

Abstract
In the human umbilical artery (HUA) pre-contracted with the thromboxane mimetic U46619 or with 5-hydroxytryptamine (5-HT), (and pretreated with indomethacin (3 μM) to suppress the synthesis of prostanoids), authentic nitric oxide (NO) evoked concentration-dependent relaxation (pEC50 7.05 and 5.99, respectively). In contrast, sodium nitroprusside (SNP) induced relaxation only in U46619 pre-contracted HUA (pEC50 6.52).
At high (>300 mmHg) vs low (<55 mmHg) oxygen tension the dose-response curves to NO- and SNP-induced relaxations were biphasic and shifted leftward.
Preincubation of the arterial rings with the soluble guanylyl cyclase (sGC) inhibitor 1H[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; 10 μM) shifted the concentration-response curve to NO, reduced the maximal relaxation response to NO (Emax 71%) and to SNP (Emax 10%).
Pre-exposure of HUA rings to high extracellular K+ (50 mM) reduced Emax relaxation responses to NO (36%) and SNP (1%).
Pretreatment of the HUA with the K+ channel inhibitors, tetraethylammonium (TEA, 1 mM), 4-aminopyridine (4-AP, 0.5 mM), charybdotoxin (0.1 μM) or iberiotoxin (0.1 μM) increased the pEC30 for NO and SNP and changed the shape of the dose-response curves from biphasic to monophasic.
Pre-incubation of HUA rings with TEA (1 mM), 4-AP (0.5 mM) and ODQ (10 μM) significantly reduced the NO-induced maximal relaxation (Emax 26%) but not the pEC50 (5.60).
These data indicate that SNP-induced relaxation in the HUA is primarily mediated via sGC-cyclic GMP whereas NO-induced relaxation also involves the activation of KV and KCa channels and a cyclic GMP/K+ channel-independent mechanism(s).
Keywords: Nitric oxide, sodium nitroprusside, guanylyl cyclase, potassium channels, human umbilical artery
Introduction
Nitric oxide (NO) plays an important role in the regulation of vascular tone (Furchgott & Zawadzki, 1980), non-vascular smooth muscle tone (Kannan & Johnson, 1995), inhibition of platelet aggregation (Azuma et al., 1986) and is also the mediator of a number of immunological responses (Moncada et al., 1991). Most of these biological actions of NO can be attributed to the stimulation of soluble guanylyl cyclase (sGC) and the subsequent production of cyclic 3′: 5′ guanosine monophosphate (cyclic GMP) and activation of G kinase (Ignarro, 1990). Feto-placental tissue expresses endothelial NO synthase and releases NO (Myatt et al., 1993). Furthermore, the NO-mediated relaxation system is enhanced during pregnancy and inhibits uterine contractility until term (Yallampalli et al., 1994).
The human umbilical artery (HUA) has been shown to release endothelium-derived relaxing factor (EDRF) and respond in an endothelium-dependent manner to histamine, bradykinin and adenosine triphosphate (Van de Voorde et al., 1987; Chaudhuri et al., 1991). However, the HUA response to EDRF is comparatively weak (Chandhuri et al., 1991) and the HUA appears to lack an endothelium-dependent relaxation response to acetylcholine and the Ca2+ ionophore A23187 (Xie & Triggle, 1994). Indirect evidence suggests that acetylcholine-initiated relaxation is mediated by activation of a Na+, K+-ATP-ase, and subsequent hyperpolarization via K+ efflux, whereas A23187 mediated relaxation results from the synthesis of an indomethacin-resistant product (Xie & Triggle, 1994). Although endothelial cells derived from the HUA release EDRF, the artery itself has been described as being relatively refractory to the relaxant effects of both endogenously produced NO or exogenously applied nitrovasodilators and despite an increase in cyclic nucleotide levels, no corresponding reduction in vascular tone was reported (Renowden et al., 1992; Bergh et al., 1995).
Studies have also shown that NO can cause hyperpolarization of smooth muscle cells in the rat mesenteric and rabbit femoral artery (Garland & McPherson, 1992; Plane et al., 1994). In the guinea-pig uterine artery. NO, acetylcholine and nitrovasodilators also cause hyperpolarization and relaxation (Tare et al., 1990). Several studies have indicated an effect of NO on different types of K+ channels. Thus, in the rat mesenteric artery, NO-evoked hyperpolarization of the resting membrane potential is inhibited by glibenclamide, thus suggesting that NO activates a KATP channel (Garland & McPherson, 1992). However, in the rabbit mesenteric artery, the effects of NO could be inhibited by the small conductance Ca2+-activated K+ channel blocker, apamin (Murphy & Brayden, 1995). NO has also been reported to directly stimulate charybdotoxin sensitive K+ channels in the rabbit aorta (Bolotina et al., 1994) and in rat mesenteric arteriole cells (Mistry & Garland, 1998). There are, however, no functional or electrophysiological studies that have investigated the effects of exogenously applied NO on human umbilical vessels. Therefore, our study was designed to assess the contribution of cyclic GMP and K+ channel activation to NO-induced relaxation of the HUA.
Methods
Human umbilical artery tissue
Umbilical cords were cut from the placenta as soon as possible after delivery but normally within 1 h. The cord was placed in oxygenated (95% oxygen/5% carbon dioxide) physiological salt solution, PSS, (composition, mM: NaCl 118; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4 1.2; NaHCO3 25; glucose 11.1) at 4°C. Arteries were dissected free of surrounding Wharton's jelly and either utilized the same day or stored at 4°C and studied the next day. Previous studies (Xie & Triggle, 1994) have shown that overnight storage at 4°C does not affect the responsiveness of the HUA. Six to 10 rings, 3–5 mm in length, obtained from the same cord were carefully suspended in 25 ml organ baths containing PSS with 3 μM indomethacin at 37°C bubbled with 95% oxygen/ 5% carbon dioxide (actual oxygen tension >300 mmHg) under a preload of 2 g. Some studies were carried out under low, oxygen tension produced by bubbling with 2.5% oxygen/8% carbon dioxide 89.5% nitrogen (actual oxygen tension <55 mmHg) and with arteries dissected in de-oxygenated PSS (Templeton et al., 1991; Xie & Triggle, 1994). The oxygen tension was measured with a 1306 pH/blood gas analyser (Instrumentation Laboratory, Lexington, MA, U.S.A.).
In all experiments, tissues were equilibrated for 3–4 h prior to any experimental protocols with the bath solution being changed every 30 min. Isometric force was monitored using Grass FT-03 transducers and a Grass model 7D polygraph.
The results described in this manuscript were obtained with endothelium-intact preparations of the HUA. In most arterial preparations it is possible to assess endothelial cell integrity by determining the endothelial-cell dependent relaxation response to ACh and, although the pre-contracted HUA does not relax in response to ACh, it is possible to assess the structural integrity of the endothelium in HUA using scanning electron microscopy (Xie & Triggle, 1994). In the present study we did not routinely use scanning electron microscopy to determine the presence or absence of the endothelial cell layer, but care was taken not to touch the endothelial cell layer during the preparation of the HUA rings and thus it was assumed that the endothelium remained undamaged.
Preparation of NO solution
NO was prepared in double-distilled water after de-oxygenation by bubbling the water with argon for 45 min and then with NO for at least 20–30 min to produce a saturated NO stock solution. The NO concentration in the stock solution was 0.9–1.0 mM which was measured using a chemiluminescence technique (O'Neill et al., 1993).
