

Abstract
We utilized a rat model of myocardial infarction to investigate whether manganese superoxide dismutase (Mn-SOD), an intrinsic radical scavenger, and tumour necrosis factor- α (TNF-α) and/or interleukin-1β (IL-1β) are involved in the late phase of ischaemic preconditioning (IP).
IP was induced in anaesthetized rats by four 3-min left coronary artery (LCA) occlusions, each separated by 10 min of reperfusion. Twenty-four hours after the repetitive brief ischaemia, the LCA was occluded for 20 min followed by reperfusion for 48 h. IP reduced the infarct size by approximately 46% as determined after 48 h of reperfusion.
Antisense oligodeoxynucleotides to Mn-SOD inhibited the increases in Mn-SOD content and activity, and abolished the expected decrease in myocardial infarct size. Sense or scrambled oligodeoxynucleotides did not abolish either Mn-SOD induction or tolerance to ischaemia/reperfusion.
The simultaneous administration of the antibodies to TNF-α (0.5 ml) and IL-1β (0.5 mg) prior to IP abolished the cardioprotection and the increase in Mn-SOD activity induced by IP.
We conclude that the induction and activation of Mn-SOD, mediated by TNF-α and IL-1β after IP, plays an important role in the acquisition of late-phase cardioprotection against ischaemia/reperfusion injury in rats.
Keywords: Mn-SOD, TNF-α, IL-1β, ischaemic preconditioning, cardioprotection, antisense oligodeoxynucleotides, myocardium
Introduction
Ischaemic preconditioning is the phenomenon in which non-lethal brief ischaemia increases the tolerance of the heart to subsequent lethal ischaemia. The time course of the cardioprotection induced by ischaemic preconditioning exhibits a biphasic pattern. The early form of preconditioning, referred to as classic preconditioning, is observed immediately following brief sublethal ischaemia and disappears within 3 h (Murry et al., 1986; 1991). A delayed form of preconditioning is manifested sub-acutely around 24 h following preconditioning (Kuzuya et al., 1993; Marber et al., 1993; Baxter et al., 1994; 1995; 1997; Yamashita et al., 1998a). This reappearance of the cardioprotective effect is named as the ‘second window of ischaemic preconditioning', ‘delayed preconditioning' or ‘late preconditioning'.
This second window of ischaemic preconditioning is closely related to de novo synthesis of proteins such as heat shock proteins (HSPs) (Marber et al., 1993). We reported that an endogenous free radical scavenger, mitochondrial manganese superoxide dismutase (Mn-SOD), was induced in heart tissue coincident to the reappearance of cardioprotection in the late phase of ischaemic preconditioning in rat (Yamashita et al., 1998a) and canine (Hoshida et al., 1993) model. In isolated neonatal cardiomyocytes, we and others showed that hypoxia-reoxygenation preconditioning resulted in the increase of de novo synthesis of Mn-SOD 24 h later with marked protection against prolonged hypoxic insult (Yamashita et al., 1994; Zhou et al., 1996). However, the direct evidence that the induction of this protein is responsible for the acquisition of the tolerance to ischaemia/reperfusion injury in vivo has not been presented yet.
Cardiac resistance to ischaemia/reperfusion injury is increased by exposure to such sublethal stress as a brief period of ischaemia, exercise, and whole-body hyperthermia in a biphasic manner (Kuzuya et al., 1993; Marber et al., 1993; Yamashita et al., 1998b; 1999a). Tumour necrosis factor-α (TNF-α) and interleukin 1 (IL-1) are known as potent inducers of Mn-SOD mRNA (Wong & Goeddel, 1988; Masuda et al., 1988; Fujii & Taniguchi, 1991) and these cytokines are also reported to be involved in the delayed cardioprotection against ischaemia/reperfusion injury (Brown et al., 1990; Eddy et al.; 1992; Nelson et al., 1995; Yamashita et al., 1999a). Akashi et al. (1995) showed that the induction of Mn-SOD after irradiation, which induces the production of superoxide radicals (Oberley et al., 1987), is regulated by IL-1 production. TNF and IL-1 are both reported to be involved in the radioresistance induced by sublethal ionizing radiation or lipopolysaccharide (Neta et al., 1991). We also reported that TNF-α or IL-1β induces a biphasic cardioprotection, which corresponds to the activation of Mn-SOD (Yamashita et al., 1999a). Moreover, the induction of both TNF-α and IL-1β during exercise plays an important role in the exercise-induced cardioprotection through the activation and/or induction of Mn-SOD (Yamashita et al., 1999a).
In the present study, we attempted to demonstrate a direct association between Mn-SOD induction in myocardium and the acquisition of tolerance to ischaemia/reperfusion injury at the late phase of ischaemic preconditioning, using a rat model of occlusion-reperfusion in the left coronary artery (LCA). We also examined whether the cytokine-mediated mechanisms were involved in the delayed cardioprotective effect induced by ischaemic preconditioning.
