

Abstract
The effects of caffeine on both levcromakalim-induced macroscopic and unitary currents in pig proximal urethra were investigated by the use of patch-clamp techniques (conventional whole-cell configuration and cell-attached configuration). The effects of caffeine were also examined on currents in inside-out patches of COS7 cells expressing carboxy terminus truncated inwardly rectifying K+ channel (Kir6.2) subunits (i.e. Kir6.2ΔC36) which form ATP-sensitive K+ channels (KATP channels).
In conventional whole-cell configuration, the levcromakalim (100 μM)-induced inward current (symmetrical 140 mM K+ conditions) was inhibited by caffeine (⩾1 mM) at a holding potential of −50 mV. In contrast, ryanodine (10 μM) caused no significant inhibitory effect on the gradual decay of the levcromakalim-induced current at −50 mV.
The amplitude of the 30 μM levcromakalim-induced current was enhanced by 3-isobutyl-1-methylxanthine (IBMX, 100 μM).
In cell-attached configuration, the levcromakalim-induced K+ channel openings were inhibited by subsequent application of 10 mM caffeine, decreasing the channel open probability at −50 mV.
Reverse transcriptase-polymerase chain reaction (RT–PCR) analysis revealed the presence of Kir6.2 transcript in pig urethra.
Caffeine (⩾3 mM) inhibited the channel activity of Kir6.2ΔC36 expressed in COS7 cells (3 mM caffeine, 65±6%, n=4; 10 mM caffeine, 29±2%, n=4).
These results suggest that caffeine can inhibit the activity of KATP channels through a direct blocking effect on the pore-forming Kir subunit.
Keywords: ATP-sensitive K+ channel, caffeine, carboxy terminus truncated Kir6.2, glibenclamide, levcromakalim, phosphodiesterase inhibitor
Introduction
Caffeine elicits physiological responses in a wide variety of cells by triggering the mobilization of Ca2+ from intracellular organelles, and it has often been used as a pharmacological tool for facilitating Ca2+-induced Ca2+ release (CICR) (reviewed by Fleischer & Inui, 1989). It has also been shown that caffeine (100–300 μM) inhibits not only phosphodiesterase activity, leading to a gradual increase in intracellular cyclic AMP (Butcher et al., 1968), but also adenosine receptors as a competitive antagonist (Fredholm, 1982). Furthermore, Islam et al. (1995) have reported that high concentrations of caffeine (10 mM) suppressed the activity of ATP-sensitive K+ channels (KATP channels) in mice pancreatic β-cells. Recent molecular biological studies have revealed that KATP channels are composed of at least two subunits: an inwardly-rectifying K+ channel (Kir) which forms the ion pore and a sulphonylurea receptor (SUR), a member of the ATP-binding cassette superfamily (reviewed by Quayle et al., 1997). However, it is still not clear which subunit(s) in KATP channels may be regulated by the high concentrations of caffeine. Furthermore, there are no published single-channel data concerning the inhibitory effects of caffeine on K+ channels in smooth muscle cells.
Multiple effects of high concentrations of caffeine (⩾10 mM) have been reported on urethral smooth muscle cells. It not only abolished the slow waves and spontaneous transient depolarizations (guinea-pig, Hashitani & Edwards, 1999) but also induced Ca2+-activated Cl− currents (sheep, Cotton et al., 1997). Moreover, it is believed that caffeine can regulate urethral muscle tone in vivo (Palermo & Zimskind, 1977). Thus, it is of importance to elucidate the direct effects of caffeine on ion channels in urethral myocytes. We previously demonstrated the presence of KATP channels in pig urethra (Teramoto et al., 1997), concluding that the KATP channels play an important role in regulating both the resting urethral muscle tone and the resting membrane potential. In the present experiments, we have investigated the inhibitory effects of caffeine on glibenclamide-sensitive K+ currents (KATP currents) in pig urethra induced by the KATP channel openers, and have studied the blocking mechanisms of caffeine on currents in COS7 cells expressing a carboxy terminus truncated Kir6.2 (Kir6.2ΔC36) which can form K+ channels in the absence of SURs. A preliminary account of the present results has been communicated to the 72nd annual meeting of the Japanese Pharmacological Society (Teramoto et al., 2000b).
Methods
Cell dispersion
Fresh urethra from female pigs was collected from a local abattoir. Pig urethral myocytes were freshly isolated by the gentle tapping method (Teramoto & Brading, 1996). Relaxed spindle-shaped cells, with length varying between 400 μm and 500 μm, were isolated and stored at 4°C. The dispersed cells were normally used within 2 h for experiments.
Molecular biology
Kir6.2ΔC36, in which the last 36 amino acids were truncated from the carboxy terminus (C-terminus), was constructed by PCR, inserting a stop codon at the appropriate position. Kir6.2ΔC36 was subcloned into the pCl vector which had the CMV-IE promoter/enhancer (Promega, Madison, WI, U.S.A.). pEGFP-N1, enhanced fluorescent mutant green fluorescent protein (GFP) driven by CMV-IE promoter/enhancer, (Clontech, Laboratories, Inc., Palo Alto, CA, U.S.A.) was co-transfected as a maker and the whole nucleotide sequence of the PCR clone was confirmed by DNA sequencing.
Cell culture and transfection
COS7 cells were plated on coverslips at a density of 1×105 per dish (35 mm in diameter) and cultured in Dulbecco's modified Eagle's medium supplemented with 10% foetal calf serum. Two days later, a cocktail of pCl-Kir6.2ΔC36 and pEGFP-N1 was co-transfected into the COS7 cells using LipofectAMINE and Opti-MEM (GIBCO BRL, Life Technologies, Inc., Rockville, MD, U.S.A.) according to the manufacturer's instruction.
Electrophysiological measurements were usually conducted from 2 to 4 days after transfection.
Recording procedure
Patch-clamp experiments were performed at room temperature (21–23°C) as described previously (Teramoto et al., 2000a). Junction potentials between bath and pipette solutions were measured with a 3 M KCl reference electrode and were <2 mV, so that correction for these potentials was not made. Noise associated with capacitance was kept to a minimum by maintaining the test solution in the electrode (2–3 MΩ) as low as possible. At the beginning of each experiment, the series resistance was compensated. Transfected cells were identified by green fluorescence under a microscope.