Experimental protocols
After the equilibration period, the rings were contracted with a high concentration of KCl (50 mM). Following a further 1 h period of recovery, with repeated washing every 15 min, either the thromboxane mimetic U 46619 (0.1 nM–10 μM) or 5-HT (0.1 nM–10 μM) was used to induce contraction in the HUA. After establishing concentration-response curves to U46619 or 5-HT, the tissues were contracted with EC70 concentrations of either U46619 (1–10 nM) or 5-HT (0.1–1 μM) and NO (0.03–30 μM) or SNP (0.01–10 μM) was then added cumulatively to elicit relaxations and concentration-relaxation curves were constructed. The concentration-response curves were then repeated after a 30 min incubation of the tissue with a sGC, potassium channel or voltage-gated calcium channel inhibitor. In a separate series of experiments NO- and SNP-induced relaxations were tested in KCl (50 mM) pre-contracted HUA. All experiments were performed in the presence of indomethacin (3 μM) to suppress the synthesis of prostanoids.
Under low oxygen tension and in the presence of indomethacin (3 μM) NO (0.03–30 μM)- and SNP (0.01–10 μM)- induced relaxations were also studied but only in the vessels pre-contracted with of EC70 concentration of U46619. All other experiments were carried out under high oxygen tension.
Each ring preparation was exposed to only one vasodilator and one inhibitor or combination of K+ channel/sGC inhibitors. In any given protocol, a cumulative concentration-response curve to a single vasodilator was obtained and then repeated in the presence of a K+ channel and/or sGC inhibitor. In all experiments, one HUA ring served as a time control and was only exposed to the vasodilator.
Drugs
Ultrapure grade argon and pure NO were obtained from Linde div. Union Carbide (Calgary, AB, Canada). The following compounds were used: ODQ (1H[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one; Tocris Inc., Ballwin, MO, U.S.A.), dendrotoxin and nifedipine (RBI, Natick, MA, U.S.A.) and all other compounds were from Sigma Chemical Co. (St. Louis MO, U.S.A.).
Data analysis
The EC50 and the EC30 values for NO and SNP were calculated using nonlinear regression analysis and reported as the negative logarithm of the mean of individual estimation for each vessel (pEC50 and pEC30). The shift of concentration-response curves to SNP along the x-axis were also measured at the 30% level of the contraction because it was at this level that some of the treatment protocols appeared to have their greatest effect. Emax refers to the maximal relaxation where 100% denotes a complete reversal of the 5-HT or U 46619 induced contraction. Values are presented as mean±s.e. mean, and n reflects the number of cords from which umbilical arteries were obtained. Statistical analysis was performed by Student's t-test (paired). Statistical significance was accepted when P<0.05.
Results
Effects of vasoactive agents on NO- and SNP-induced relaxation
In the HUA, both 5-HT and U46610 were potent contractile agents producing a concentration-dependent increase in tone with pEC50 values of 7.56±0.08 and 8.56±0.05, respectively, n=10 (Figure 1). Following incubation with nifedipine (0.3 μM), the maximal contractions and pEC50 values for U46619 and 5-HT were reduced with Emax values 62±2 and 27±3%, and pEC50s of 7.48±0.04 and 6.15±0.02, respectively, n=10 (Figure 1).
Figure 1.
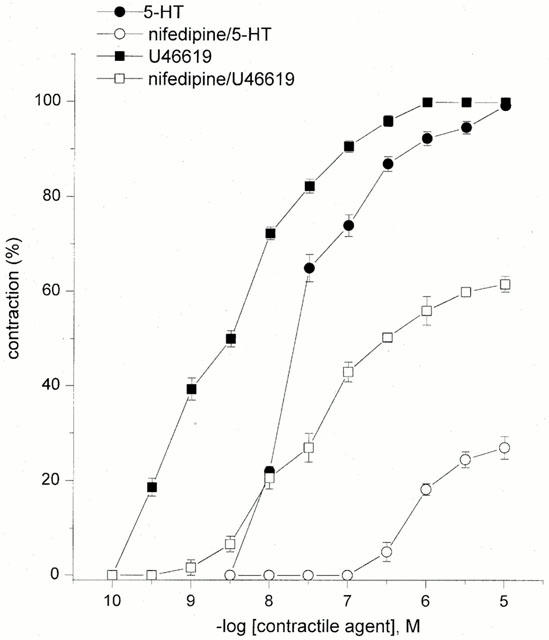
Concentration-response curves for 5-HT and U46619 alone and after incubation with 0.3 μM nifedipine. Each point represents the arithmetic mean±s.e.mean of n=10 experimental determinations.
U46619 was applied in various concentrations (1–10 nM) in order to produce a comparable and similar level of contraction in each experiment. NO or SNP was added against a mean background contraction of 2.75±0.5 mN (n=31) corresponding to 70±5% of the high K+ (50 mM)-induced maximum contraction.
In the HUA pre-contracted with the thromboxane mimetic U46619 (1–10 nM), NO (0.03–30 μM) initiated relaxations in a concentration-dependent manner, pEC30 7.43±0.11, pEC50 7.05±0.49 and Emax 95±5%, n=10. However, in 5-HT (0.01–1 μM) pre-contracted vessels, the NO-induced relaxation was attenuated (pEC50 5.99+0.07 and Emax 62±3%, n=8, P<0.05; Figure 2).
Figure 2.
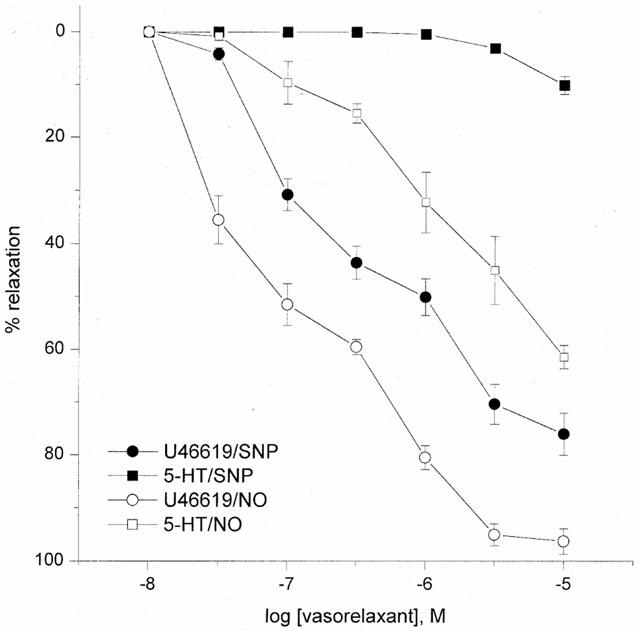
Concentration-response curves for NO- and SNP-induced relaxations in 5-HT and U46619 pre-contracted tissue. Each point represents the arithmetic mean±s.e.mean of n=5 and 6 experimental determinations.
SNP (0.01–10 μM)-induced relaxations were observed in U46619 pre-contracted vessels, pEC30 6.52±0.12, pEC50 6.27±0.04 and Emax 76±4%, n=15, but not in 5-HT pre-contracted vessels (Emax 10±2%, n=10) (Figure 2). The SNP-induced relaxation was found to be distinctly biphasic. The first phase of the response was between 0.1 nM and 0.5 μM, and the second phase between 0.5 and 10 μM.
In all subsequent experiments U46619 was used to pre-contract the vessels.
