

Abstract
The atria and ventricles of transgenic mice (TGβ2) with cardiac overexpression of the human β2-adrenoceptor (β2AR) were initially reported to show maximum contractility in the absence of β-AR stimulation. However, we have previously observed a different phenotype in these mice, with myocytes showing normal contractility but reduced βAR responses. We have investigated the roles of cyclic AMP and Gi in basal and βAR function in these myocytes.
ICI 118,551 at inverse agonist concentrations decreased contraction by 32%. However, the cyclic AMP antagonist Rp-cAMPS had no effect on contraction in TGβ2 myocytes, indicating that there was no tonic influence of raised cyclic AMP. These findings cannot be explained by the proposed model for inverse agonism, where the activated receptor (R*) raises cyclic AMP levels and so increases contraction in the absence of agonist.
After pertussis toxin (PTX) pretreatment to produce inactivation of Gi, the basal contraction in 1 mM Ca2+ was increased in TGβ2 mice (7.82±0.47%, n=23) compared to LM mice (3.60±0.59%, n=11) (P<0.001). The contraction amplitude of myocytes to the maximal concentration of isoprenaline was also increased significantly by PTX in TGβ2 mice (9.40±1.22%, n=8) and was no longer reduced compared to LM mice (8.93±1.50%, n=11). Both β1- and β2AR subtypes were affected both by the original desensitization and by the resensitization with PTX.
PTX treatment has therefore restored the original phenotype, with high basal contractility and little further effect of isoprenaline. We suggest that both β-AR desensitization and lack of increased basal contraction in ventricular myocytes from our colony of TGβ2 mice were due to increased activity of PTX-sensitive G-proteins.
Keywords: β2-adrenoceptor overexpression, inhibitory G-protein, myocyte shortening, transgenic mice
Introduction
β-Adrenoceptors (βARs) are integral membrane proteins mediating the effects of catecholamines on heart rate and force. β1 and β2 subtypes increase contractile force through an action on adenylyl cyclase, coupled via the stimulatory guanine nucleotide binding protein (Gs) (Kaumann & Molenaar, 1997). The β1AR subtype is the predominant influence on ventricular contraction, but the β2ARs assume greater importance in man, especially during the development of heart failure (del Monte et al., 1993; Harding et al., 1994). The β1AR-selective β-blockers now being used for heart failure (as well as angina and hypertension) can upregulate the β2AR subtype in atria (Hall et al., 1990). Recent evidence suggests that a polymorphism in the β2AR subtype has a strong prognostic effect for mortality in patients with cardiomyopathies (Liggett et al., 1998).
Because of this interest in the β2AR subtype, a transgenic mouse was created with a large (200 fold) overexpression of the human β2AR (TGβ2) (Milano et al., 1994). The TGβ2 phenotype itself initially showed high basal contractility, with little further stimulation by isoprenaline. Certain βAR antagonists, termed inverse agonists (Bond et al., 1995), produced negative inotropic effects and reduced the high basal contractility in isolated atria. Neutral antagonists blocked this negative inotropic effect. This led to the suggestion that a proportion of the β2ARs were spontaneously active in the absence of agonists: in the overexpressing mice this proportion was sufficient to increase contraction to levels equivalent to those produced by maximum isoprenaline stimulation.
However, later studies confirmed the lack of response to isoprenaline but did not detect the raised basal contractility (Heubach et al., 1999). It was suggested that an adaptation had occurred to oppose the effect of the spontaneously active β2ARs. β2ARs in a cell line have been shown to couple to both Gs and Gi (the inhibitory guanine nucleotide-binding protein), with phosphorylation by protein kinase A (PKA) switching the β2AR towards Gi (Daaka et al., 1997). It would be predicted that PKA activity would be high in TGβ2 myocardium, predisposing to Gi coupling. Additionally, Gi levels are reported to be increased in the TGβ2 mouse (Nagaraja et al., 1997). Activation of Gi by muscarinic or adenosine A1 receptors is known to suppress the effects of simultaneous stimulation through Gs. It was therefore hypothesized that excess Gi coupling was decreasing the observed effects of the overexpressed β2AR in the hearts of TGβ2 mice.
In this investigation we have confirmed that basal contractility is not increased in the myocytes from TGβ2 mice at either low or high Ca2+ levels, and that cyclic AMP does not support contraction in the absence of βAR stimulation. By using isolated ventricular myocytes we have excluded the possibility that endogenously released catecholamines might be modulating contractility. We show that pertussis toxin treatment, which inactivates Gi, restores responses to βAR stimulation in myocytes from TGβ2 ventricle. In this we agree with another study, which has been published while the experiments reported here were in progress (Xiao et al., 1999). We further show here that both overexpressed β2AR and the native β1AR are involved in the spontaneous down-regulation and pertussis toxin-induced recovery of response. Additionally, we demonstrate that pertussis toxin treatment increases basal contraction of TGβ2 myocytes towards levels seen during agonist stimulation, thus completely restoring the original phenotype.