Drugs and solutions
The following solutions were used when recording glibenclamide-sensitive membrane currents: physiological salt solution (PSS) containing (mM): Na+ 140, K+ 5, Mg2+ 1.2, Ca2+ 2, glucose 5, Cl− 151.4, HEPES 10, titrated to pH 7.35–7.40 with Tris base (sometimes 140 mM K+ PSS was obtained by replacing 135 mM Na+ with equimolar K+); high K+ pipette solution containing (mM): K+ 140, Cl− 140, glucose 5, EGTA 5, and HEPES 10/Tris (pH 7.35–7.40). Occasionally, 5 mM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′,-tetraacetic acid (BAPTA) was further added to the above pipette solution (pH 7.35–7.40). For single-channel recordings, the pipette and bath solution were high K+ solution (mM): K+ 140, Cl− 140, EGTA 5, glucose 5, HEPES 10/Tris (pH 7.35–7.40) producing symmetrical 140 mM K+ conditions. Cells were allowed to settle in the small experimental chamber (80 μl in volume). The bath was superfused by gravity throughout the experiments at a rate of 2 ml min−1 from a solution reservoir driven by gravity. The following chemicals were used: BAPTA, collagenase (type I), dimethysulphoxide (DMSO), EGTA, glibenclamide, HEPES, 3-isobutyl-1-methylxanthine (IBMX), papain, ryanodine and TrisCl (Sigma-Aldrich Japan K.K., Tokyo, Japan). Pinacidil was purchased from RBI Chemicals Ltd. (Natick, MA, U.S.A.). Levcromakalim was kindly provided by SmithKline Beecham Pharmaceuticals (Harlow, U.K.). Pinacidil, levcromakalim and glibenclamide were prepared daily as 100 mM stock solutions in DMSO. The final concentration of DMSO was less than 0.1% and this concentration was shown not to affect K+ channels.
Data analysis
The data recording system used was essentially the same as that described previously (Teramoto et al., 2000a). In short, generation of voltage pulses was performed using a L/M-EPC 8 patch-clamp amplifier (HEKA Elektronik Corp., Lambrecht, Germany) in conjunction with a circuit board which has both an analogue to digital and a digital to analogue conversion function (ITC-16, Instrutech Corp., NY, U.S.A.), using a ‘Pulse' software package (HEKA Elektronik Corp., Lambrecht, Germany). The sampled current data were filtered at 10 kHz and stored together with potential records on videotape using a digital data recorder (VR-10B, Instrutech Corp., NY, U.S.A.) coupled to a video recorder (Panasonic, Tokyo, Japan) for subsequent off-line analysis. The whole-cell current data were low-pass filtered at 500 Hz (continuous traces) or 2 kHz (ramp current) by an 8 pole Bessel filter, sampled at 25 ms intervals (continuous traces) or 1 ms (ramp current) and analysed on a PowerMac G3 computer (Apple Computers, Tokyo, Japan) using commercial software ‘MacLab 3.5.6' (ADInstruments Pty Ltd., Castle Hill, Australia). For single-channel recordings, the stored data were low-pass filtered at 2 kHz (−3 dB) and sampled into the computer with a digitalized interval of 80 μs using ‘PAT' program (kindly provided by Dr J. Dempster, University of Strathclyde, U.K.); events briefer than 80 μs were not included in the evaluation. Continuous traces in the figures were obtained from records filtered at 500 Hz for presentation (digital sampling interval, 25 ms). Values for the channel open state probability (Popen) were measured at −50 mV for 1 min according to the equation,
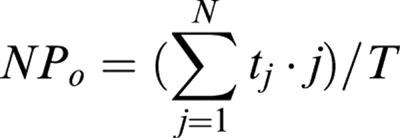 |
where tj is the time spent at each current level corresponding to j=0, 1, 2,...N, T is the duration of the recording, and N was the maximum number of channels observed but the minimal number of channels in the patch. Data points were fitted using a least-squares fitting.
RNA preparation and reverse transcription-polymerase chain reaction analysis
For RNA isolation, total RNA from both pig urethral smooth muscle and rat ventricle was isolated using TRIzol reagent according to the manufacturer's instructions (Life Technologies). First-strand synthesis of cDNA using random hexamers was prepared as follows: total RNA isolated from tissues was incubated with random hexamers at 70°C for 10 min and then with PCR buffer (20 mM Tris/HCl, pH 8.4, 50 mM KCl), 2.5 mM MgCl2, 0.5 mM deoxynucleoside-5′-triphosphate, and 10 mM dithiothreitol at 25°C for 5 min. RT–PCR was initiated by the addition of Superscript II RT (200 U) at 25°C for 10 min followed by incubation at 42°C for 50 min. The reaction was terminated by incubation at 70°C for 15 min, before chilling on ice. PCR was performed using 1 μl of cDNA in 20 μl reaction containing 0.5 μM concentration of each primer, 200 μM concentration of each deoxynucleoside-5′triphosphate, and 0.5 units of Taq polymerase (Takara Co. Ltd., Japan). The cycling conditions were 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s for 40 cycles. An aliquot (10 μl) of the RT–PCR product was analysed on a 2% Tris-borate-EDTA polyacrylamide gel. Since no sequence information is available about KATP channel subunits in pig, generic subunit-specific primers were designed based on information from rat, mouse, and human sequences (Gopalakrishnan et al., 1999). The locations of the primers indicated are based on the subunit sequence information obtained from GenBank: Kir6.2 (rat, U73626; mouse, U73626; human, D50582). The sequences of the primers for amplification of the novel Kir6.2 were as follows: 5′-GGCTCCTAGTGACCTGCACCA-3′ (forward) and 5′-CCACAGCCACACTGCGCTTGCG-3′ (reverse), corresponding to nucleotide positions 810–830 and 1126–1105, respectively. Control reactions were carried out where samples in the absence of reverse transcriptase were amplified to ensure that the detected product was not the result of possible DNA contamination and by use of corresponding templates as positive controls to ensure that the primers were annealing successfully. These primers gave products of the expected sizes that were confirmed by DNA sequence analysis.
Statistics
Statistical analyses were performed with Student's t-test for paired values. Changes were considered significant at P<0.01 and data are expressed as mean±standard error (s.e.).