Effects of oxygen tension on NO- and SNP-induced relaxation
Under low oxygen tension (2.5% oxygen/8% carbon dioxide/ 89.5% nitrogen), HUA pre-contracted with the thromboxane mimetic U46619 (1–10 nM) responded to NO (0.03–30 μM) and SNP (0.01–10 μM) with concentration-dependent relaxations. Increasing the oxygen tension resulted in a significant leftward shift and a change in shape of the NO and SNP concentration-relaxation curves from monophasic to biphasic. NO induced a similar maximal relaxation (Emax) under low and high oxygen tension but with significantly decreased pEC50 values (6.25±0.09 vs 6.93±0.12, respectively, n=5, P<0.05, Figure 3). However, the maximal relaxation induced with SNP was significantly attenuated under low oxygen tension (62±2 vs 77±4% under high oxygen tension, n=5, P<0.05) with pEC50 values of 5.78±0.19 vs 6.52±0.14, n=5, P<0.05 (Figure 3).
Figure 3.
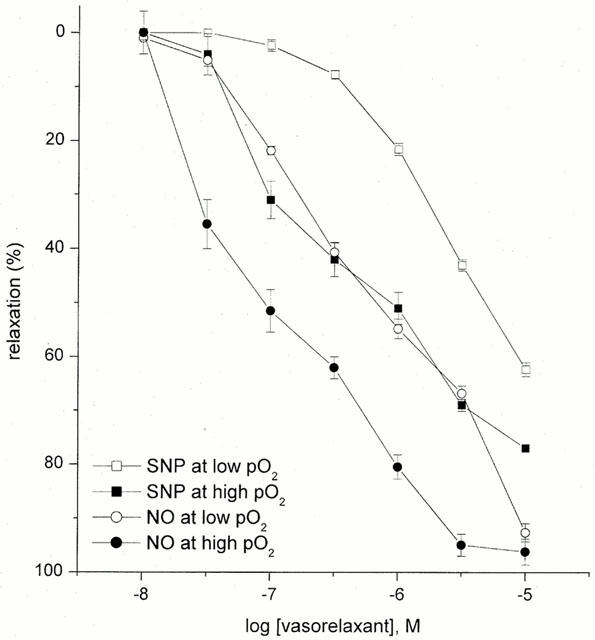
Concentration-response curves for NO- and SNP-induced relaxation in U46619 pre-contracted tissue at low (<55 mmHg) versus high (>300 mmHg) oxygen tension. Each point represents the arithmetic mean±s.e.mean of n=5 experimental determinations.
Role of high K+ concentration and K+-channel inhibitors on NO- and SNP-induced relaxation
In the HUA pre-contracted with 50 mM KCl NO (0.03–30 μM)-induced relaxations were substantially inhibited from 95±5 to 36±5% (Figure 4, n=5, P<0.05).
Figure 4.
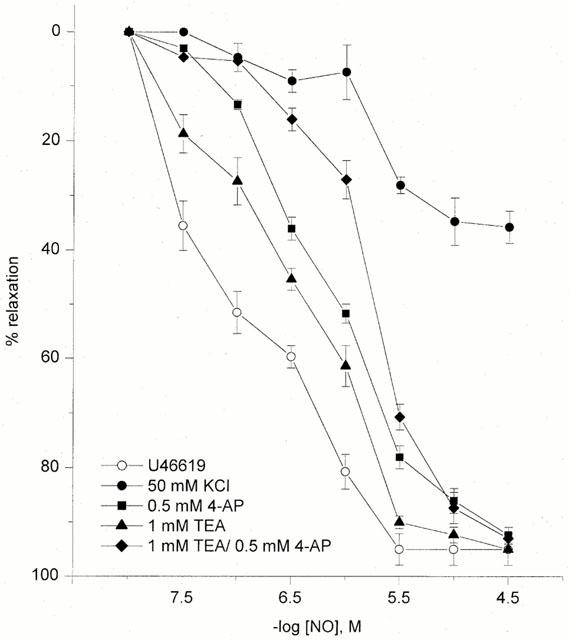
NO-induced relaxation in U46619- or KCl (50 mM)- precontracted HUA. Effects of 0.5 mM 4-AP, 1 mM TEA or 4-AP and TEA together on NO-induced relaxation in U46619- precontracted vessels at high oxygen tension (300 mmHg). Each point represents the arithmetic mean±s.e.mean of n=4–7 experimental determinations.
In U46619-precontracted vessels, pretreatment with the KATP inhibitor glibenclamide (10 μM) did not affect the relaxation induced by NO (n=4, P>0.05) or SNP (n=3, P>0.05) (data not shown). However, 4-aminopyridine (4-AP; 0.5 mM) an inhibitor of Kv, suppressed and produced a rightward shift of NO-evoked relaxations with pEC30 values of 6.62±0.27, pEC50 5.80±0.40 and Emax 86±5, n=7, P<0.05 (Figure 4), whereas dendrotoxin (0.5 μM), a selective Kv 1.1 and Kv 1.6 inhibitor, failed to significantly inhibit the effects of NO (n=3, P>0.05) (data not shown).
Pretreatment with tetraethylammonium (TEA; 1 mM) resulted in a rightward shift of the concentration-response curve for NO without affecting the maximal response; pEC30 6.90±0.15, pEC50 6.58±0.44 and Emax 90±5%, n=5, P>0.05 (Figure 4). The combination of TEA (1 mM) and 4-AP (0.5 mM) produced no greater inhibition or additional shift in the dose-response curve to NO than that observed for 4-AP alone at the pEC50 level (pEC50 5.72±0.22 and Emax 93±2%, n=5, P>0.05, Figure 4). However, the combination of the above-mentioned two K+ channel inhibitors did significantly shift the dose response curve to NO at the pEC30 level (5.95±0.26 vs 6.62±0.27 for 4-AP alone; n=5; P<0.05). Pretreatment with charybdotoxin (0.1 μM) evoked a parallel rightward displacement of the response curve to NO without attenuation of the maximum response (n=3); pEC50 6.19±0.28 vs 7.05±0.49 in controls, and Emax 89±7 vs 95±5%, respectively, P<0.05 (Figure 5). Iberiotoxin (0.1 μM), a selective inhibitor of large conductance Ca2+-activated K+ channels (BKCa) also produced a rightward shift of the dose-response for NO-evoked relaxation; pEC50 and Emax in the presence of iberiotoxin were 5.95±0.26 and 95±5% (n=3), respectively, P>0.05 (Figure 5). Apamin (1 μM) was devoid of inhibitory activity on NO induced relaxation (n=4, P>0.05) (data not shown). In the presence of 4-AP, TEA, charybdotoxin or iberiotoxin the dose-response curve to NO, which was biphasic under control conditions, became monophasic in the presence of K+ channel blockers.
Figure 5.
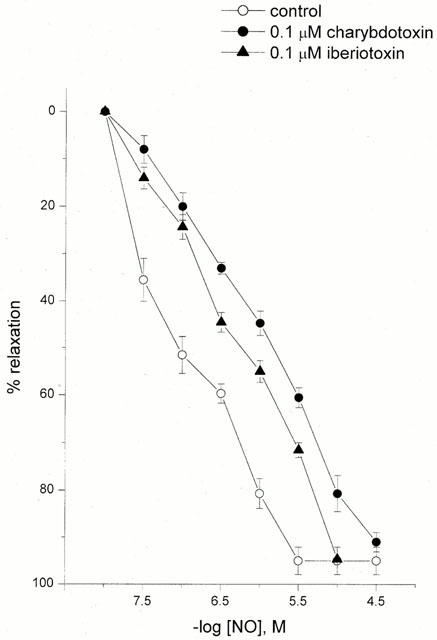
The effect of selective KCa channel inhibitors on NO-induced relaxation in U46619 pre-contracted HUA: 0.1 μM charybdotoxin, and 0.1 μM iberiotoxin. Each point represents the arithmetic mean±s.e.mean of n=3–5 experimental determinations.