Methods
Confirmation of transgenic status
All studies complied with the United Kingdom Home Office Regulation Governing the Care and Use of Laboratory Animals. TGβ2 mice and non-transgenic littermate mice had a mixed genetic background and their age ranged from 8 weeks to 12 months. Animals were derived from transgenic/transgenic or littermate/littermate pairings. The genetic status of the offspring was checked prior to experimentation. Mouse genomic DNA was extracted from tail clips by the Genera Purgene system with DNeasy Tissue Kit (Qiagen Ltd., U.K.). TGβ2 mice were identified by Southern blot analysis with an 850 bp SV-40 DNA probe (Milano et al., 1994). The method did not distinguish between homo-and heterozygous genotype for the human β2AR transgene.
Isolation of ventricular myocytes
Ventricular myocytes were isolated by enzymatic dissociation as previously described (Heubach et al., 1999). Mice were killed by cervical dislocation and the heart quickly removed and placed into ice-cold Krebs-Henseleit (KH) solution (composition in mM: NaCl 119, KCl 4.7, MgSO4 0.94, KH2PO4 1.2, NaHCO3 25, glucose 11.5, CaCl2 1), previously bubbled with 95% O2/5% CO2 to bring the pH to 7.4. The heart was then mounted on a needle canal (23G 1 1/4′′) attached to a Langendorff perfusion apparatus and equilibrated with KH solution containing 1 mM Ca2+ for 5 min at 37°C. The perfusate was then changed to a low calcium (LC) medium of the following composition (mM): NaCl 120, KCl 5.4, MgSO4 5, pyruvate 5, glucose 20, taurine 20, HEPES 10, nitrilotriacetic acid 5, containing 12–15 μM, calcium as measured with a calcium electrode, also at 37°C and bubbled with 100% O2 at pH 6.95, for 5 min. This was followed by 1 min perfusion with the same low calcium solution (with nitrilotriacetic acid omitted) at pH 7.4 with 200 μM calcium and 2 I.U. (Sigma P-8038) protease type XXIV added. This enzyme solution was then switched to one containing no added protease but 1.0 mg ml−1 (Worthington 4376 CLS-2) collagenase and 0.6 mg ml−1 (Sigma H-3506) hyaluronidase for a further 10 min, after which the ventricles were chopped and incubated in the same enzyme solution for 2×10 min. The medium was shaken gently throughout this incubation at 35°C, and kept under an atmosphere of 100% O2, the dispersed cells were then strained through gauze of mesh size 300 μm and supernatant was centrifuged at 40×g for 1 min at room temperature. Cells were washed and resuspended in KH solution at room temperature until used.
Pertussis toxin treatment
Myocytes were incubated with pertussis toxin (PTX) (1.5 μg ml−1) at 35°C for at least 3 h. PTX-treated myocytes were compared with cells that had been kept at 35°C in the absence of PTX for an equal time. After PTX treatment, both PTX-treated and non-treated cells were kept at room temperature until the time of experiments.
Measurement of cell contraction
The contraction of single ventricular myocyte was recorded as previously described (Harding et al., 1988). Myocytes were placed into a Perspex cell bath on the stage of an inverted microscope (Nikon or Zeiss IM), and superfused continuously with KH solution (32°C) equilibrated with 95% O2/5% CO2 at 1.8 to 2 ml min−1. Cells were stimulated at a basal rate of 1 Hz by bipolar pulses through platinum electrodes placed along the bath. Unloaded cell shortening was measured with a video-camera/length detection system, with spatial resolution 1 to 512 and time resolution 10 ms. Cells were allowed to stabilize for at least 10 min at 1 mM Ca2+, before they were challenged with increasing concentrations of Ca2+ or cumulative concentrations of ISO (3×10−11∼10−6) until either a maximum response was reached, or the myocyte developed arrhythmias.
For the studies with Rp-cAMPS or inverse agonist, myocytes were contracting in near-physiological (2 mM) or high Ca2+ (4 mM) Rp-cAMPS (100 μM final concentration) was added to the reservoir and allowed to recirculate for 40 min. For the antagonist studies myocytes were preincubated with either 50 nM ICI 118,551 (selective β2AR antagonist) or CGP 20712A (selective β1AR antagonist) for 20–30 min as previously described (del Monte et al., 1993): at these concentrations there was no effect of the antagonist on basal contraction. When inverse agonist effects of ICI 118,551 were required, the concentration used was 1 or 3 μM, which was the maximum used in the original report (Milano et al., 1994). Inverse agonist effects were maximal after 5 min exposure and reversed with approximately the same time course.
Radioligand receptor binding assay
Myocytes were suspended in 25 mM Tris-HCl buffer (pH 7.4) containing 0.32 mM sucrose, and ventricles (trimmed free of connective tissue, great vessels and valves) were suspended in buffer containing (mM): Tris-HCl 50, EGTA 5, EDTA 5, MgCl2 4, ascorbic acid 1, phenylmethylsulphonylfluoride (PMSF) 0.5 (pH 7.4). Both were homogenized at 4°C with a Polytron homogenizer, then myocytes were further disrupted by repeated passage through a sterile 21G 1.5 needle. The homogenate was then centrifuged at low speed (myocytes; 1000×g, ventricle; 175×g) for 10 min at 4°C to remove unhomogenized debris, and the supernatant centrifuged at 40,000×g for 20 min (myocytes) or 50,000×g for 15 min (ventricle) at 4°C. Aliquots of membrane were incubated with [125I]-iodocyanopindolol (ICYP) (specific activity: 2000 Ci mmol−1) in the absence and presence of 200 μM ISO (to define non-specific binding). Incubation was carried out at 37°C for 120 min in duplicate or triplicate. The incubation was terminated by rapid vacuum filtration through glass-fibre filters using a cell harvester (Brandel). Filters were rapidly washed with 3×5 ml ice-cold assay buffer and counted in a Gamma counter (Packard Instruments, Downers Grove, IL, U.S.A) at an efficiency of 80%. Protein concentration was determined by the method of Bradford (Bradford, 1976) with IgG used as the standard. The experiments were analysed for one binding site by non-linear curve fitting with the equation:
where Beq is the amount of ICYP binding at equilibrium, Bmax is the maximum density of β-ARs and KD is the equilibrium dissociation constant of ICYP.
