phosphates reduction may reflect in part the difference in the affinities of the antagonist. However, because of their marked differences in affecting receptor density and basal inositol phosphates level irrespective of their high affinities to both the wild-type and mutant receptors (Table 1), we selected prazosin and KMD-3213 for a further study of their potent inverse agonist and/or neutral antagonist activity.
Table 1.
Binding affinity of wild-type and mutant alpha-1a AR
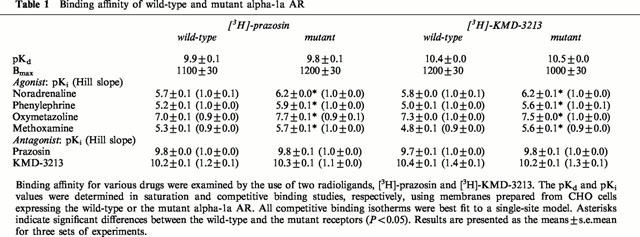
Figure 1.
Up-regulation of receptor density and reduction of inositol phosphates accumulation by a range of alpha-1 AR antagonists. In (A) sustained treatment with antagonists resulted in up-regulation of the mutant receptor but not the wild-type receptor. CHO clones were treated for 72 h with no drug or with 100 nM of prazosin, KMD-3213, BMY7378, WB4101, phentolamine or HV723. Cells were extensively washed to remove the antagonist and receptor density was estimated in the binding study with [3H]-prazosin as outlined under Methods. Results are presented as the means±s.e.mean for three sets of experiments. In (B) CHO cells expressing the mutant were treated with 100 nM drugs as in (A) and accumulated inositol phosphates in the presence of LiCl was measured as described in Methods. The value of scintillation counts of inositol phosphates in mock cells without antagonist (8600±100 d.p.m.) was subtracted from those of other preparations. Resulting values were normalized against that in CHO cells expressing the mutant receptor without antagonist treatment (3400±100 d.p.m.). All antagonists except KMD-3213 produced significant (P<0.05) changes in values from that of cells without drug treatment in both assays.
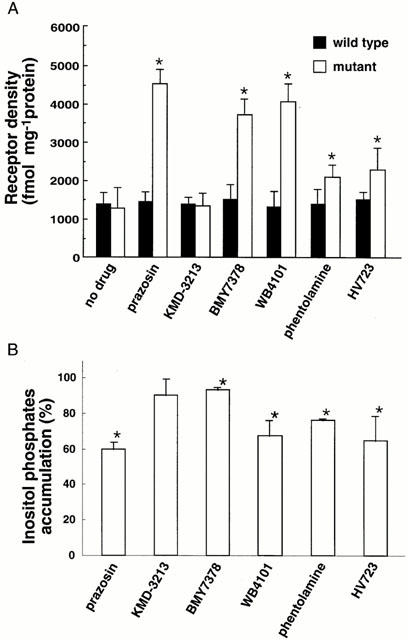
Up-regulation of the alpha-1a ARs by prazosin and KMD-3213
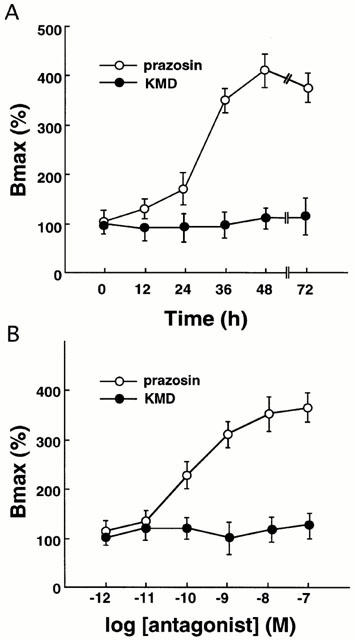
As shown in Figure 2, prazosin on its own caused receptor up-regulation in a time- and concentration-dependent manner. On the other hand, KMD-3213 had no effect on the level of the receptor density by itself (Figure 2) and caused a rightward shift of the concentration-response curve for prazosin (Figure 3A). The pEC50 values for prazosin were 9.6±0.2 and 8.9±0.3 in the absence and presence of 1 nM KMD-3213, respectively. The up-regulation of receptor density caused by 1 nM prazosin was reversed by KMD-3213 in a concentration-dependent manner with pIC50 value of 9.4±0.3 (Figure 3B).