Results
Effects of caffeine on the glibenclamide-sensitive membrane currents in pig proximal urethra
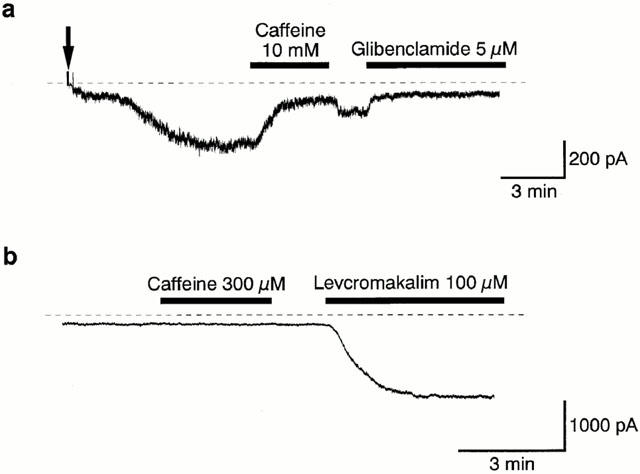
Levcromakalim (100 μM) evoked an inward current under symmetrical K+ conditions (pipette solution, 140 mM KCl containing EGTA 5 mM; bath solution, PSS 140 mM K+) at a holding potential of −50 mV in pig proximal urethra. This levcromakalim-induced current was rapidly inhibited by 10 mM caffeine (Figure 1a). On removal of caffeine, the amplitude of the levcromakalim-induced current showed a quick recovery towards the value just before application of caffeine. Subsequent application of 5 μM glibenclamide suppressed the inward current to the control current level. In the same experiment, six triangular ramp potential pulses (see inset in Figure 1a) were applied in order to obtain current-voltage (I–V) relationships under each condition. Figure 1b shows the averaged membrane currents during the falling phase of the ramp pulses under the various experimental conditions of the experiment. The levcromakalim-induced membrane current was obtained by subtracting the averaged control current from the membrane current in the presence of 100 μM levcromakalim, demonstrating an inwardly rectifying property at positive membrane potentials (Figure 1c). The caffeine-sensitive membrane current in the presence of levcromakalim was obtained by subtracting the mean membrane current in the presence of 10 mM caffeine from the membrane current in its absence. In Figure 1c, the I–V relationships between the caffeine-sensitive current and the levcromakalim-induced current are virtually identical from −120 mV to +10 mV. The effect of caffeine, when additionally applied after the levcromakalim-induced current was maximally evoked, was concentration-dependent above 1 mM (Figure 2c). Caffeine (10 mM) inhibited the levcromakalim-induced current to 21±2%, (n=18), but the inhibition was partially irreversible (65±4%, n=18).
Figure 1.
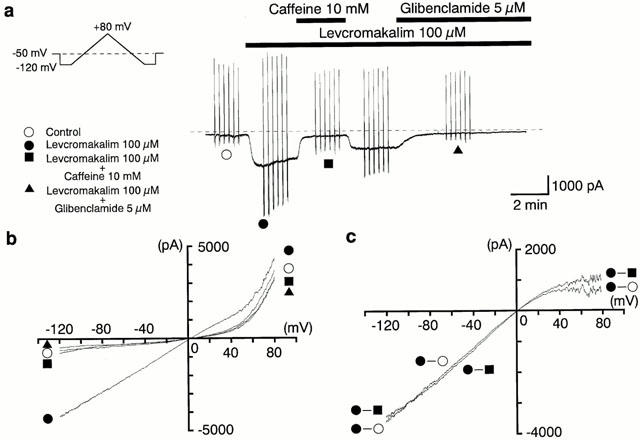
Inhibitory effects of caffeine on the levcromakalim-induced glibenclamide-sensitive inward membrane current at −50 mV. Whole-cell recording, bath solution 140 mM K+ PSS, pipette solution 140 mM K+ containing 5 mM EGTA. (a) Current trace. The vertical lines are responses to triangular ramp potential pulses of 200 mV s−1 from −120 mV to +80 mV, applied after an initial 300 ms conditioning pulse to −120 mV (see inset). Levcromakalim (100 μM) caused an inward membrane current (peak amplitude about 1.8 nA) which gradually decayed. The current was inhibited by application of 10 mM caffeine, recovered to a steady state amplitude, and was then suppressed to the same level by 5 μM glibenclamide. The dashed line indicates zero current. (b) I–V curves measured from the negative-going limb (the falling phase) of the ramp pulse. Each symbol is the same as in the current trace (a). The lines are mean membrane currents from the six ramps in each condition. (c) Net membrane currents. The levcromakalim-induced membrane current was obtained by subtraction of the mean ramp current recorded before from that during application of 100 μM levcromakalim. The caffeine-sensitive membrane current was obtained by subtraction of the membrane currents in the absence and presence of 10 mM caffeine when 100 μM levcromakalim was present in the bath solution.
Figure 2.
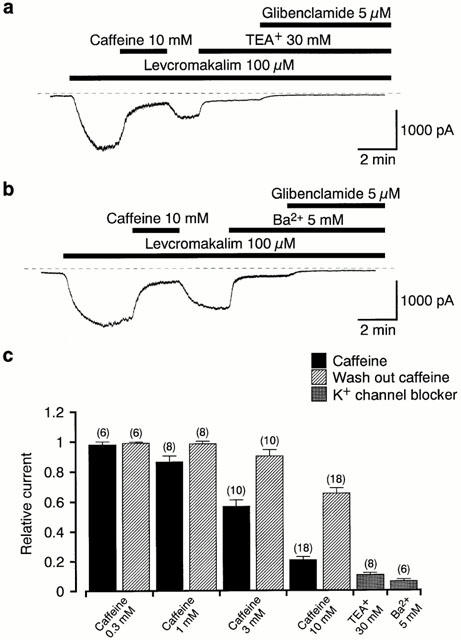
Effects of caffeine, TEA+ (30 mM) and Ba2+ (5 mM) on the 100 μM levcromakalim-induced current in pig urethral myocytes. The bath solution was 140 mM K+ PSS and the pipette solution was 140 mM KCl containing 5 mM EGTA. (a), (b) Effects of 30 mM TEA+ (a) and 5 mM Ba2+ (b) on the levcromakalim-induced current at −50 mV. The dashed line indicates zero current. (c) Effects of caffeine (0.3–10 mM; presence and wash-out), TEA+ (30 mM) and Ba2+ (5 mM) on the 100 μM levcromakalim-induced membrane current at a holding potential of −50 mV. Each column shows the relative amplitude of the levcromakalim-induced current at −50 mV (mean value with±s.e.) when the amplitude of the levcromakalim-induced current was taken as one just before application of each drug. The membrane current was measured from the current level in the presence of 5 μM glibenclamide. The number of observations (n) is shown above each column.