In the HUA pre-contracted with 50 mM KCl SNP (0.01–10 μM)-induced relaxations were abolished (Figure 6). In U46619-precontracted vessels, 4-AP (0.5 mM) pretreatment shifted the first phase of the relaxation-response curve to SNP without altering the second phase, pEC30 6.27±0.04 vs 6.90±0.12 in control (P<0.05) (Figure 6). Pretreatment of the tissues with TEA (1 mM) for 30 min was found to produce a significant inhibitory effect (P<0.05) on the first phase of the concentration-response curve to SNP with the pEC30 and Emax values being 6.29±0.05 and 72±1% vs 6.90±0.12 and 76±4% in control (Figure 6). The control concentration-response curves to SNP were biphasic and the responses to low concentrations of SNP were blocked with TEA or 4-AP and in the presence of these K+ channel inhibitors the concentration-response curve became monophasic.
Figure 6.
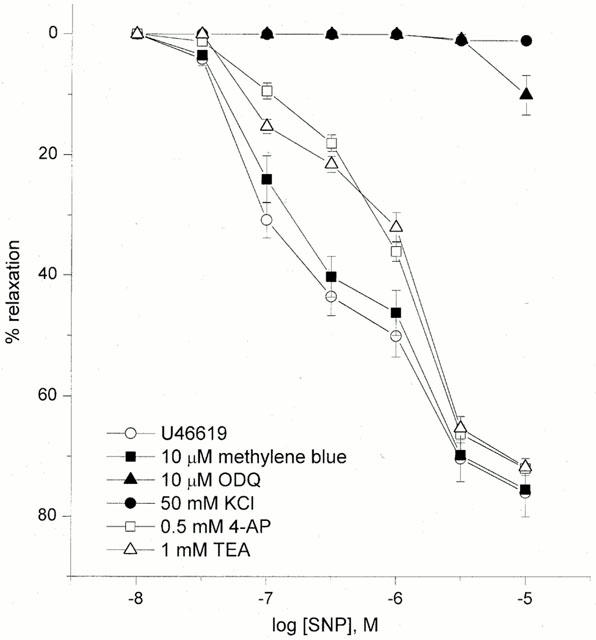
SNP-induced relaxation in U46619- and KCl (50 mM)-precontracted vessels. Effects of guanylyl cyclase inhibitors or potassium channel inhibitors on SNP-induced relaxation in U46619 pre-contracted tissue at high oxygen tension (>300 mmHg). Each point represents the arithmetic mean±s.e.mean of n=5–7 experimental determinations.
Effects of sGC inhibitors alone or in the combination with K+-channel inhibitors on NO- and SNP-induced relaxation.
Pretreatment of the tissues with the sGC inhibitor methylene blue (10 μM) for 30 min did not affect SNP-induced relaxation while ODQ pretreatment significantly decreased SNP-induced relaxation, Emax 10±3%, n=7, P<0.05 (Figure 6).
Pretreatment of the HUA with methylene blue was found to displace the concentration-response curve for NO with pEC50 and Emax values in the presence or absence of methylene blue (n=5) of 6.47±0.45 and 7.05±0.49, and 95±3 and 95±5%, respectively (P<0.05). In the presence of 10 μM of the sGC inhibitor ODQ (n=5), the relaxation responses to NO were partially inhibited with pEC50 and Emax values significantly lower, 5.87±0.28 and 71±5%, P<0.05 (Figure 7).
Figure 7.
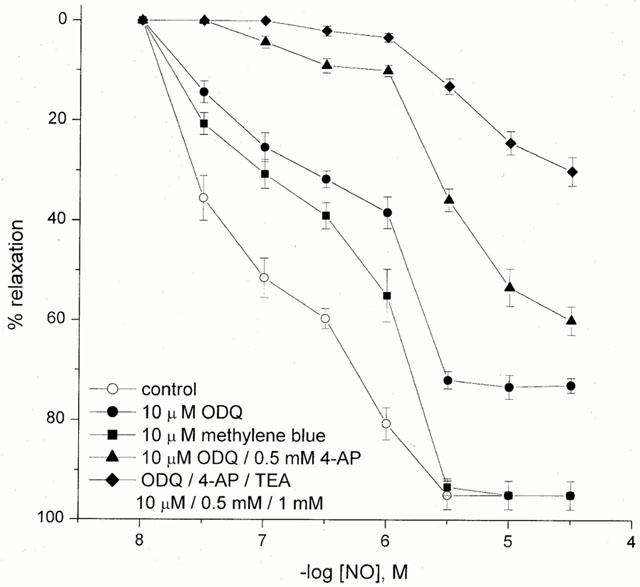
Concentration-response curves for NO-induced relaxation in U46619 pre-contracted tissue in the absence and presence of guanylyl cyclase inhibitors: 10 μM methylene blue and 10 μM ODQ. Effect of ODQ in the presence of 4-AP or 4-AP and TEA on NO-induced relaxation in U46619 pre-contracted HUA. Each point represents the arithmetic mean±s.e.mean of n=5–7 experimental determinations.
Pre-incubation of the HUA for 30 min with the sGC inhibitor ODQ (10 μM) induced a transient contraction of the HUA and vascular tone induced by U46619 (1–10 nM) increased by 7±5% (P>0.05).
A combination of the sGC inhibitor ODQ (10 μM) and 4-AP (0.5 mM) caused no further inhibition in maximal NO-induced relaxation vs ODQ alone (Emax 65±3 and 71±5%, respectively, n=5, P>0.05), but significantly shifted the concentration-response curve (pEC50 5.60±0.11 vs 5.87±0.28, P<0.05, Figure 7). Although TEA (1 mM), in the presence of either ODQ (10 μM) or 4-AP (0.5 mM), failed to produce any additional inhibition of the NO-induced vasorelaxation to that already seen with ODQ or 4-AP alone, a combination of TEA (1 mM) with 4-AP (0.5 mM) and ODQ (10 μM) did significantly reduce the Emax (26±6%, n=5, P<0.05, Figure 7). The combination of apamin with either 4-AP or the KCa inhibitors (TEA, charybdotoxin, iberiotoxin) produced no additional antagonism to that seen with 4-AP or the large conductance KCa inhibitors alone (data not shown).
Effect of nifedipine on NO- and SNP-induced relaxation
In the HUA pre-contracted with U46619 (0.1–10 μM), nifedipine treatment (0.3 μM) produced no significant effect (P>0.05) on either the pEC50 (6.85±0.18) or Emax (94±6%), n=5, for NO-induced relaxation and, in addition, the SNP-induced relaxation was unaffected (pEC30 6.97±0.06 and Emax 74±5%, n=6 versus pEC30 6.90±0.12 and Emax 76±4%, n=13 for control) (Figure 8).
Figure 8.