Figure 2.
Increased receptor density in alpha-1a AR mutant by treatment with prazosin but not with KMD-3213 in time- (A) and concentration- (B) dependent manner. (A) CHO cells expressing the alpha-1a AR mutant were treated with 100 nM prazosin or KMD-3213 for the time indicated and the specific binding of [3H]-prazosin were measured as outlined under Methods. (B) CHO cells expressing the mutant alpha-1a AR was treated for 72 h with various concentrations of prazosin or KMD-3213 and the specific binding of [3H]-prazosin was measured. Data represent mean±s.e.mean from three experiments.
Figure 3.
Antagonism of prazosin-induced upregulation by KMD-3213 in mutant alpha-1a AR. (A) CHO cells expressing the mutant alpha-1a AR were treated for 48 h with various concentrations of prazosin in the absence or presence of 1 nM KMD-3213, and the specific binding of [3H]-prazosin was measured. Bmax values were expressed as per cent of the Bmax in the membrane from the cells treated with no drug and represented mean±s.e.mean from five independent experiments. (B) CHO cells expressing the mutant alpha-1a AR were co-incubated for 48 h with 1 nM prazosin in the presence of various concentrations of KMD-3213 and the specific binding of [3H]-prazosin was measured. The up-regulation of the receptor density is expressed as per cent of the up-regulation in the membrane from the cells treated with prazosin only and represented mean±s.e.mean from four experiments.
Basal GTPγS binding
As shown in Figure 4, prazosin reduced the basal [35S]-GTPγS binding to membranes prepared from the CHO clone expressing the mutant alpha-1a AR in a concentration-dependent manner. KMD-3213 did not alter basal GTPγS binding by itself, but caused a rightward shift of the concentration-response curve of prazosin. The pEC50 values for prazosin were 8.64±0.20 and 8.04±0.31 in the absence and presence of 1 nM KMD-3213, respectively. One μM prazosin had no effect on GTPγS binding in the membranes prepared from the CHO clone expressing the wild-type alpha-1a AR (data not shown).
Figure 4.
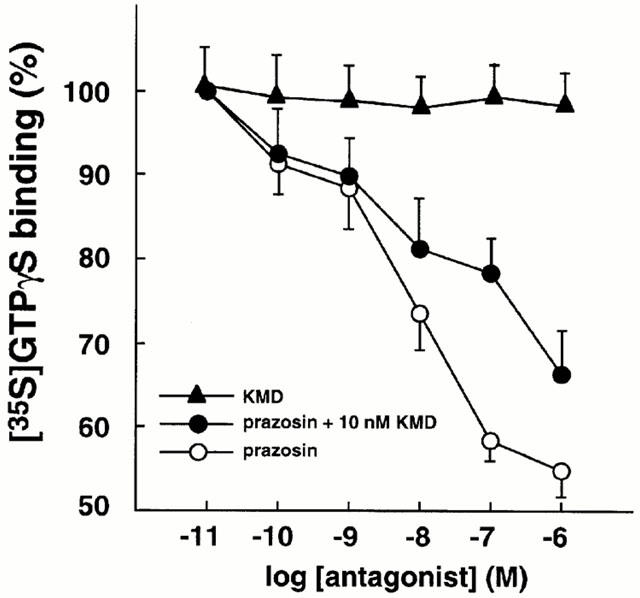
Antagonism of prazosin-induced decrease of [35S]-GTPγS binding activity by KMD-3213 in the mutant alpha-1a AR. Specific binding of [35S]-GTPγS to the membrane from the CHO cells expressing the mutant alpha-1a AR was measured and various concentrations of KMD-3213 or prazosin with or without 10 nM KMD-3213 was added in the reaction mixture as described under Methods. Values were expressed as per cent of the binding obtained in the absence of the drug and are represented mean±s.e.mean from six experiments.