Immunoblotting techniques
Immunoblotting techniques were performed as previously described (Gierschik et al., 1985). The polyclonal antiserum (MB1) was raised in rabbits against the terminal decapeptide of retinal transducin α (KENLKDCGLF) coupled to keyhole limpet haemocyanin as described (Goldsmith et al., 1987). The antiserum recognized Giα-1 and Giα-2. Blots were stained with an HRP-labelled goat anti IgG antiserum. The immunoreactive bands were visualized with the ECL detection system (Amersham).
Statistical analyses
Numbers of myocytes and numbers of hearts are given in parentheses. Where only one number is given this indicates the number of hearts, with only one cell from each heart being used for that experimental protocol. Results were expressed as the means±standard error of the mean (s.e.mean). Between group comparison was made by unpaired Student's t-test and paired t-test was applied for within group comparison. Agonist (ISO) EC50 values were calculated by nonlinear regression analysis using the HYPMIC program. A value of P<0.05 was considered to be statistically significant.
Drugs
Pertussis toxin and (−)-ISO were obtained from RBI (Natick, MA, U.S.A.). Other drugs were obtained from Sigma (Pool, Dorset, U.K.).
Results
β-AR binding and Giα expression
Radioligand binding studies confirmed the increase in β-AR number in the transgenic mice, which are directly descended from the TG4 mice used in the initial report (Milano et al., 1994). Bmax values in whole ventricle were 5120±580 fmol mg protein−1 for TGβ2 mice (n=10) compared with 13.1±0.7 fmol mg−1 for LM controls (n=7), an approximately 400 fold overexpression. KD values were 6.1±0.9 pM for TGβ2 and 9.0±1.4 pM for LM. In isolated myocyte preparations the overexpression was even more pronounced, with TGβ2 Bmax at 8158±3467 (n=7) and LM having 11.8±2.3 fmol·mg protein−1 (n=10). Expression of Giα-1 and 2 was tested at two different ages by Western blotting of membranes. Levels were increased significantly by 60 days (Figure 1), and the alteration was maintained up to 120 days.
Figure 1.
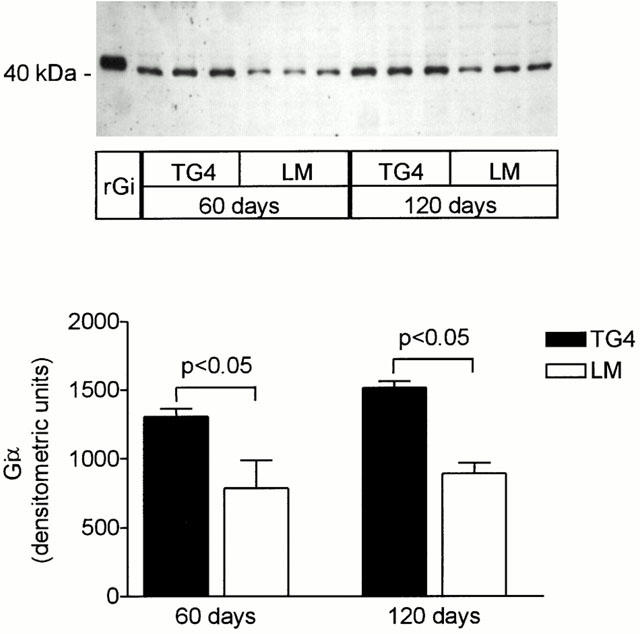
Gia expression in left ventricular myocardium from 60 and 120-day-old LM and TG4 mice. The upper panel shows a representative Western blot. For comparison, immunoreactive signal of recombinant Giα-2 and Giα-3 (rGi) is shown. The bar graph represents the densitometric quantification (n=5 for each group).
Concentration-response curves to the non-selective β-AR agonist (−)-isoprenaline in ventricular myocytes
Single ventricular myocytes isolated from TGβ2 mice or LM mice were superfused with normal KH buffer solution containing 1.0 mM Ca2+. The concentration-response curve of myocytes to isoprenaline (ISO) with changes in contraction amplitude expressed as percentage of the maximum response is shown in Figure 2. There was no significant difference in the EC50 value (concentration for half-maximal response) between TGβ2 mice (1.92±0.37 nM, n=6; mean±s.e.mean) and LM mice (2.69±0.31 nM, n=19).
Figure 2.