In order to compare the potency of caffeine on the levcromakalim-induced current with other types of K+ channel blockers, the effects of TEA+ and Ba2+ were tested (Figure 2a,b). Figure 2c summarizes the residual current in each K+ channel blocker relative to the 100 μM levcromakalim-induced current at −50 mV, which was measured as the difference between the current just before the blocker was applied and the current level in the presence of 5 μM glibenclamide. The inhibitory potency of 10 mM caffeine on the 100 μM levcromakalim-induced current was somewhat weaker than that of either TEA+ (30 mM, 10±1%, n=8) or Ba2+ (5 mM, 6±2%, n=6) at −50 mV.
Effects of ryanodine and BAPTA on the levcromakalim-induced current at −50 mV
A similar inhibitory effect of caffeine (10 mM) was observed on the inward membrane current elicited by 100 μM pinacidil at −50 mV (16±4%, n=4, Figure 3a). Applying 10 μM ryanodine, which releases Ca2+ from intracellular stores, caused no significant effect on the normal gradual decay of the levcromakalim-induced current (Figure 3b, 2.5 min application n=5). Figure 3c shows the relative time course of inward currents induced by 100 μM levcromakalim in the absence and presence of ryanodine at −50 mV. After reaching a peak, the levcromakalim-induced K+ current gradually declined and the rate of this decline was not affected by ryanodine (3 min later, control, 68±7%, n=5; ryanodine, 65±7%, n=5). When 5 mM BAPTA was further included in the 140 mM K+ pipette solution containing 5 mM EGTA, application of 10 mM caffeine inhibited the levcromakalim-induced membrane current to a similar level (18±3%, n=4, Figure 3d) at a holding potential of −50 mV.
Figure 3.
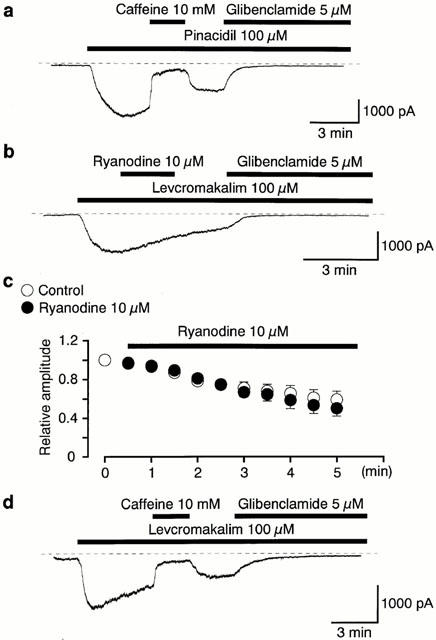
Whole-cell recording of KATP currents at −50 mV. Bath solution 140 mM K+ PSS, pipette solution 140 mM K+ containing 5 mM EGTA. The dashed lines indicate the zero current level. (a) Application of caffeine 10 mM inhibited the 100 μM pinacidil-induced glibenclamide-sensitive inward current at −50 mV. (b) Application of ryanodine (10 μM, 2 min duration) did not suppress the amplitude of the 100 μM levcromakalim-induced membrane current. (c) The time course of the relative amplitude of inward currents induced by 100 μM levcromakalim in the absence (control) and presence of 10 μM ryanodine. The peak amplitude of the levcromakalim-induced K+ current was taken as one. Time 0 indicates the time when the levcromakalim-induced inward current reached its maximal value. Each symbol indicates the mean of four observations determined at 30 s intervals. (d) When 5 mM BAPTA was further added to the pipette solution, application of 10 mM caffeine inhibited the levcromakalim-induced membrane current.
Effects of caffeine on the UDP-induced inward currents
When UDP (10 mM) was included in the pipette solution in order to stimulate the glibenclamide-sensitive components, a significant inward current was recorded, that developed slowly at a holding potential of −50 mV and was well maintained (Figure 4a, Teramoto et al., 1997). The maximum current recorded in the presence of UDP was measured from the current level in the presence of 5 μM glibenclamide (the peak amplitude, approximately 260 pA). Caffeine (10 mM) caused a reversible inhibition of the UDP-induced inward current (15±5%, n=4; wash-out, 52±5%, n=4). In contrast, a lower concentration of caffeine (300 μM) did not induce an obvious inward current, and in the same cells, a levcromakalim-induced current could be subsequently elicited at −50 mV (Figure 4b, n=4). Similarly, in the absence of UDP, application of caffeine (100–300 μM) alone caused no inward current at −50 mV (n=6, data not shown). These results suggest that caffeine may not antagonize the actions of KATP channel openers on the glibenclamide-sensitive K+ currents.
Figure 4.
Effects of caffeine on the 10 mM UDP-induced current in a conventional whole-cell configuration at −50 mV. UDP (10 mM) was included in the pipette solution. Bath solution 140 mM K+ PSS, pipette solution 140 mM K+ containing 5 mM EGTA. The dashed lines indicate the zero current level. (a) When a small inward current developed slowly after the established of the conventional whole-cell configuration (indicated by the filled arrow), caffeine (10 mM) reversibly inhibited the 10 mM UDP-induced inward current. Subsequently, application of 5 μM glibenclamide suppressed the UDP-induced inward current. (b) When the amplitude of the UDP-induced current became stable at −50 mV, application of 300 μM caffeine caused no effect on the UDP-induced membrane current.