Basal IP3 level
As shown in Figure 5, the CHO clone expressing the mutant alpha-1a AR showed a significant increase in basal IP3 level compared to the wild-type (P<0.05). The basal production of IP3 was significantly reduced by prazosin but not by KMD-3213. When co-administered, KMD-3213, which had no effect on its own, reversed the activity of prazosin to reduce IP3 levels. The equivalent treatment of the CHO clone expressing the wild-type AR by either prazosin or KMD-3213 showed no change in basal IP3 levels. These results indicated that in the mutant alpha-1a AR displaying constitutive activity, prazosin behaved as an inverse agonist while KMD-3213 behaved as a neutral antagonist, reversing the inverse agonist activity of prazosin.
Figure 5.
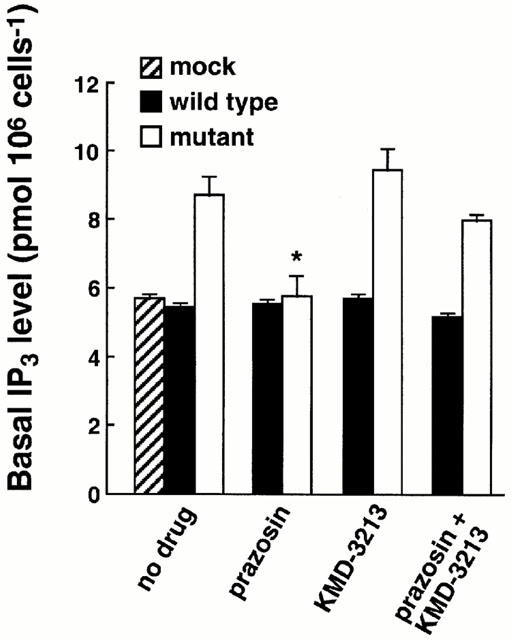
Effects of alpha-1a AR antagonists on the basal level of IP3 production in wild-type and mutant alpha-1a AR. CHO cells expressing the wild-type and mutant alpha-1a AR were treated for 10 min with 10 nM prazosin, 100 nM KMD-3213 or 10 nM prazosin plus 100 nM KMD-3213, and the basal IP3 productions were measured as described under Methods. The asterisk indicates significant reduction of basal IP3 level in prazosin treatment from those in other treatments of mutant alpha-1a AR. Data represent the mean±s.e.mean from three experiments.
Pharmacological characterization of mutant alpha-1a AR
To characterize the mutant alpha-1a AR further, the ligand binding profile was compared with that of the wild-type (Table 1). The binding affinities for prazosin and KMD-3213 in the wild-type and mutant receptors did not differ either as radiolabelled or unlabelled ligands. In contrast, the binding affinities for a number of agonists were 3–10 fold higher in the mutant receptor (P<0.05) compared with the wild-type receptor. This increased affinity of agonists for the mutant receptor was observed using either [3H]-prazosin or [3H]-KMD-3213 as receptor probes (Table 1). All saturation and competitive binding isotherms were best fit to a single-site model.
Effects of KMD-3213 on IP3 production, intracellular [Ca2+] and extracellular acidification rate in prazosin-treated mutant clone
We further investigated the interaction between an inverse agonist prazosin and a neutral antagonist KMD-3213 under conditions where the mutant clone had first been pre-incubated with 10 nM prazosin for 48 h. In such cells, the addition of 100 nM KMD-3213 caused a transient increase in IP3 content and in intracellular [Ca2+] even in the continued presence of prazosin (Figure 6). Next, the extracellular acidification rates were assessed using a microphysiometer to estimate cellular responses through alpha-1a AR activation. As shown in Figure 7, addition of 10 nM KMD-3213 after 48 h preincubation of cells with 10 nM prazosin resulted in a rapid increase in the extracellular acidification rate of the mutant clone in the continued presence of prazosin. KMD-3213 did not induce such increases either in the mutant clone without prazosin pre-treatment or in the wild-type clone irrespective of the pre-treatment with prazosin (Figures 6B, 7 and data not shown). These results demonstrated further speculation that the activity of the neutral antagonist, KMD-3213, reversed the action of the inverse agonist, prazosin, on the mutant receptor.