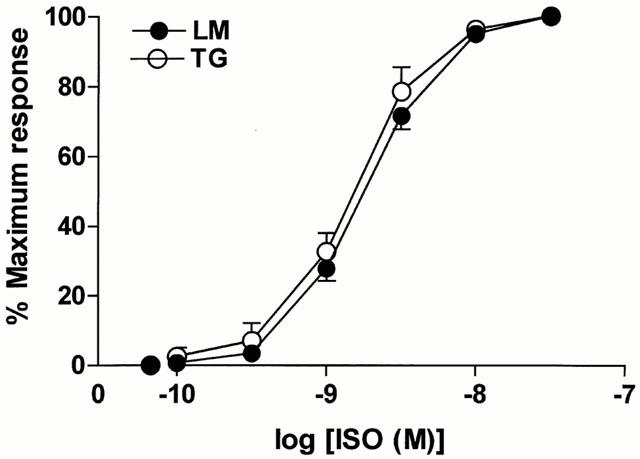
Comparison of the normalized concentration-response curves to (−)-isoprenaline (ISO) in myocytes from TGβ2 (open circles, n=6) and LM (solid circles, n=19) mice. The range of concentrations of ISO was from 3×10−11 to 3×10−8. The concentration–response curves were not significantly different between TGβ2 and LM mice.
Basal, ISO- and calcium-stimulated contraction of ventricular myocytes
Figure 3 shows that baseline contraction amplitude in 1 mM Ca2+ was not different between TGβ2 (3.54±0.25%, n=36 cells/23 hearts) and LM mice (3.63±0.28%, n=41 cells/35 hearts). Although ISO could directly enhance contraction amplitude in ventricular myocytes from TGβ2 mice or LM mice, the ISO-induced maximum contraction amplitude was decreased in TGβ2 mice (5.65±0.34%, n=36 cells/23 hearts) relative to LM mice (9.09±0.52%, n=41 cells/35 hearts) (P<0.001). The decreased positive inotropic response indicates that there is a functional βAR desensitization in TGβ2 mice overexpressing the β2AR. Contraction of myocytes in maximum Ca2+ (4–6 mM) was not different between TGβ2 mice (11.9±1.1%, n=16 cells/11 hearts) and LM mice (13.1±0.5%, n=23 cells/18 hearts). This suggests that the contractile function of myocytes mediated by Ca2+ was not affected in TGβ2 mice.
Figure 3.
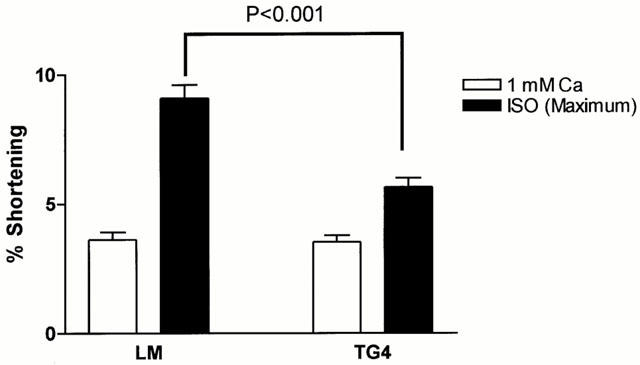
ISO (3×10−8–10−6) stimulated maximum contraction amplitude in ventricular myocytes of TGβ2 mice (36 cells/23 hearts) and LM mice (41 cells/35 hearts). Unfilled columns: baseline, 1 mM Ca2+; solid columns: baseline plus maximal concentration of ISO.
Inverse agonism in TGβ2 myocytes: a comparison with a cyclic AMP antagonist
The lack of a tonic enhancement in contraction contradicts the original hypothesis that the overexpressed β2ARs are stimulating cyclic AMP and so increasing contractility in the absence of agonist (Milano et al., 1994). However, it is possible that mechanisms distal to the cyclic AMP/protein kinase A interaction are decreasing contraction in a compensatory manner (e.g. changes in myofilament sensitivity). TGβ2 myocytes were therefore challenged with an inverse agonist concentration of ICI 118,551 (1 μM) to discover whether tonic stimulation through the β2AR was occurring. The amplitude of contraction was decreased significantly (Figure 4), although the magnitude of the decrease (32%) was less than the 80% in the original report (Bond et al., 1995). All decreases were fully reversible on washout of ICI 118,551, or on addition of 10 μM ISO in the presence of the β1AR antagonist CGP20712A (300 nM) (Basal; 9.63±1.23%, +ICI; 6.09±0.68%, +ICI+ISO/CGP; 12.5±1.3%, n=7 cells/four hearts). The experiments shown in Figure 4 were performed in 4 mM Ca2+. Similar results were obtained in 2 mM Ca2+ (basal; 9.34±0.60%,+ICI; 6.57±0.40% shortening, P<0.001, n=14 cells/eight hearts). A small (<10%) but significant decrease in amplitude was also seen in control myocytes at 4 mM Ca2+.
Figure 4.