Effects of IBMX on the levcromakalim-induced hyperpolarization and membrane current
In order to study whether or not caffeine could inhibit the glibenclamide-sensitive K+ currents by acting as a phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine (IBMX), a non-selective phosphodiesterase inhibitor, was used to compare the actions on the levcromakalim-induced currents and hyperpolarization. In Figure 5a, application of 100 μM IBMX had no effect on the membrane current at −50 mV. Subsequently, application of levcromakalim (100 μM) caused a glibenclamide-sensitive inward current in the presence of IBMX. The peak amplitude of the levcromakalim-induced current in the presence of IBMX was much larger than that recorded in its absence (2780±133 pA, n=5 with IBMX versus 1455±94 pA, n=10 without IBMX). In current-clamp mode, 100 μM levcromakalim induced a stable hyperpolarization (to −78±1 mV, n=5), which was close to the theoretical K+ equilibrium potential (EK=−84.2 mV) under the present experimental conditions. Additional application of IBMX had no effect (Figure 5b, n=5). As shown in Figure 6a, application of levcromakalim (30 μM) caused an inward current which, after reaching a peak value, gradually decreased in amplitude at −50 mV. When 100 μM IBMX was additionally applied, the amplitude of the levcromakalim-induced inward current increased. On removal of IBMX, the current gradually declined. Subsequent application of 5 μM glibenclamide suppressed the inward current to a value close to that seen in the control conditions. In the same experiment, four triangular ramp potential pulses (see inset in Figure 6a) were applied in order to obtain I–V relationships under each condition. Figure 6b shows the averaged membrane currents during the falling phase of the ramp pulses under the various experimental conditions. The IBMX-sensitive membrane current was obtained by subtracting the mean membrane current in the presence of IBMX from the membrane current in its absence, not only changing sign at approximately 0 mV (−3±1 mV, n=5) but also exhibiting an inwardly rectifying property at positive membrane potentials (Figure 6c).
Figure 5.
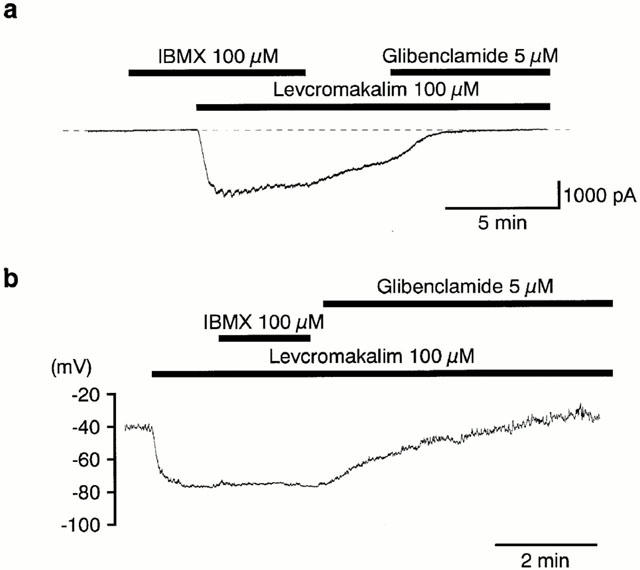
Effects of IBMX on the 100 μM levcromakalim-induced current and hyperpolarization in a conventional whole-cell recording. (a) Although no membrane current was evoked by application of IBMX 100 μM, subsequent application of 100 μM levcromakalim caused a large inward current at −50 mV. Bath solution 140 mM K+ PSS, pipette solution 140 mM K+ containing 5 mM EGTA. The dashed lines indicate the zero current level. (b) 100 μM IBMX had no effect on the 100 μM levcromakalim-induced membrane hyperpolarization in current-clamp mode. Bath solution 5 mM K+ PSS, pipette solution 140 mM K+ containing 5 mM EGTA.
Figure 6.
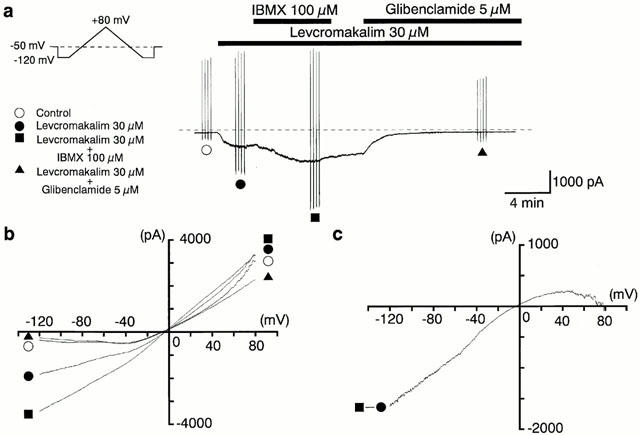
IBMX enhanced the amplitude of the 30 μM levcromakalim-induced membrane current at a holding potential of −50 mV. Whole-cell recording, bath solution 140 mM K+ PSS, pipette solution 140 mM K+ containing 5 mM EGTA. (a) Current trace. The vertical lines are responses to triangular ramp potential pulses of 200 mV s−1 from −120 mV to +80 mV, applied after an initial 300 ms conditioning pulse to −120 mV (see inset). Levcromakalim (30 μM) caused an inward membrane current (peak amplitude about 0.7 nA). The current was enhanced by application of 100 μM IBMX, gradually recovered to a steady state amplitude after IBMX was removed, and was then suppressed by 5 μM glibenclamide. The dashed line indicates zero current. (b) I–V curves measured from the negative-going limb (the falling phase) of the ramp pulse. Each symbol is the same as in (a). The lines are mean membrane currents from the four ramps in each condition. (c) The IBMX-sensitive membrane current obtained by subtraction of the membrane current in the absence from that in the presence of 100 μM IBMX when 30 μM levcromakalim was in the bath.
Effects of caffeine on the levcromakalim-induced K+ channels at −50 mV in cell-attached configuration
The effects of caffeine (10 mM) on the K+ channels activated by 100 μM levcromakalim were investigated in the cell-attached patches when small numbers of the 2.1 pA channels were opened. In Figure 7a, a 2.1 pA K+ channel was activated by application of 100 μM levcromakalim (NPo value, 0.2). Application of 10 mM caffeine reversibly inhibited the channel activity to an NPo value of 0.02. When the control 100 μM levcromakalim-induced NPo value was taken as one, the relative NPo value was 0.12±0.02 (n=5) in the presence of 10 mM caffeine. On some occasions (five out of 15 patches), the unitary current during channel opening was transiently reduced in the presence of 10 mM caffeine (2.1±0.04 pA, control versus 0.8±0.04 pA, caffeine 10 mM, n=5, Figure 7c). Note that the apparent reduction in urinary current amplitude was never observed in the absence of caffeine (control). On removal of caffeine, both the channel activity and the unitary amplitude completely recovered to the control level. Approximately 7 min later, bath application of glibenclamide (5 μM) reversibly abolished the channel activity.
Figure 7.