Figure 6.
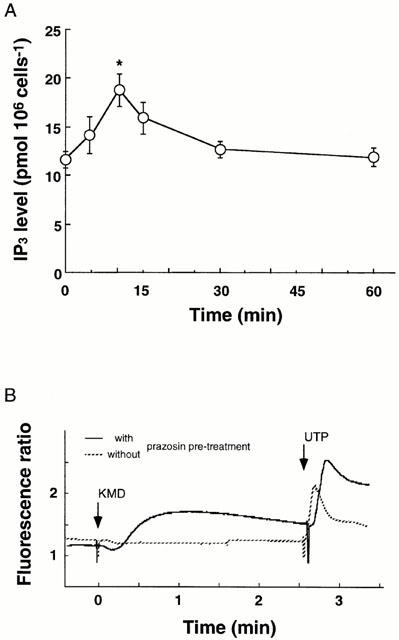
Apparent activation of IP3 production (A) and intracellular Ca2+ recruitment by KMD-3213 in prazosin-pretreated CHO cells expressing the mutant alpha-1a AR. CHO cells expressing the mutant alpha-1a AR were preincubated for 48 h with 10 nM prazosin and then 100 nM KMD-3213 was added under the continued presence of prazosin. Level of IP3 and intracellular [Ca2+] were estimated as described under Methods. In (A) data represent the mean±s.e.mean from three experiments. The asterisk indicates a significant (P<0.05) increase of IP3 from the value at 0 time. In (B) a representative result of three experiments was shown. The response of the mutant clone without the prazosin pretreatment and in the absence of prazosin was depicted with dotted line. At the end of experiments, 10 μM UTP was administered to confirm the ability of the cells to increase intracellular [Ca2+].
Figure 7.
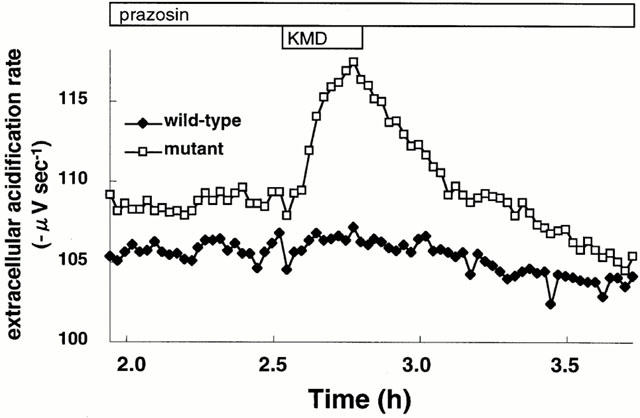
Apparent activation of extracellular acidification by KMD-3213 in prazosin-pretreated CHO cells expressing the mutant alpha-1a AR. CHO cells expressing the wild-type or mutant alpha-1a AR were preincubated for 48 h with 10 nM prazosin. After basal extracellular acidification rate were stable in the microphysiometer as described under Methods, cells were challenged with 10 nM KMD-3213 for 15 min. This is a representative result of six experiments.