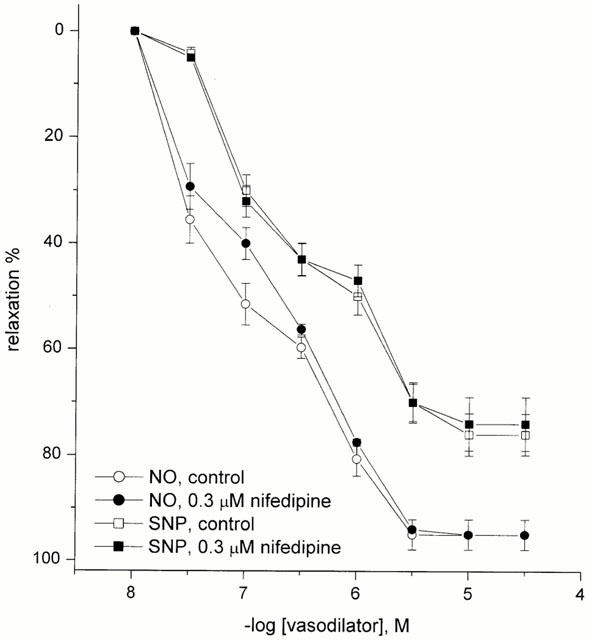
Effect of nifedipine on NO- and SNP-induced relaxation in U46619 pre-contracted HUA. Each point represents the arithmetic mean±s.e.mean of n=5–6 experimental determinations.
Discussion
Contribution of oxygen tension and voltage-operated calcium channels on the HUA responsiveness
In the present study, both NO and the NO-donor SNP evoked relaxation of the pre-contracted HUA, but with different sensitivity. NO-induced relaxation was complete and SNP-induced less than maximal relaxation of the vessel. The differences in the relaxation response to these two compounds in the HUA may reflect differences in the mechanism whereby each of them generates NO. Exogenously applied solutions of NO gas readily provides NO in the physiological bathing solution whereas SNP, although subject to photolysis (O'Neill et al., 1993), does not decompose spontaneously to release NO and has been reported to release only small amounts of NO over comparatively long time periods (Bates et al., 1992; Rao et al., 1991). SNP requires cellular metabolism to produce NO (Kowaluk et al., 1992). The requirement for SNP metabolism may result in a lower maximal relaxation response. Murphy (1999) has reported that variations in the metabolism of vasodilators may underlie differences in their regional specificity or tolerance development. In a recent comparative study of the vasculature from eNOS knockout (−/−) and control mice a supersensitivy to NO and SNP was reported for aortic tissues from eNOS (−/−) mice and, furthermore, the sensitivity increase to SNP was greater than that to NO (Waldron et al., 1999). It was argued that this differential sensitivity to SNP versus NO may reflect differences in the chemical form of NO and, perhaps, differences related to the local release of NO from SNP versus the global cellular effects of a solution of NO gas as well as differing degrees of sGC activation (Waldron et al., 1999). In the HUA we have demonstrated that the relaxation-response to NO compared to SNP is less sensitive to ODQ indicating that cellular mechanisms, other than those mediated by activation of sGC, are more important in determining the sensitivity of the HUA to NO and SNP.
It has been shown that changes in oxygen tension affects the response to agonists in the HUA. A thromboxane A2 receptor antagonist, or blockade of the synthesis of thromboxane, selectively attenuates oxygen-induced contractions, indicating that the elevation of the tone induced by high oxygen tension is likely mediated by the release of endogenous thromboxane A2 (Templeton et al., 1991). To block synthesis of prostanoids at high oxygen tension, all of our experiments were performed in the presence of indomethacin (3 μM). In addition, it has been reported that prostanoid formation in the HUA is altered at high oxygen tension (Bjøro et al., 1987) and that prostanoids are formed endogenously by a cyclo-oxygenase-independent mechanism in vivo in humans (Morrow et al., 1990) and in vitro in human arteries (femoral, tibialis anterior, poplitea, aorta, vertebralis) and umbilical vein (Gniwotta et al., 1997). It is not known whether the production of prostanoids by this pathway contributed to the responses obtained in the HUA.
There are no reports of whether changes in oxygen tension affects vasorelaxation induced with NO or NO donor compounds in the HUA. Our study demonstrates that increasing oxygen tension from the low, but physiological levels, to higher levels obtained with the use of the 95% O2, 5% CO2 gas mixture resulted in an increased vasodilatory potency of NO and SNP. Furthermore, at high oxygen tension the concentration-relaxation curves for both NO and SNP became biphasic. The increased potency at high oxygen tension might reflect the involvement of two distinct cellular mechanisms mediating the vasodilatory effects of NO and SNP. In our study, most of the experimental protocols described were pursued at high oxygen tension and the data indicate that differences between NO- and SNP-mediated relaxation at the high oxygen tension levels generated by the use of 95% O2/5% CO2 gas mixture and PSS, however, it is possible that the cellular mechanism that mediates NO- versus SNP-induced relaxation differs at high versus low oxygen tension.
The vascular receptors mediating 5-HT-induced contraction in the HUA has been shown to be 5-HT2A and 5-HT1B subtypes (Lovren et al., 1999). It is well established that 5-HT2 receptors are coupled to phospholipase C, and inositolphospholipid hydrolysis and calcium mobilization linked to phosphatidylinositol turnover and 5-HT1 receptors are negatively coupled to adenylyl cyclase (Hoyer et al., 1994). In the HUA, contractions evoked following stimulation of 5-HT receptors primarily result from an enhanced influx of extracellular Ca2+ via voltage- or receptor-operated Ca2+ channels with a lesser involvement of mobilization of Ca2+ from intracellular stores (Medeiros & Calixto, 1991). 5-HT-induced contractions in HUA vessels are dependent on extracellular calcium since contractions were greatly attenuated following Ca2+ withdrawal from the physiological solution (Wylam et al., 1993). Moreover, 5-HT evoked contractions were depressed in the presence of nifedipine, a blocker of voltage-gated L-type calcium channels, indicating that calcium entry via nifedipine-sensitive channels plays a major role in 5-HT-mediated contraction (Mederios & Calixto, 1991).
In our studies we found that SNP-induced relaxation in 5-HT pre-contracted HUA vessels is only observed in the presence of calcium-free PSS (personal observation), whereas SNP relaxed the HUA in PSS containing Ca2+ when the thromboxane analogue, U46619 was used to induce contraction. U46619 initiates a contraction in the HUA which is largely independent of voltage-operated Ca2+ channels and depends primarily on the release of intracellular Ca2+ (Toyofuku et al., 1995) and this is supported by the data that we have presented in the current study.
It has been reported that NO modulates Ca2+-channel activity in vascular smooth muscle and thus induces relaxation (Blatter & Wier, 1994; Clapp & Gurney, 1991). However, in the presence of 0.3 μM nifedipine, which should inhibit L-type Ca2+ channel activity, NO and SNP still induced maximum relaxation in the HUA pre-contracted with U46619. Thus, NO- and SNP-mediated relaxations in the HUA are nifedipine-resistant indicating that the vasorelaxant action is independent of the closure of voltage-operated Ca2+-channels.
Role of sGC activation in relaxation response to NO and SNP
Activation of sGC and subsequent accumulation of cyclic GMP is thought to be the principal mechanism that ultimately leads to NO-induced vasorelaxation. In the present study, we report that whereas the presence of the sGC inhibitor ODQ completely inhibited the relaxation response to SNP that to NO was, even in the presence of K+-channel blockers, not completely eliminated. Although there are many other potential molecular targets for NO, sGC is probably the most sensitive (Stamler, 1994).