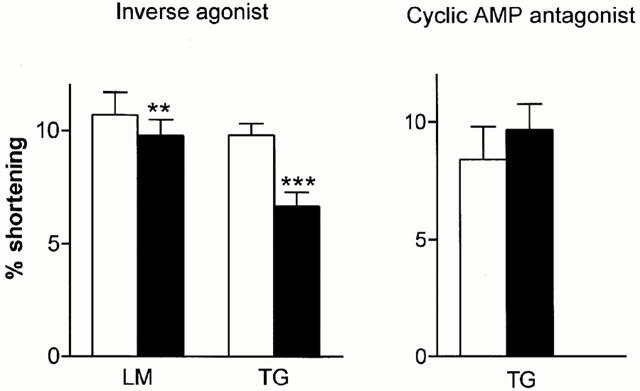
Left panel: effect on contraction amplitude of 5–10 min exposure to the inverse agonist ICI 118,551 (1 μM) in myocytes from LM (n=7 cells/six hearts) and TG (n=10 cells/eight hearts) rats. All effects were reversible. Right panel: effect on contraction amplitude of 40 min exposure to 100 μM Rp-cAMPS in myocytes from TG rats (n=9 cells/seven hearts). Open bars, control conditions; solid bars, presence of inverse agonist/Rp-cAMPS. **P<0.01, ***P<0.001 compared to control, paired t-test.
If the action of inverse agonists is to decrease tonic production of cyclic AMP, then a similar effect should be obtained using Rp-cAMPS, an inactive analogue of cyclic AMP which competes for binding at protein kinase A (PKA). However, no significant decrease in contraction in TGβ2 myocytes was observed using 100 μM Rp-cAMPS (Figure 4). This concentration is sufficient to inhibit responses to maximal concentrations of ISO in guinea-pig or human myocytes (Money-Kyrle et al., 1998), or in myocytes from LM controls (ISO; 5.89±0.64%, ISO+Rp-cAMPS; 3.29±0.65%, P<0.01, basal; 3.31±1.29, n=4 myocytes/hearts).
Enhancement of basal and ISO-stimulated contraction amplitude of myocytes from TGβ2 mice following pertussis toxin (PTX) treatment
The effectiveness of PTX to inactivate Gi was confirmed by loss of the anti-adrenergic effect of carbachol. Before PTX treatment 1 μM carbachol reduced contraction stimulated by 10 nM ISO from 6.99±0.88 to 3.80±0.68% shortening (basal 3.29±0.65%) (P<0.02, n=7 myocyte, four hearts). After PTX treatment the contraction amplitude in ISO was unchanged by carbachol (ISO alone; 7.43±1.32%, ISO+carbachol; 7.37±1.24%, basal; 3.71±0.32%, n=6 myocytes, four hearts).
After PTX pretreatment for inactivation of inhibitory G-protein (Gi), basal contractility of TGβ2 heart cells in 1 mM Ca2+ was increased more than 2 fold (P<0.001). The basal contractility of LM heart cells was not changed by PTX treatment (Figure 5). Maximum contraction amplitude to ISO was also increased significantly in TGβ2 myocytes (9.40±1.22%, n=8 cells/six hearts) relative to that in non PTX-treated TGβ2 myocytes (5.79±0.40%, n=29 cells/18 hearts) and was not different from that of LM mice before (8.93±1.50%, n=11 cells/eight hearts) or after (9.89±0.73%, n=19 cells/12 hearts) PTX treatment. This indicates that PTX treatment has restored the original phenotype. After PTX pretreatment the contraction of myocytes to maximum Ca2+ was not different in TGβ2 mice or LM mice (LM: 12.4±1.3% shortening, n=11 cells/nine hearts, TGβ2: 11.9±0.9%, n=8 cells/seven hearts).
Figure 5.
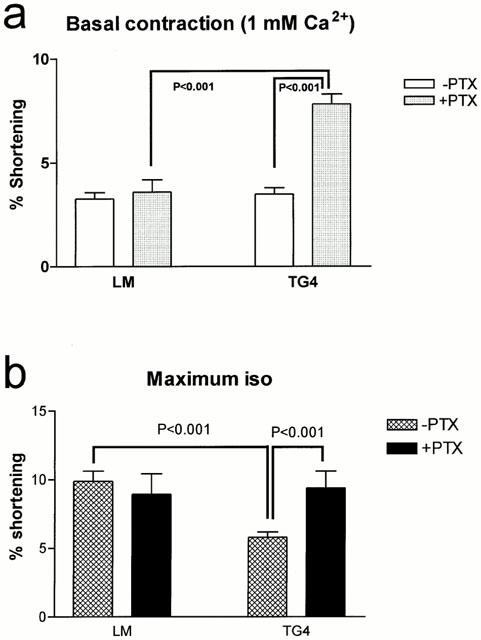
Effect of pertussis toxin on (a) basal (LM 11 cells/eight hearts, TGβ2 23 cells/18 hearts) and (b) maximum ISO-stimulated (LM 11 cells/eight hearts, TGβ2 8 cells/six hearts) contraction amplitude of myocytes.