Discussion
Constitutive-active mutant of alpha-1a AR
Spontaneous constitutive activity has been reported for wild-type alpha-1 AR (Garcia-Sainz & Torres-Padilla, 1999; Rossier et al., 1999). However, when expressed in CHO cells, the wild-type alpha-1a AR did not display any detectable constitutive activity in the assay systems employed in this study. Therefore, we introduced a substitution of amino acid at 271 position, from Ala to Thr, in the C-terminal region of the third intracellular loop of alpha-1a AR. The resulting receptor mutant was constitutively active as evidenced by an increased basal level of IP3 (Figure 5), upregulation of the receptor level by inverse agonists (Figures 1 and 2) and higher binding affinity for agonists (Table 1) compared with the wild-type receptor. This Ala271 is equivalent with Ala293 in alpha-1b AR for which substitution also resulted in constitutive activation (Kjelsberg et al., 1992). A number of point mutations in this region, which result in constitutive receptor activity, have also been described in other receptors such as the beta-2 AR (Samama et al., 1993), the alpha-2 AR (Ren et al., 1993) and the thyrotropin receptor (Parma et al., 1993). These results suggest that the C-terminal region of the third intracellular loop is probably involved in a common mechanism that regulates the coupling of GPCRs with their respective G-proteins.
Inverse agonists and neutral antagonists of the mutant alpha-1a AR
The conclusion that prazosin is an inverse agonist whereas KMD-3213 is a neutral antagonist, was supported by the three following findings. First, prazosin caused a time- and concentration-dependent up-regulation of receptor density, whereas KMD-3213 did not (Figure 2). Second, prazosin (but not KMD-3213) decreased the basal binding of GTPγS in a concentration-dependent manner (Figure 4). Third, there was a reduction of the basal IP3 level (Figure 5) caused by prazosin but not by KMD-3213. None of these results were observed with the wild-type receptor. Furthermore, KMD-3213 shifted the prazosin-induced response curves to the right in an almost parallel fashion (Figures 3A and 4) and reversed the inverse agonist action of prazosin in a concentration-dependent manner (Figure 3B). The data indicated that the antagonism between prazosin and KMD-3213 was competitive in nature.
As shown in Figure 1, the other antagonists also have inverse agonist activity to some extent. However, there were some discrepancy in two assays, especially in the case of BMY7378 which showed small reduction in basal inositol phosphate level in spite of high up-regulation of receptor density. At present, it is difficult to draw a certain conclusion regarding this discrepancy because their experimental conditions as well as their mechanisms of observed responses were different in the assays. Further investigation is required to determine the definition as inverse agonist of these drugs.
Interaction of prazosin and KMD-3213 at the mutant alpha-1a AR
As shown in Figures 6 and 7, the administration of KMD-3213 to the CHO clone expressing the mutant alpha-1a AR resulted in positive responses of the cells only when the cells had been pretreated with prazosin. In these cases the neutral antagonist, KMD-3213, was able to reverse the action of the inverse agonist, prazosin. The mechanism whereby KMD-3213 reverses the inverse activity of prazosin merits comment. One possibility is that KMD-3213 possesses a weak partial agonist activity which would be evident in up-regulated receptor condition. However, this seems unlikely because of the following two reasons: first that KMD-3213 by itself did not induce any agonist response in both the wild-type- and mutant-AR-expressing cells before prazosin treatment (Figures 2, 4, 5, 6B and 7); and second, that these positive responses were observed in the presence of 10 nM prazosin which should be high enough to suppress a weak partial-agonist activity. Another possibility is that the neutral antagonist competes at a key receptor binding into allowing for the dissociation of the inverse agonist. The neutral antagonist remaining bound to the receptor thereby uncovers the intrinsic activity of the receptor per se, promoting a timed response. It is worth to point out that the time courses of the cellular responses shown in Figures 6 and 7 were generally slower than those seen when induced by noradrenaline; a peak within 30 s for IP3 (Suzuki et al., 2000), around 20 s for intracellular [Ca2+] (Suzuki et al., 2000) or within 4.5 min for extracellular acidification rate (Taniguchi et al., 1999). We are speculating that this is likely to reflect a dissociation time course of prazosin in the interaction with KMD-3213 on the receptor. This result may have important implications for clinical pharmacotherapy. Wherein different antagonists may be selected, some of which do and some of which may not posses inverse agonist activity. The combined use and/or switching of such antagonists in a clinical setting may cause unexpected side effects. Based on the evidence presented here, it may be necessary in the future to establish guidelines for the clinical use of inverse agonists and neutral antagonists.