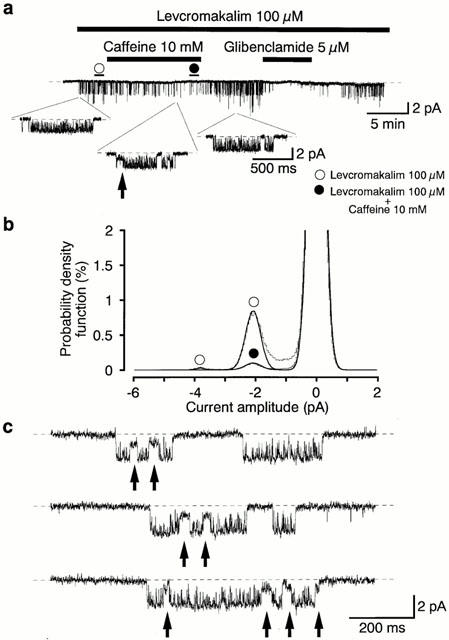
Effects of 10 mM caffeine on the 100 μM levcromakalim-induced K+ channel activity recorded under symmetrical 140 mM K+ conditions. Cell-attached patch at −50 mV. Levcromakalim 100 μM was added to the bath solution. (a) Application of 10 mM caffeine (11 min duration) reversibly reduced the activity of the 2.1 pA K+ channel. After removal of caffeine, 7 min later, application of glibenclamide (6 min duration) abolished channel activity. The dashed line indicates the current when the channel is not open. Lower traces show expansions of the upper trace (2 kHz filtration; 80 μs digital sampling interval). (b) All-point amplitude histograms in the absence (obtained during the last 1 min just before application of caffeine) and presence of 10 mM caffeine (obtained during the last 1 min of a 11 min application) in the presence of 100 μM levcromakalim. Continuous lines in the histograms are theoretical curves fitted with the Gaussian distribution, by the least-squares method. The abscissa scales show the amplitude of the current (pA) and the ordinate scales show the percentage value of the probability density function (%) for the recording period (1 min). (c) The open channel blocking effects of 10 mM caffeine on the 2.1 pA K+ channel at −50 mV. These events are marked by arrows. The expanded traces were obtained from trace (a) from the last 2 min of an 11 min application. The same caffeine-induced blocking effects were recorded in 42 events in the presence of 10 mM caffeine. Similar caffeine-induced blocking effects were observed in five other membrane patches. Note that the open channel blocks are never observed in the absence of caffeine. The dashed line indicates the current when the channel is not open.
Molecular expression of inwardly rectifying K+ channels in pig urethra
In order to determine tissue distributions of Kir6.2, RT–PCR was performed. The specific primers for amplification of Kir6.2 mRNA were designed to produce a cDNA fragment of 316 bp. As shown in Figure 8a, Kir6.2 mRNA was expressed in rat Kir6.2 cDNA, pig urethra and rat ventricle with the expected fragment size for Kir6.2.
Figure 8.
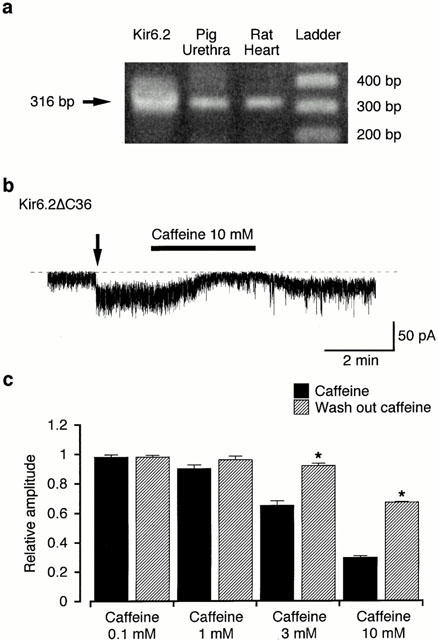
Effects of caffeine on channel activity of Kir6.2ΔC36. (a) RT–PCR detection of Kir6.2. RT–PCR was performed as described in Methods and size makers were used to indicate the size of the experimental fragments (lane 4). RT–PCR yielded visible amounts of Kir6.2 (316 bp fragment) in mRNAs from rat cDNA of Kir6.2 (lane 1), pig urethra (lane 2) and rat heart (lane 3). (b) Current trace of the inhibitory effects of caffeine (10 mM) on channel activity of Kir6.2ΔC36 in inside-out configuration at a holding potential of −50 mV. The arrow indicates the time when the inside-out patch was established by excision from the cell. The dashed line indicates the current when the channel is not open. (c) Effects of caffeine (0.1–10 mM, presence and wash-out) on channel activity of Kir6.2ΔC36 at 50 mV. Each column shows the relative level of the channel activity of Kir6.2ΔC36 (mean value with+s.e.) when the mean amplitude of the channel activity of Kir6.2ΔC36 was normalized as one just before application of caffeine (n=4). The mean membrane current was measured from the current level for 30 s duration when the channel was not open. *Significantly different from the relative amplitude in the presence of caffeine (t-test, P<0.01).
Effects of caffeine and glibenclamide on channel activity of Kir6.2ΔC36
Activity of Kir6.2ΔC36 was consistently observed in cell-attached patches from GFP-positive COS7 cells at −50 mV (Figure 8b). When the membrane patch was excised in inside-out configuration, the channel activity was enhanced. Application of 10 mM caffeine caused a reversible inhibition of channel activity. However, glibenclamide (10 μM) had no significant effect (data not shown). Figure 8c shows that caffeine (⩾3 mM) produces a concentration-dependent inhibition of Kir6.2ΔC36 channel activity at −50 mV (n=4).
Discussion
In the present experiments, we have demonstrated that application of caffeine substantially inhibited membrane currents induced by KATP channel openers (such as levcromakalim, pinacidil etc.) by reducing the open probability of KATP channels.
Mechanisms by which caffeine blocks ATP-sensitive K+ currents in pig urethra
It is possible that the inhibitory effects of caffeine that we have shown on the levcromakalim-induced K+ currents in the pig urethral myocytes are indirect, and due to either its inhibitory effect on phosphodiesterase or its ability to activate Ca2+-induced Ca2+ release mechanisms. We will consider each of these possibilities separately.