Stimulation through β1- and β2AR subtypes
Concentration-response curves to ISO were constructed on myocytes in the presence and absence of the specific antagonists ICI 118,551 (β2AR) and CGP 20712A (β1AR). ICI 118,551 at the concentration used (50 nM) would be predicted to produce a 2 log unit shift in a purely β2AR-mediated effect, while 300 nM CGP 20712A would shift a pure β1AR effect by >3 log units (del Monte et al., 1993). Smaller shifts would indicate mixed responses. In myocytes from LM animals, the initial EC50 (concentration to produce half-maximal response) was 3.22±0.83 nM (n=12). Addition of 50 nM ICI 118,551 produced little shift, increasing the EC50 to only 4.48±1.72 nM (n=7): subsequent addition of CGP 20712A as well as ICI 118,551 to four of these cells gave a 2–3 log unit shift. Addition of CGP 20712A alone increased the EC50 to 39.8±2.9 nM (n=5, P<0.05). This indicates that the response of ventricular myocytes to ISO in LM mice is largely β1AR-mediated. Because repeated concentration-response curves are more difficult in mouse myocytes than in other species, due to their high rate of spontaneous arrhythmia, we also performed the experiments by using a maximal concentration of ISO and adding the antagonists when a steady increase in contraction had been reached. In LM myocytes, addition of 50 nM ICI 118,551 increased (rather than decreased) contraction by 3.4±6.2% (n=3, NSD), confirming the lack of β2AR contribution.
Fewer TGβ2 myocytes than LM could be used for experiments involving repeated concentration-response curves, because of the decreased effect of ISO. When these could be performed, results indicated a mixed β1- and β2AR response. Initial addition of ICI 118,551 increased the EC50 from 2.73±1.34 to 8.70±2.89 nM, with an average log shift of 0.80±0.21 units (n=5, P<0.05). This indicates a significant effect mediated through β2ARs. However, some β1AR contribution was still evident first, because the shift was less than predicted for a pure β2AR-mediated response, and second, because addition of CGP 20712A also produced a shift either before (n=3, 0.25–3.4 log units) or in the presence of ICI 118,551 (n=3, 0.1–2.95 log units). Addition of antagonist during exposure to a single concentration of ISO gave a similar result, with five of seven myocytes (six hearts) decreasing contraction (>10% change) in response to ICI 118,551 and six of eight myocytes (seven hearts) decreasing contraction with CGP 20712A. An example of a complete concentration-response curve experiment is given in Figure 6 and several traces showing sequential addition of antagonists are displayed in Figure 7.
Figure 6.
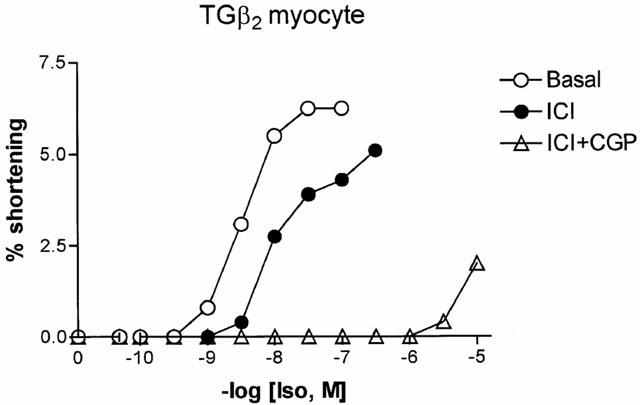
Sequential concentration-response curves to isoprenaline in a single myocyte from a TGβ2 mouse. The first curve was constructed in the absence of antagonists, the second after 30 min exposure to ICI 118,551 (50 nM) and the third after 30 min exposure to both ICI 118,551 and 300 nM CGP 20712A. The antagonists remained in contact with the cell throughout the subsequent exposure to isoprenaline.
Figure 7.
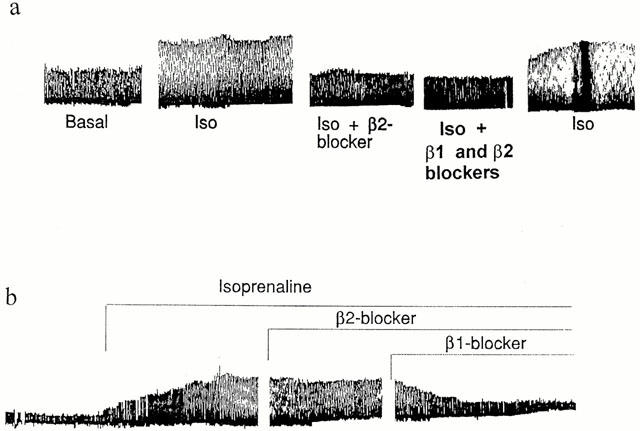
Examples traces of sequential additions of antagonists (β1AR=300 nM CGP20712A, β2AR-blocker=50 nM ICI 118,551) in the presence of isoprenaline (10−7 M). The effect of isoprenaline in cell (a) is predominantly mediated by β2ARs, since ICI 118,551 abolishes the response with little further effect of CGP 20712A. The effect in cell (b) is mediated predominantly by β1ARs.
Following pertussis toxin treatment in TGβ2 myocytes, the mixed β1- and β2AR response to ISO was still evident, with ICI 118,551 decreasing the response in four of five myocytes and CGP 20712A decreasing it in three of four cells (two hearts). Although this does not give as accurate a measure of the β1:β2AR ratio as full concentration-response curves, the result suggests that pertussis toxin treatment up-regulates both β1- and β2AR responses.