In conclusion, we have reported that prazosin acts as an inverse agonist and KMD-3213 functions as a neutral antagonist at the alpha-1a AR in which Ala271 was mutated to Thr. In addition, we have indicated the potential clinical impact that might occur with use of inverse agonists and neutral antagonists in combination or in an overlapping manner.
Acknowledgments
This work was supported in part by grants from the Smoking Research Foundation of Japan and from Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan. We would like to appreciate Dr M.D. Hollenberg, University of Calgary, for his editorial assistance.
Abbreviations
- AR
adrenoceptor
- CHO
Chinese hamster ovary
- GPCRs
G protein coupled receptors
- IP3
inositol-1,4,5-trisphosphate
References
- BARKER E.L., WESTPHAL R.S., SCHMIDT D., SANDERS-BUSH E. Constitutively active 5-hydroxytryptamine2C receptors reveal novel inverse agonist activity receptor ligands. J. Biol. Chem. 1994;269:11687–11690. [PubMed] [Google Scholar]
- BOND R.A., LEFF P., JOHNSON T.D., MILANO C.A., ROCKMAN H.A., MCMINN T.R., APPARSUNDARAM S., HYEK M.F., KENAKIN T., ALLEN L.F., LEFKOWITZ R.J. Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the β2-adrenoceptor. Nature. 1995;374:272–276. doi: 10.1038/374272a0. [DOI] [PubMed] [Google Scholar]
- CHIDIAC P., HEBERT T.E., VALIQUETTE M., DENNIS M., BOUVIER M. Inverse agonist activity of β-adrenergic antagonists. Mol. Pharmacol. 1994;45:490–499. [PubMed] [Google Scholar]
- COSTA T., OGINO Y., MUNSON P.J., ONARAN H.O., RODBARD D. Drug efficacy at guanine nucleotide-binding regulatory protein-linked receptors: Thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol. Pharmacol. 1992;41:549–560. [PubMed] [Google Scholar]
- GARCÍA-SÁINZ J.A., TORRES-PADILLA M.E. Modulation of basal intracellular calcium by inverse agonists and phorbol myristate acetate in rat-1 fibroblasts stably expressing alpha1d-adrenoceptors. Febs Lett. 1999;443:277–81. doi: 10.1016/s0014-5793(98)01738-4. [DOI] [PubMed] [Google Scholar]
- GRAHAM R.M., PEREZ D.M., HWA J., PIASCIK M.T. α1-adrenergic receptor subtypes Molecular structure, function, and signaling. Circ. Res. 1996;78:737–749. doi: 10.1161/01.res.78.5.737. [DOI] [PubMed] [Google Scholar]
- HWA J., PEREZ D.M. The unique nature of the serine interactions for α1-adrenergic receptor agonist binding and activation. J. Biol. Chem. 1996;271:6322–6327. doi: 10.1074/jbc.271.11.6322. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. The classification of seven transmembrane receptors in recombinant expression systems. Pharmacol Rev. 1996;48:413–463. [PubMed] [Google Scholar]
- KJELSBERG M.A., COTECCHIA S., OSTROWSKI J., CARON M.G., LEFKOWITZ R.J. Constitutive activation of the α1B-adrenergic receptor by all amino acid substitutions at a single site. J. Biol. Chem. 1992;267:1430–1433. [PubMed] [Google Scholar]
- KUNKEL T.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. U.S.A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE T.W., COTECCHIA S., MILLIGAN G. Up-regulation of the levels of expression and function of a constitutively active mutant of the hamster α1B-adrenoceptor by ligands that act as inverse agonists. Biochem. J. 1997;325:733–739. doi: 10.1042/bj3250733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFKOWITZ R.J., COTECCHIA S., SAMAMA P., COSTA T. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol. Sci. 1993;14:303–307. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- MACEWAN D.J., MILLIGAN G. Inverse agonist-induced up-regulation of the human β2-adrenoceptor in transfected neuroblastoma X glioma hybrid cells. Mol. Pharmacol. 1996;50:1479–1486. [PubMed] [Google Scholar]