First, blocking KATP channels in pig urethra might be caused by an increase of cyclic AMP following inhibition of intracellular phosphodiesterase, which directly or indirectly could switch off the levcromakalim-activated KATP channels. However, in the present experiments, additional application of IBMX, a non-selective phosphodiesterase inhibitor, not only increased the levcromakalim-induced membrane currents at −50 mV but also had no inhibitory effect on the 100 μM levcromakalim-induced hyperpolarization, suggesting that phosphodiesterase inhibition by caffeine may not be responsible for inhibiting the KATP channels. In contrast, neither application of IBMX (100 μM) nor caffeine (100–300 μM) alone activated a significant membrane current in pig urethral myocytes. These results suggest that KATP current is not evoked by inhibition of phosphodiesterase. Similar results were observed in pig coronary artery, suggesting that cyclic AMP-dependent protein kinase (protein kinase A) activated a glibenclamide-sensitive K+ current, not through inhibition of phosphodiesterase but by a direct increase in cyclic AMP concentration (Wellman et al., 1998).
Second, possible involvement of Ca2+ released from the intracellular stores was investigated in the present experiments by applying ryanodine (10 μM). This showed even in the presence of 5 mM EGTA, no effect on the levcromakalim-induced membrane current in pig urethra or its decline. In order to ensure that any Ca2+ released from the stores was completely chelated, BAPTA (5 mM) was added to the pipette solution in which EGTA (5 mM) had been included. Again, there was no significant difference in the caffeine-induced inhibitory ratio of the levcromakalim-induced membrane current whether or not BAPTA was present in the pipette solution. These results suggest that Ca2+ release mechanisms are not responsible for the inhibitory effect of caffeine on the levcromakalim-induced K+ current. These present pharmacological observations, which suggest that changes in intracellular Ca2+ concentration are not involved in this action of caffeine, are strongly supported by our previous observations that neither the levcromakalim-induced glibenclamide-sensitive K+ currents nor the levcromakalim-induced KATP channels exhibit intracellular Ca2+ sensitivity in pig proximal urethra (Teramoto & Brading, 1996; 1998; Teramoto et al., 1997).
On the other hand, it is equally possible that caffeine might directly block the K+ channels in a non-selective manner. It has been reported that extracellular application of caffeine (⩾10 mM) inhibited voltage-dependent Ca2+ currents and voltage-dependent K+ currents in a wide variety of smooth muscle cells (rat myometrium, Martin et al., 1989; rabbit ear artery, Hughes et al., 1990; guinea-pig ileum, Zholos et al., 1991; guinea-pig urinary bladder, Yoshino et al., 1996), indicating that caffeine itself may directly block the pore of several types of ion channels in a non-selective manner. In the cell-attached patches, caffeine is effective when applied extracellularly (presumably by diffusion through the membrane), reducing not only the NPo value of channel activity but also the mean unitary amplitude of KATP channel in pig urethra. A similar inhibition of the KATP channels was reported in frog skeletal muscle through TEA+, in which intracellular application of TEA+ reduced the amplitude of the single channel current due to multiple binding sites (at least four sites) of K+ channel pore (Davies et al., 1989). Thus, the results from the present experiments in pig urethra suggest that the last possibility is most likely. We have also shown using RT–PCR the presence of Kir6.2 in pig urethra as well as rat ventricular myocytes. Kir6.2 is dominantly expressed in several types of smooth muscle (murine colon, Koh et al., 1998; A-10 cells, Miller et al., 1999; guinea-pig urinary bladder, Gopalakrishnan et al., 1999).
Although this type of study addresses only the expression of the mRNA transcript, it is tempting to speculate that the pore of the KATP channels in pig urethra may be composed of Kir 6.2. Tucker et al. (1997) have reported that significant currents were recorded from Xenopus oocytes injected with mRNA encoding a truncated form of Kir6.2 in which the last 36 amino acids of the C-terminus had been deleted (i.e. Kir6.2ΔC36) even in the absence of SURs. This enabled the direct effects of certain drugs to be tested on the pore forming Kir6.2 subunits. In the present experiments, we have demonstrated the inhibitory effects of caffeine on the channel activity of Kir6.2ΔC36 in the absence of SURs. The inhibitory potency of caffeine on the channel activity was remarkably similar to that observed for levcromakalim- and UDP-induced KATP currents in pig urethra. These results strongly suggest that higher concentrations of caffeine may block the pore forming Kir6.2 subunits of KATP channels directly. Islam et al. (1995) have reported that 10 mM caffeine completely suppressed the channel activity of KATP channels in β-cells in inside-out patches without demonstrating any precise inhibitory mechanisms of caffeine. In the present study, however, in single-channel recordings some 30% of the channel activity remained in both levcromakalim-induced KATP channels in pig urethra and in the channel activity of Kir6.2ΔC36, after the application of 10 mM caffeine. Similar results were observed for levcromakalim- and UDP-induced KATP currents in pig urethra when a conventional whole-cell recording was performed. We are not sure whether or not this significant discrepancy regarding the effects of caffeine is due to the characterization of KATP channels from different tissues or to the different experimental conditions used.
In conclusion, we have been able to demonstrate that high concentrations of caffeine inhibit KATP channel opener-induced glibenclamide-sensitive K+ currents through a direct blockade of K+ channels in smooth muscle myocytes from pig urethra.
Acknowledgments
We thank Prof A.F. Brading and Dr T.C. Cunnane (University Department of Pharmacology, Oxford, U.K.) for their critical reading of the manuscript. This work was supported by the grants from the Kaibara Morikazu Medical Science Promotion Foundation (Fukuoka, Japan), awarded to Noriyoshi Teramoto.