In the HUA smooth muscle cyclic GMP levels were increased in response to an NO donor such as SNP (Bergh et al., 1995). Furthermore, it was reported that A23187-evoked relaxation of the HUA was accompanied by an increase in cyclic GMP (Chaudhuri et al., 1993). However, HUA vessels pre-contracted with 5-HT exhibited little if any response to nitrovasodilators (White, 1988; Chaudhuri et al., 1991) leading to the suggestion that the HUA either lacks sGC or lacks the cellular targets for cyclic GMP (Renowden et al., 1992; Bergh et al., 1995). Nonetheless, in vessels pre-contracted with 5-HT in the absence of extracellular Ca2+, or when the entry of extracellular Ca2+ is prevented, nitrovasodilators produced dose-dependent relaxation (Sugawara et al., 1997; Wylam et al., 1993). These findings are compatible with the view that cyclic GMP relaxes the HUA by reducing intracellular Ca2+ concentration and that the 5-HT-mediated influx of extracellular Ca2+, via the nifedipine-sensitive process, overwhelms the cyclic GMP-dependent cellular events. Our demonstration of the greater ability of ODQ to inhibit SNP- versus NO-mediated relaxation indicated that NO, as applied to the cell as a solution of the gas, has cellular targets in addition to sGC whereas the NO released from SNP mediates relaxation of the HUA entirely via an ODQ-sensitive mechanism(s).
Recently, Feelisch et al. (1999) have reported that ODQ lacks specificity for sGC and interferes with other haeme-dependent processes. They showed that ODQ affects SNP-mediated relaxation by inhibiting reductive bioactivation via cytochromes P450. Therefore, the higher sensitivity of SNP versus NO to inhibition by ODQ that we report in our study might be result not only of the sGC inactivation with ODQ, but also an impairment of SNP biotransformation in the presence of ODQ. Another possibility is that the concentration range of SNP used in our experiments were insufficient to relax the vessel when ODQ was applied. Of interest is the report that after pretreatment of the rat aorta with ODQ, SNP- induced relaxation only in very high concentrations (>1 mM) (Feelisch et al., 1999).
Role of K+ - channel activation to relaxation of HUA by NO and SNP
SNP-induced relaxations in the HUA were completely inhibited and those to NO were significantly reduced (by approximately 60%) by increasing the extracellular concentration of K+, thus suggesting that relaxation involves smooth muscle hyperpolarization. K+ channels and the transmembrane K+ gradient modify the membrane potential, thus affecting the tone of the smooth muscle cells (Adeagbo & Triggle, 1993) and the opening of K+ channels results in K+ efflux, hyperpolarization and relaxation (Edwards & Weston, 1994). We examined the contribution of K+ channels to SNP-induced vasodilation using selective inhibitors of K+ channels in the HUA pre-contracted with U46619. SNP failed to induce relaxation when the extracellular concentration of K+ was increased. We attribute this inhibition to the predicted membrane depolarization elicited by elevated extracellular K+ that interferes with SNP-induced relaxation in a similar manner as we also report for HUA pre-contracted with 5-HT. SNP-induced relaxations in the HUA were found to be noticeably biphasic suggesting the role for two cellualar sites of action. The relaxations induced with low concentrations of SNP were significantly reduced by 4-AP or TEA treatment at pEC30 but not pEC50 levels (pEC30 6.27 and 6.29, respectively, versus control 6.90) and were abolished by the sGC inhibitor, ODQ. Recent studies have shown that NO can stimulate charybdotoxin-sensitive K+ channels, either directly (Bolotina et al., 1994) or via a cyclic GMP-dependent mechanism (George & Shikata, 1995) and that cyclic GMP itself can directly activate K+ channels (Robertson et al., 1993). Our data indicates that at low concentrations, SNP induced relaxation occurs through sGC and activating K+ channels. In contrast, at higher concentrations, SNP-induced relaxation is mediated through a sGC-cyclic GMP pathway, but independent of K+ -channel activation. In addition, the involvement of two cellular mechanisms of action may also explain the biphasic nature of the relaxation to SNP and our observation that in the presence of K+ channel inhibitors the relaxation becomes monophasic. These data indicate that a cyclic GMP-dependent K+ channel-sensitive pathway can mediate smooth muscle relaxation in the HUA.
We examined the contribution of K+ channels to NO-induced vasorelaxation using selective inhibitors of K+ channels in the HUA pre-contracted with U46619. Comparative studies with TEA and the selective blockers, iberiotoxin, charybdotoxin, apamin and glibenclamide, suggest that the activation of a large conductance BKCa channel and 4-AP-sensitive voltage-gated KV channels may contribute to NO-evoked relaxation, but not small conductance Ca2+-activated nor glibenclamide sensitive KATP channels. The inhibition of NO-induced relaxation by both charybdotoxin and iberiotoxin indicates that NO can activate BKCa (Giangiacomo et al., 1992; Kaczorowski et al., 1996), but charybdotoxin also inhibits Kv 1.2 and Kv 1.3 (Kaczorowski et al., 1996). 4-AP also inhibited NO-induced relaxation in the HUA. 4-AP can, at high concentrations, also inhibit BKCa and KATP channels (Hermann & Gorman, 1981; Nelson & Quayle, 1995) but at the concentration of 0.5 mM used in the present study, it is likely selective for Kv channels. Thus, the inhibitory effects of both 4-AP and charybdotoxin on NO-induced relaxation is suggestive of a role for Kv channels. High extracellular K+ concentration also produces a greater inhibition of NO-induced relaxation than that reported for a combination of K+ channel inhibitors (Figure 4). This may suggest that an additional TEA and 4-AP insensitive, K+ channel(s) contributed to the NO- mediated vasorelaxation of the HUA and/or that high extracellular K+ produces effects other than eliminating the role of K+-channel-mediated hyperpolarization.
In addition, neither the sGC inhibitor ODQ nor K+ channel blockers alone completely eliminated NO-induced relaxation. We have, as already noted, reported similar findings for the rat renal afferent arterioles (Trottier et al., 1998). The cellular basis for the cyclic GMP/K+-channel-independent action of NO is unknown, but may involve an action on cellular metabolism (Brüne et al., 1994; Cleeter et al., 1994), activation of heat-shock protein 70 (Xu et al., 1997) and/or an action subsequent to the formation of peroxynitrite (Beckman & Koppenol, 1996).
No attempt was made to remove the endothelial cell layer from the HUA since it was previously reported that the endothelium of the HUA does not respond to endothelium-dependent vasorelaxants with the release of an endothelium-derived relaxing factor (EDRF) (Xie & Triggle, 1994). It is thus unlikely that the presence of the endothelium and the release of an EDRF affects the relaxation responses to NO and SNP.
Conclusions
In conclusion, the present study provides evidence that relaxation induced by authentic NO in the HUA is mediated via both sGC-cyclic GMP-dependent and independent mechanisms as well as K+ channel activation that, presumably, leads to hyperpolarization, whereas SNP-induced relaxation is primary mediated via a cyclic GMP-dependent mechanism and, at low concentrations, a cyclic GMP-dependent activation of a TEA-sensitive K+ channel(s). Based on studies of the effects of K+ channel inhibitors, we conclude that NO-mediated hyperpolarization is partly due to the opening of Kv and KCa channels. In addition, since none of the tested inhibitors could completely eliminate relaxation induced by higher concentrations of NO (⩾1 μM), we suggest that an additional, as yet unidentified mechanism(s) may be also involved in NO-induced relaxation in the HUA. Furthermore, we conclude that the relative contribution of voltage-dependent and -independent mechanisms to relaxation is influenced by the cellular processes mediating contraction. Thus, the vasorelaxation response to NO in the presence of 5-HT induced vascular tone is significantly lower than that produced in the presence of an equivalent level of tone induced by U46619. Moreover, HUA is unresponsive to SNP when contracted with 5-HT, but relaxes when contracted with U46619. These data may reflect the greater dependence of 5-HT-induced contraction of the HUA on membrane depolarization and the opening of voltage-gated Ca2+ -channels.