Discussion
This study presents strong evidence for the role of increased Gi in the spontaneous deviation of the TGβ2 mice from the original phenotype. Initially, it had been reported that ventricular contraction in TGβ2 mice was maximally activated in the absence of βAR agonist stimulation, and this was thought to be due to the presence of tonically active β2ARs. Adenylate cyclase activity was increased, and the consequent rise in cyclic AMP was the suggested mechanism for the raised contractility. In contrast, we have shown that basal contraction in ventricular myocytes from TGβ2 mice was not supported tonically by cyclic AMP, since Rp-cAMPS did not decrease contraction amplitude. Nor was contraction in basal (1 mM) or high (4–8 mM) Ca2+ different between LM and TGβ2 myocytes. We have shown for the first time that treatment with pertussis toxin, to inactivate Gi, increased basal contraction until it reached levels similar to those seen with high ISO in LM mice.
Inactivation of Gi also restored the response to ISO in ventricular myocytes from TGβ2 mice. These data agree well with those of Xiao et al. (1999) who were the first to show the functional relevance of raised Gi in the TGβ2 mice. We have investigated the contribution of the β1- and β2AR subtypes to ISO-stimulated contraction in LM and TGβ2 mice under the various conditions. More of the response to ISO was mediated by β2AR in TGβ2 mice than in LM, which are almost entirely under β1AR control. However, there was still a significant contribution from the β1AR even in TGβ2 myocytes, with the concentration-response curve shifted less than predicted by ICI 118,551 and additionally shifted by CGP 20712A. This indicates that despite the extreme excess of β2AR, the contribution of β1AR was still detectable, and implies that the efficiency of the overexpressed receptors was lower than that of the native ones.
In a proportion of myocytes, responses to ISO were lost completely (previously reported: Heubach et al., 1999) which indicates that both β1- and β2AR had been down-regulated by the increase in Gi. After pertussis toxin treatment the ISO-stimulated responses were again mediated by both receptor subtypes. This argues against an exclusive relationship between the β2AR subtype and Gi, as has been suggested (Xiao et al., 1999). We have found that β1ARs in guinea-pig myocytes also show PTX-sensitive changes in contractility during desensitization, again indicating an interaction of Gi with this subtype (Ranu et al., 2000).
Taking into account both our studies and those of Xiao et al. (1999) it appears that there is a gradual modification of the initial phenotype of the TGβ2 mouse, in which basal activity is maximal due to a large excess of the activated form of the β2AR (R*) and cannot be increased further by ISO. Gi is up-regulated, probably in response to the continual activation of the adenylyl cyclase pathway: there is a site for the cyclic AMP-activated transcription factor AP-2 on the promoter region of the Giα-2 gene (Imagawa et al., 1987). Additionally, the high cyclic AMP levels encourage the switch of β2AR-coupling from Gs to Gi, by PKA-dependent phosphorylation of the receptor (Daaka et al., 1997). Increased coupling through Gi suppresses the effect of R*, decreasing first the basal activation and finally the response to ISO through β2AR. The high levels of Gi also suppress in addition the response to ISO through the β1AR. Treatment with pertussis toxin reverses this sufficiently to allow the recovery of the βAR responses, as in the work of Xiao et al. (1999) and can also restore the activation of basal activity (present study).
Several intriguing questions remain. First, why does this change occur? We have shown that it is not age of the mouse per se that produces the change in phenotype (Heubach et al., 1999), which implies that it must be due to some genetic drift. It has recently been shown that overexpression of the β2AR above a certain level (60 fold) results in development of a cardiomyopathy, and that this is more rapidly fatal as the receptor level increases (Liggett et al., 2000). We have paired transgenic to transgenic for mating, and the hearts now have a 400 fold or greater excess of the β2AR compared to 200 fold in the original report. It may be we have a higher mortality rate due to cardiomyopathy, and that animals which survive to the time of experiment are those in which Gi has up-regulated in a protective fashion. Second, why is the alteration seen in ventricle but not in right atria, which retain the original increased chronotropic activity (Heubach et al., 1999)? Third, what exactly is inverse agonism? We observe a 32% decrease in contraction amplitude in these myocytes with 1 μM ICI 118,551 (an inverse agonist concentration) despite the observations that basal contraction was not higher than that in LM and that the cyclic AMP antagonist Rp-cAMPS did not decrease contraction. This is not consistent with the hypothesis that inverse agonism represent a reduction in the tonic stimulation of adenylyl cyclase by R* (Milano et al., 1994). This model is worth continuing investigation, because of the strong parallels between spontaneous desensitization due to up-regulation of Gi in TGβ2 mice and the natural history of human heart failure.
Acknowledgments
This study was supported by the Wellcome Trust, the British Heart Foundation (FS/97035), the National Heart Research Fund and the British Council. The work of J.F. Heubach and U. Ravens was supported by a grant of the Deutsche Forschungsgemeinschaft (Ra 222/8-1). We would like to thank Prof R. Lefkowitz for his continued interest and support.