Abbreviations
- CICR
Ca2+-induced Ca2+ release
- EK
theoretical K+ equilibrium potential
- GFP
green fluorescence protein
- KATP channels
ATP-sensitive K+ channels
- Kir
inwardly-rectifying K+ channel
- Kir6.2ΔC36
carboxy terminus truncated Kir6.2
- NPo
channel open state probability
- PSS
physiological salt solution
- RT–PCR
reverse transcriptase-polymerase chain reaction
- SUR
sulphonylurea receptor
- TEA+
tetraethylammonium
- UDP
uridine 5′-diphosphate
References
- BUTCHER R.W., BAIRD C.E., SUTHERLAND E.W. Effects of lipolytic and antilipolytic substances on adenosine 3′,5′-monophosphate levels in isolated fat cells. J. Biol. Chem. 1968;243:1705–1712. [PubMed] [Google Scholar]
- COTTON K.D., HOLLYWOOD M.A., MCHALE N.G., THORNBURY K.D. Ca2+ current and Ca2+-activated chloride current in isolated smooth muscle cells of the sheep urethra. J. Physiol. 1997;505:121–131. doi: 10.1111/j.1469-7793.1997.121bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIES N.W., SPRUCE A.E., STANDEN N.B., STANFIELD P.R. Multiple blocking mechanisms of ATP-sensitive potassium channels of frog skeletal muscle by tetraethylammonium ions. J. Physiol. 1989;413:31–48. doi: 10.1113/jphysiol.1989.sp017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLEISCHER S., INUI M. Biochemistry and biophysics of excitation-contraction coupling. Annu. Rev. Biophys. Biophys. Chem. 1989;18:333–364. doi: 10.1146/annurev.bb.18.060189.002001. [DOI] [PubMed] [Google Scholar]
- FREDHOLM B.B. Adenosine actions and adenosine receptors after 1 week treatment with caffeine. Acta. Physiol. Scand. 1982;115:283–286. doi: 10.1111/j.1748-1716.1982.tb07078.x. [DOI] [PubMed] [Google Scholar]
- GOPALAKRISHNAN M., WHITEAKER K.L., MOLINARI E.J., DAVIS-TABER R., SCOTT V.E., SHIEH C.C., BUCKNER S.A., MILICIC I., CAIN J.C., POSTL S., SULLIVAN J.P., BRIONI J.D. Characterization of the ATP-sensitive potassium channels (KATP) expressed in guinea pig bladder smooth muscle cells. J. Pharmacol. Exp. Ther. 1999;289:551–558. [PubMed] [Google Scholar]
- HASHITANI H., EDWARDS F.R. Spontaneous and neurally activated depolarizations in smooth muscle cells of the guinea-pig urethra. J. Physiol. 1999;514:459–470. doi: 10.1111/j.1469-7793.1999.459ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGHES A.D., HERING S., BOLTON T.B. The action of caffeine on inward barium current through voltage-dependent calcium channels in single rabbit ear artery cells. Pflügers Arch. 1990;416:462–466. doi: 10.1007/BF00370755. [DOI] [PubMed] [Google Scholar]
- ISLAM M.S., LARSSON O., NILSSON T., BERGGREN P.O. Effects of caffeine on cytoplasmic free Ca2+ concentration in pancreatic beta-cells are mediated by interaction with ATP-sensitive K+ channels and L-type voltage-gated Ca2+ channels but not the ryanodine receptor. Biochem. J. 1995;306:679–686. doi: 10.1042/bj3060679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOH S.D., BRADLEY K.K., RAE M.G., KEEF K.D., HOROWITZ B., SANDERS K.M. Basal activation of ATP-sensitive potassium channels in murine colonic smooth muscle cell. Biophys. J. 1998;75:1793–1800. doi: 10.1016/S0006-3495(98)77621-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN C., DACQUET C., MIRONNEAU C., MIRONNEAU J. Caffeine-induced inhibition of calcium channel current in cultured smooth cells from pregnant rat myometrium. Br. J. Pharmacol. 1989;98:493–498. doi: 10.1111/j.1476-5381.1989.tb12622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER T.R., TABER R.D., MOLINARI E.J., WHITEAKER K.L., MONTEGGIA L.M., SCOTT V.E., BRIONI J.D., SULLIVAN J.P., GOPALAKRISHNAN M. Pharmacological and molecular characterization of ATP-sensitive K+ channels in the TE671 human medulloblastoma cell line. Eur. J. Pharmacol. 1999;370:179–185. doi: 10.1016/s0014-2999(99)00128-4. [DOI] [PubMed] [Google Scholar]
- PALERMO L.M., ZIMSKIND P.D. Effect of caffeine on urethral pressure. Urol. 1977;10:320–324. doi: 10.1016/0090-4295(77)90159-5. [DOI] [PubMed] [Google Scholar]
- QUAYLE J.M., NELSON M.T., STANDEN N.B. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol. Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- TERAMOTO N., BRADING A.F. Activation by levcromakalim and metabolic inhibition of glibenclamide-sensitive K channels in smooth muscle cells of pig proximal urethra. Br. J. Pharmacol. 1996;118:635–642. doi: 10.1111/j.1476-5381.1996.tb15448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERAMOTO N., BRADING A.F. The effects of nifedipine and other calcium antagonists on the glibenclamide-sensitive currents in smooth muscle cells from pig urethra. Br. J. Pharmacol. 1998;123:1601–1608. doi: 10.1038/sj.bjp.0701777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERAMOTO N., MCMURRAY G., BRADING A.F. Effects of levcromakalim and nucleoside diphosphates on glibenclamide-sensitive K+ channels in pig urethral myocytes. Br. J. Pharmacol. 1997;120:1229–1240. doi: 10.1038/sj.bjp.0701033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERAMOTO N., NAKASHIMA T., ITO Y. Properties and pharmacological modification of ATP-sensitive K+ channels in cat tracheal myocytes. Br. J. Pharmacol. 2000a;130:625–635. doi: 10.1038/sj.bjp.0703333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TERAMOTO N., YUNOKI T., ITO Y. The inhibitory effects of caffeine on the glibenclamide-sensitive K+ currents in smooth muscle cells from pig urethra. Jpn. J. Pharmacol. 2000b;94P:O–213. doi: 10.1038/sj.bjp.0703586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., ZHAO C., TRAPP S., ASHCROFT F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- WELLMAN G.C., QUAYLE J.M., STANDEN N.B. ATP-sensitive K+ channel activated by calcitonin gene-related peptide and protein kinase A in pig coronary arterial smooth muscle. J. Physiol. 1998;507:117–129. doi: 10.1111/j.1469-7793.1998.117bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHINO M., MATSUFUJI Y., YABU H. Voltage-dependent suppression of calcium current by caffeine in single smooth muscle cells of the guinea-pig urinary bladder. Naunyn Schmiedeberg's Arch. Pharmacol. 1996;353:334–341. doi: 10.1007/BF00168637. [DOI] [PubMed] [Google Scholar]
- ZHOLOS A.V., BAIDAN L.V., SHUBA M.F. The inhibitory action of caffeine on calcium currents in isolated intestinal smooth muscle cells. Pflügers Arch. 1991;419:267–273. doi: 10.1007/BF00371106. [DOI] [PubMed] [Google Scholar]