- MEWES T., DUTZ S., RAVENS U., JAKOBS K. Activation of calcium currents in cardiac myocytes by empty β-adrenoreceptors. Circulation. 1993;88:2916–2922. doi: 10.1161/01.cir.88.6.2916. [DOI] [PubMed] [Google Scholar]
- MILANO C.A., DOLBER P.C., ROCKMAN H.A., BOND R.A., VENABLE M.E., ALLEN L.H., LEFKOWITZ R.J. Myocardial expression of a constitutively active α1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10109–10113. doi: 10.1073/pnas.91.21.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINNEMAN K.P., THEROUX T.L., HOLLINGER S., HAN C., ESBENSHADE T.A. Selectivity of agonists for cloned alpha 1-adrenergic receptor subtypes. Mol. Pharmacol. 1994;46:929–936. [PubMed] [Google Scholar]
- MURAMATSU I., MURATA S., ISAKA M., PIAO H.L., ZHU J., SUZUKI F., MIYAMOTO S., OSHITA M., WATANABE Y., TANIGUCHI T. Alpha1-adrenoceptor subtypes and two receptor systems in vascular tissues. Life Sciences. 1998;62:1461–1465. doi: 10.1016/s0024-3205(98)00090-3. [DOI] [PubMed] [Google Scholar]
- NEWMAN-TANCREDI A., CONTE C., CHAPUT C., SPEDDING M., MILLAN M. Inhibition of the constitutive activity of human 5-HT1A receptors by the inverse agonist, spiperone but not the neutral antagonist WAY 100,635. Br. J. Pharmacol. 1997;120:737–739. doi: 10.1038/sj.bjp.0701025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARMA J., DUPREZ L., VAN SANDE J., COCHAUX P., GERVY C., MOCKEL J., DUMONT J., VASSART G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365:649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- REN Q., KUROSE H., LEFKOWITZ R.J., COTECCHIA S. Constitutively active mutants of the α2-adrenergic receptor. J. Biol. Chem. 1993;268:16483–16487. [PubMed] [Google Scholar]
- ROSSIER O., ABUIN L., FANELLI F., LEONARDI A., COTECCHIA S. Inverse agonism and neutral antagonism at α1a- and α1b-adrenergic receptor subtypes. Mol. Pharmacol. 1999;56:858–866. doi: 10.1124/mol.56.5.858. [DOI] [PubMed] [Google Scholar]
- SAMAMA P., COTECCHIA S., COSTA T., LEFKOWITZ R.J. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- SHRYOCK J.C., OZECK M.J., BELARDINELLI L. Inverse agonists and neutral antagonists of recombinant human A1 adenosine receptors stably expressed in Chinese hamster ovary cells. Mol. Pharmacol. 1998;53:886–893. [PubMed] [Google Scholar]
- SMIT M.J., LEURS R., ALEWIJNSE A.E., BLAUW J., VAN NIEUW AMERONGEN G.P., VAN DE VREDE Y., ROOVERS E., TIMMERMAN H. Inverse agonism of histamine H2 antagonist accounts for upregulation of spontaneously active histamine H2 receptors. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6802–6807. doi: 10.1073/pnas.93.13.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI F., TANIGUCHI T., TAKAUJI R., MURATA S., MURAMATSU I. Splice isoforms of α1a-adrenoceptor in rabbit. Br. J. Pharmacol. 2000;129:1569–1576. doi: 10.1038/sj.bjp.0703242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANIGUCHI T., INAGAKI R., MURATA S., AKIBA I., MURAMATSU I. Microphysiometric analysis of human α-1a adrenoceptor expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1999;127:962–968. doi: 10.1038/sj.bjp.0702609. [DOI] [PMC free article] [PubMed] [Google Scholar]