Abbreviations
- β2AR
β2-adrenoceptor
- Gi
inhibitory G-protein
- ISO
isoprenaline
- LM
littermate
- PTX
Pertussis toxin
- TG
transgenic
References
- BOND R.A., LEFF P., JOHNSON T.D., MILANO C.A., ROCKMAN H.A., MCMINN T.R., APPARSUNDARAM S., HYEK M.F., KENAKIN T.P., ALLEN L.F., LEFKOWITZ R.J. Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the beta 2-adrenoceptor. Nature. 1995;374:272–276. doi: 10.1038/374272a0. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid a sensitive method for the quantification of microgram quantities of protein utilising the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- DAAKA Y., LUTTRELL L.M., LEFKOWITZ R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- DEL MONTE F., KAUMANN A.J., POOLE-WILSON P.A., WYNNE D.G., HARDING S.E. Coexistence of functioning β1- and β2-adrenoceptors in single myocytes from human ventricle. Circulation. 1993;88:854–863. doi: 10.1161/01.cir.88.3.854. [DOI] [PubMed] [Google Scholar]
- GIERSCHIK P., CODINA J., SIMONS C., BIRNBAUMER L., SPIEGEL A. Antisera against a guanine nucleotide binding protein from retina cross-react with the β-subunit of the adenylyl cyclase-associated guanine nucleotide binding protein, Ns and Ni. Proc. Natl. Acad. Sci. U.S.A. 1985;82:727–731. doi: 10.1073/pnas.82.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDSMITH P., GIERSCHIK P., MILLIGAN G., UNSON C.G., VINITSKY R., MALECH H.I., SPIEGEL A.M. Antibodies directed against synthetic peptides distinguish between GTP-binding proteins in neutrophils and brain. J. Biol. Chem. 1987;262:14683–14688. [PubMed] [Google Scholar]
- HALL J.A., KAUMANN A.J., BROWN M.J. Selective beta 1-adrenoceptor blockade enhances positive inotropic responses to endogenous catecholamines mediated through beta 2- adrenoceptors in human atrial myocardium. Circ. Res. 1990;66:1610–1623. doi: 10.1161/01.res.66.6.1610. [DOI] [PubMed] [Google Scholar]
- HARDING S.E., BROWN L.A., WYNNE D.G., DAVIES C.H., POOLE-WILSON P.A. Mechanisms of beta-adrenoceptor desensitisation in the failing human heart. Cardiovasc. Res. 1994;28:1451–1460. doi: 10.1093/cvr/28.10.1451. [DOI] [PubMed] [Google Scholar]
- HARDING S.E., JONES S.M., POOLE-WILSON P.A.Contractile responses of myocytes isolated from human atria to isoprenaline, calcium, forskolin and histamine J. Physiol. 1988406219P(Abstract) [Google Scholar]
- HEUBACH J.F., TREBEB I., WETTWER E., HIMMEL H.M., MICHEL M.C., KAUMANN A.J., KOCH W.J., HARDING S.E., RAVENS U. L-type calcium current and contractility in ventricular myocytes from mice overexpressing the cardiac β2-adrenoceptor. Cardiovasc. Res. 1999;42:173–182. doi: 10.1016/s0008-6363(98)00262-4. [DOI] [PubMed] [Google Scholar]
- IMAGAWA M., CHIU R., KARIN M. Transcription factor AP-2 mediates induction by two different signal-transduction pathways: protein kinase C and cAMP. Cell. 1987;51:251–260. doi: 10.1016/0092-8674(87)90152-8. [DOI] [PubMed] [Google Scholar]
- KAUMANN A.J., MOLENAAR P. Modulation of human cardiac function through 4 beta-adrenoceptor populations. Naunyn Schmiedebergs Arch. Pharmacol. 1997;355:667–681. doi: 10.1007/pl00004999. [DOI] [PubMed] [Google Scholar]
- LIGGETT S.B., TEPE N.M., LORENZ J.N., CANNING A.M., JANTZ T.D., MITARAI S., YATANI A., DORN G.W., II Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000;101:1707–1714. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- LIGGETT S.B., WAGONER L.E., CRAFT L.L., HORNUNG R.W., HOIT B.D., MCINTOSH T.C. The Ile164 β2-adrenergic receptor polymorphism adversely affects the outcome of congestive heart failure. J. Clin. Invest. 1998;102:1534–1539. doi: 10.1172/JCI4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILANO C.A., ALLEN L.F., ROCKMAN H.A., DOLBER P.C., MCMINN T.R., CHIEN K.R., JOHNSON T.D., BOND R.A., LEFKOWITZ R.J. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science. 1994;264:582–586. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- MONEY-KYRLE A.R., DAVIES C.H., HARDING S.E. The role of cyclic AMP in the frequency-dependent changes of contraction of guinea-pig cardiomyocytes. Cardiovasc. Res. 1998;37:532–540. doi: 10.1016/s0008-6363(97)00253-8. [DOI] [PubMed] [Google Scholar]
- NAGARAJA S., IYER S., EICHBERG J., BOND R.A.Cardiac β2-adrenoceptor overexpression in transgenic mice results in G protein alterations Pharmacologist 19973940(Abstract) [Google Scholar]
- RANU H.K., MAK J.C., BARNES P.J., HARDING S.E. Gi-dependent suppression of β1-adrenoceptor effects in ventricular myocytes from norepinephrine-treated guinea-pigs. Am. J. Physiol. 2000;278:H1807–H1814. doi: 10.1152/ajpheart.2000.278.6.H1807. [DOI] [PubMed] [Google Scholar]
- XIAO R.P., AVDONIN P., ZHOU Y.Y., CHENG H., AKHTER S.A., ESCHENHAGEN T., LEFKOWITZ R.J., KOCH W.J., LAKATTA E.G. Coupling of β2-adrenoceptor to G1 proteins and its physiological relevance in murine cardiac myocytes. Circ. Res. 1999;84:43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]