Acknowledgments
The support of the Heart & Stroke Foundation of Alberta (to C.R. Triggle) is gratefully acknowledged. We would like to thank the staff of the delivery room at the Foothills Hospital, Calgary for their cooperation.
Abbreviations
- 4-AP
4-aminopyridine
- cyclic GMP
cyclic 3′: 5′ guanosine monophosphate
- EDRF
endothelium-derived relaxing factor
- sGC
soluble guanylyl cyclase
- 5-HT
5-hydroxytryptamine
- HUA
human umbilical artery
- KCa
Ca2+-activated K+ channels
- KV
voltage-gated K+ channels
- NO
nitric oxide
- ODQ
1H[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- SNP
sodium nitroprusside
- TEA
tetraethylammonium
References
- ADEAGBO A.S., TRIGGLE C.R. Varying extracellular [K+]: A functional approach to separating EDHF- and EDNO-related mechanism in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- AZUMA H., ISHIKAWA M., SEKIZAKI S. Endothelium-dependent inhibition of platelet aggregation. Br. J. Pharmacol. 1986;88:411–415. doi: 10.1111/j.1476-5381.1986.tb10218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BATES J.N., BAKER M.T., GUERRA R., HARRISON D.E. Chemical release of nitric oxide from sodium nitroprusside to nitric oxide in vascular smooth muscle. J. Pharmacol. Exp. Ther. 1992;262:916–922. [PubMed] [Google Scholar]
- BECKMAN J.S., KOPPENOL W.H. Nitric oxide, superoxide, and peroxynitrate: the good, the bad and the ugly. Am. J. Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- BERGH C.M., BROPHY C.M., DRANSFIELD D.T., LINCOLN T., GOLDENRING J.R., RASMUSSEN H. Impaired cyclic nucleotide-dependent vasorelaxation in human umbilical artery smooth muscle. Am. J. Physiol. 1995;286:H202–H212. doi: 10.1152/ajpheart.1995.268.1.H202. [DOI] [PubMed] [Google Scholar]
- BJØRO K., HAUGEN G., STRAY-PEDERSEN S. Altered prostanoid formation in human umbilical vasculature in response to variations in oxygen tension. Prostaglandins. 1987;34:377–384. doi: 10.1016/0090-6980(87)90083-9. [DOI] [PubMed] [Google Scholar]
- BLATTER L.A., WIER W.G. Nitric oxide decreased [Ca2+]i in vascular smooth muscle by inhibition of the calcium current. Cell Calcium. 1994;15:122–131. doi: 10.1016/0143-4160(94)90051-5. [DOI] [PubMed] [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P.J., COHEN R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;386:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- BRÜNE B., DIMMELER S., MOLINA Y., VEDIA L., LAPETINA E.G. Nitric oxide: a signal for ADP ribosylation of proteins. Life Sci. 1994;54:61–70. doi: 10.1016/0024-3205(94)00775-6. [DOI] [PubMed] [Google Scholar]
- CHAUDHURI G., BUGA G.M., GOLD M.E., WOOD K.S., IGNARRO L.J. Characterization and actions of human umbilical endothelium derived relaxing factor. Br. J. Pharmacol. 1991;102:331–336. doi: 10.1111/j.1476-5381.1991.tb12174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAUDHURI G., CUEVAS J., BUGA G.M., IGNARRO L.J. NO is more important than PGI2 in maintaining low vascular tone in feto-placental vessels. Am. J. Physiol. 1993;265:H2036–H2043. doi: 10.1152/ajpheart.1993.265.6.H2036. [DOI] [PubMed] [Google Scholar]
- CLAPP L.H., GURNEY A.M. Outward currents in rabbit pulmonary artery cells dissociated with a new technique. Exp. Physiol. 1991;76:677–693. doi: 10.1113/expphysiol.1991.sp003535. [DOI] [PubMed] [Google Scholar]
- CLEETER M.W.J., COOPER J.M., DARLEY-USMAR V.M., MONCADA S., SCHAPIRA A.H.V. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratoty chain, by nitric oxide. FEBS. Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H.Effect of potassium channel modulating drugs on isolated smooth muscle Handbook of Experimental Pharmacology 1994Heidelberg: Springer; 469–531.eds. Szekeres, L., Papp, J.G. III, pp [Google Scholar]
- FEELISCH M., KOTSONIS P., SIEBE J., CLEMENT B., SCHMIDT H.H.H.W. The soluble guanylyl cyclase inhibitor 1H-[1,2,4] oxadiazolo [4,3,-a]quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol. Pharmacol. 1999;56:243–253. doi: 10.1124/mol.56.2.243. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GARLAND C.J., MCPHERSON G.A. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. Br. J. Pharmacol. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEORGE M.J., SHIKATA E.F. Regulation of calcium-activated potassium channels by S-nitrosothiol compounds and cyclic, guanosine monophosphate in rabbit coronary artery myocytes. J. Invest. Med. 1995;43:451–458. [PubMed] [Google Scholar]
- GIANGIACOMO K.M., GARCIA M.L., MCMANUS O.B. Mechanism of iberiotoxin block of large conductance calcium-activated potassium channel from bovine aortic smooth muscle. Biochemistry. 1992;31:6719–6727. doi: 10.1021/bi00144a011. [DOI] [PubMed] [Google Scholar]
- GNIWOTTA C., MORROW J.D., ROBERTS J.L., II, KÜHN H. Prostaglandin F2-like compounds, F2-isoprostanes, are present in increased amounts in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 1997;17:3236–3241. doi: 10.1161/01.atv.17.11.3236. [DOI] [PubMed] [Google Scholar]
- HERMANN A., GORMAN A.L.F. Effects of 4-aminopyridine on potassium currents in molluscan neuron. J. Gen. Physiol. 1981;78:63–86. doi: 10.1085/jgp.78.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOYER D., CLARKE D.E., FOZARD J.R., HARTIG P.R., MARTIN R.G., MYLECHARANE E.J., SAXENA P.R., HUMPHEREY P.A. VII. International Union of Pharmacology: Classification of receptors for 5-hydroxytriptamine (serotonin) Pharmacol. Rev. 1994;49:157–203. [PubMed] [Google Scholar]
- IGNARRO L.J. Haem-dependent activation of guanylate cyclase and cyclic GMP formation by endogenous nitric oxide: A unique transduction mechanism for transcellular signalling. Pharmacol. Toxicol. 1990;67:1–7. doi: 10.1111/j.1600-0773.1990.tb00772.x. [DOI] [PubMed] [Google Scholar]
- KACZOROWSKI G.J., KNAUS H.G., LEONARD R.J., MCMANUS O.B., GARCIA M.L. High conductance calcium activated potassium channels; structure, pharmacology and function. J. Bioenerg. Biomembr. 1996;28:255–267. doi: 10.1007/BF02110699. [DOI] [PubMed] [Google Scholar]
- KANNAN M.S., JOHNSON D.E. Modulation of nitric oxide-dependent relaxation of pig tracheal smooth muscle by inhibitors of guanylyl cyclase and calcium activated potassium channels. Life Sci. 1995;56:2229–2238. doi: 10.1016/0024-3205(95)00212-o. [DOI] [PubMed] [Google Scholar]
- KOWALUK E.A., SETH P., FUNG H.-L. Metabolic activation of sodium nitroprusside to nitric oxide in vascular smooth muscle. J. Pharmacol. Exp. Ther. 1992;262:916–922. [PubMed] [Google Scholar]
- LOVREN F., LI X.F., LYTTON J., TRIGGLE C.R. Functional characterization and m-RNA expression of 5-HT receptors mediating contraction in human umbilical artery. Br. J. Pharmacol. 1999;127:1247–1255. doi: 10.1038/sj.bjp.0702647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEDEIROS Y.S., CALIXTO J.B. Influence of calcium entry blockers and calmodulin inhibitors on 5-HT-, potassium- and calcium-induced contractions in human umbilical artery in vitro. J. Pharm. Pharmacol. 1991;43:411–416. doi: 10.1111/j.2042-7158.1991.tb03499.x. [DOI] [PubMed] [Google Scholar]
- MISTRY D.K., GARLAND C.R. Nitric oxide (NO)-induced activation of large conductance Ca2+ -dependent K+ channels (BKCa) in smooth muscle cells isolated from the rat mesenteric artery. Br. J. Pharmacol. 1998;124:1131–1140. doi: 10.1038/sj.bjp.0701940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- MORROW J.D., HILL K.E., BURK R.F., NAMMOUR T.M., BADR K.F., ROBERTS J.L., II A series of prostaglandin F2-like compounds are produced in vivo in humans by non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. U.S.A. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY E.M. Influence of redox compounds on nitrovasodilator-induced relaxations of rat coronary arteries. Br. J. Pharmacol. 1999;128:435–443. doi: 10.1038/sj.bjp.0702777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURPHY M.E., BRAYDEN J.E. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J. Physiol. 1995;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MYATT L., BROCKMANN D.E., LANGDON G., POLLOCK J.S. Constitutive calcium-dependent isoform of nitric oxide synthase in the human placental villous vascular tree. Placenta. 1993;14:373–383. doi: 10.1016/s0143-4004(05)80459-x. [DOI] [PubMed] [Google Scholar]
- NELSON M.T., QUAYLE J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- O'NEILL S.K., DUTTA S., TRIGGLE C.R. Computerized data acquisition and analysis applied to chemiluminescence detection of nitric oxide in haedspace gas. J. Pharmacol. Toxicol. Meth. 1993;29:217–221. doi: 10.1016/1056-8719(93)90028-d. [DOI] [PubMed] [Google Scholar]
- PLANE F., PEARSON T., GARLAND C.J. Multiple pathways underlying endothelium-dependent relaxation in the rabbit isolated femoral artery. Br. J. Pharmacol. 1994;115:31–38. doi: 10.1111/j.1476-5381.1995.tb16316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO D.N.R., ELGUINDI S., O'BRIEN P.J. Reductive metabolism of nitroprusside in rat hepatocytes and human erythrocytes. Arch. Biochem. Biophys. 1991;286:30–37. doi: 10.1016/0003-9861(91)90005-4. [DOI] [PubMed] [Google Scholar]
- RENOWDEN S., EDWARDS D.H., GRIFFTH T.M. Impaired cyclic nucleotide-mediated vasorelaxation may contribute to closure of the human umbilical artery after birth. Br. J. Pharmacol. 1992;106:348–353. doi: 10.1111/j.1476-5381.1992.tb14339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTSON B.E., SCHUBERT R., HESCHELER J., NELSON M.T. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle. Am. J. Physiol. 1993;265:C299–C303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- STAMLER J.S. Redox signaling: nitrozylation and related target interactions of nitric oxide (Review) Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- SUGAWARA M., TOHSE N., NAGASHIMA M., YABU H., KUDO R. Vascular reactivity of endothelium-derived relaxing factor in human umbilical artery at term pregnancy. Can. J. Physiol. Pharmacol. 1997;75:818–824. [PubMed] [Google Scholar]
- TARE M., PARKINGTON H.C., COLEMAN H.A., NEILD T.O., DUSTING G.J. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- TEMPLETON A.G.B., MCGRATH J.C., WHITTLE M.J. The role of endogenous thromboxane in contractions to U46619, oxygen, 5-HT and 5-CT in the human isolated umbilical artery. Br. J. Pharmacol. 1991;103:1079–1084. doi: 10.1111/j.1476-5381.1991.tb12303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOYOFUKU K., NISHIMURA J., KOBAYASHI S., NAKANO H., KANAIDE H. Effects of U46619 on intracellular calcium concentration and tension in human umbilical artery. Am. J. Obstet. Gynecol. 1995;172:1414–1421. doi: 10.1016/0002-9378(95)90471-9. [DOI] [PubMed] [Google Scholar]
- TROTTIER G., TRIGGLE C.R., O'NEILL S.K., LOUTZENHIESER R. Cyclic GMP-dependent and cyclic GMP-independent actions of nitric oxide on the renal afferent arteriole. Br. J. Pharmacol. 1998;125:563–569. doi: 10.1038/sj.bjp.0702090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DE VOORDE J., VANDERSTICHELE H., LENSEN I. Release of endothelium derived relaxing factor from human umbilical vessels. Circ. Res. 1987;60:517–522. doi: 10.1161/01.res.60.4.517. [DOI] [PubMed] [Google Scholar]
- WALDRON G.J., DING H., LOVREN F., KUBES P., TRIGGLE C.R. Acetylcholine-induced relaxation of peripheral nerves isolated from mice lacking endothelial nitric oxide synthase. Br. J. Pharmacol. 1999;128:653–658. doi: 10.1038/sj.bjp.0702858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE R.P. Comparision of vasorelaxants in human basilar arteries and umbilical arteries. Eur. J. Pharmacol. 1988;155:109–115. doi: 10.1016/0014-2999(88)90408-6. [DOI] [PubMed] [Google Scholar]
- WYLAM M.E., SAMSEL R.W., SCHUMACKER P.T., UMANS J.G. Extracellular calcium and intrinsic tone in the human umbilical artery. J. Pharmacol. Exper. Ther. 1993;266:1475–1481. [PubMed] [Google Scholar]
- XIE H., TRIGGLE C.R. Endothelium-independent relaxations to acetylcholine and A23187 in the human umbilical artery. J. Vasc. Res. 1994;31:92–105. doi: 10.1159/000159035. [DOI] [PubMed] [Google Scholar]
- XU Q., HU Y., KLEINDIENST R., WICK G. Nitric oxide induces heat-shock protein 70 expression in vascular smooth muscle cells via activation of heat shock factor 1. J. Clin. Invest. 1997;100:1089–1097. doi: 10.1172/JCI119619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YALLAMPALLI C., IZUMI H., BYAM-SMITH M., GARFIELD R.E. An L-arginine-nitric oxide cyclic guanosine monophosphate system exists in the uterus and inhibits contractility during pregnancy. Am. J. Obstet. Gynecol. 1994;170:175–185. doi: 10.1016/s0002-9378(94)70405-8. [DOI] [PubMed] [Google Scholar]