Methods
This study was carried out under the supervision of the Animal Research Committee in accordance with the Guideline on Animal Experiments of Osaka University and Japanese Government Animal Protection and Management Law (No. 105).
Experimental protocol
The surgical procedures of ischaemic preconditioning and occlusion-reperfusion by LCA occlusion in rats were previously described (Yamashita et al., 1998a). Male Wistar rats (300–350 g) were maintained on a 12-h dark/light cycle, housed at 23±1.5°C with 45±15% relative humidity, and fed and watered ad libitum. The animals were anaesthetized with sodium pentobarbitone (25 mg kg−1, i.p.), additional sodium pentobarbitone (5–10 mg kg−1, i.p.) was given as required. They were subsequently intubated and ventilated with a small-animal respirator using air (model SN-480-7-10; Shinano Seisakusyo, Tokyo, Japan), at a rate of 60–70 cycles min−1 and a tidal volume of 10 ml kg−1 body weight, which volume can maintain arterial pH between 7.35 and 7.50. The surgical process of left coronary artery (LCA) occlusion was previously described (Yamashita et al., 1998a). The chest was opened via a right parasternal sternomectomy. Silk thread (7-0 type) was passed around the LCA about 3–4 mm distal to the LCA origin, and an occlusive snare was placed around it.
The animals were subjected to one of six different protocols with repetitive brief ischaemia, a sham operation or control (Figure 1). Preconditioned rats received four 3-min LCA occlusions, each separated by 10 min of reperfusion. Sham operated rats were instrumented with a suture around the LCA in the same manner as the preconditioned rats. The chest wound was closed, and air was evacuated from the chest (on day 1). At 24 h after the final 10-min reperfusion, rats in these groups were again anaesthetized, and the chest was reopened (on day 2). The right femoral artery was cannulated using polyethylene tubes for the continuous measurement of arterial blood pressure with a pressure transducer (TP-300T; Nihon Kohden, Tokyo, Japan). The heart rate, the incidence of arrhythmias, and ST-segment changes were monitored. Haemodynamic variables were also continuously recorded (model WT-645G recorder; Nihon Kohden, Tokyo, Japan). The arterial pressure was measured with a transducer via the femoral artery cannula, and the LCA was ligated. After 20 min of coronary occlusion, the snare was released; reperfusion was indicated by a change in the colour of the ventricular surface. The surgical wounds were repaired 60 min after reperfusion, and the rats were returned to their cages to recover. Aseptic surgical techniques were used throughout. Benzylpenicillin (30,000 u kg−1) was injected intramuscularly as prophylaxis against infection (on day 1 and on day 2).
Figure 1.
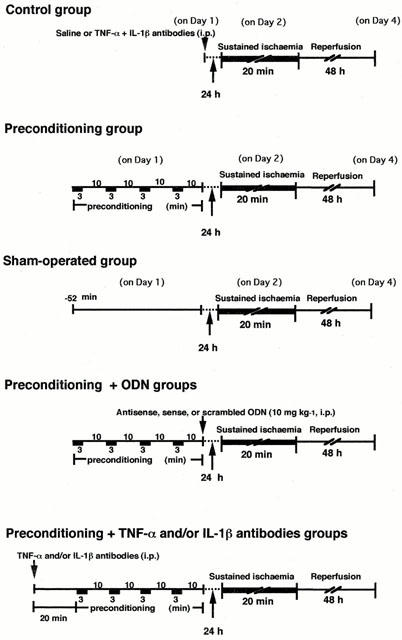
Experimental protocol. Preconditioned rats received four 3-min left coronary artery (LCA) occlusions, each separated by 10 min of reperfusion. At 24 h after the final 10-min reperfusion, the LCA was occluded for 20 min and followed by 48 h reperfusion. The animals were sacrificed 48 h after the restoration of cardiac perfusion. Antisense, sense, or scrambled oligodeoxynucleotides (ODN) was administered i.p. just after ischaemic preconditioning. TNF-α (0.1–1 ml) and/or IL-1β (0.1–1 mg) antibodies were injected i.p. 20 min before the brief repetitive ischaemia. Rats in the control group received saline or TNF-α antibody (0.5 ml) and IL-1β antibody (0.5 mg) 24 h before sustained ischaemia.
Arrhythmias were monitored by ECG. Ventricular fibrillation (VF) was defined according to the criteria of the Lambeth Conventions (Walker et al., 1988). If VF occurred and did not resolve spontaneously within 3 s, manual cardioversion was attempted by gentle flicking of the non-ischaemic region of the heart. Rats in which VF continued for more than 6 s or in which cardioversion had to be performed more than three times were excluded from infarct size analysis.
Determination of infarct size
The size of the infarct was measured by a previously reported method (Watanabe et al., 1991; Yamashita et al., 1997a). Forty-eight hours after infarct surgery (on day 4), rats were re-anaesthetized with sodium pentobarbitone (25 mg kg−1, i.p.) and were intubated and ventilated as above. After the heart was exposed and the LCA was reoccluded, 2% Evans blue dye (2 ml) was injected via the right femoral vein to estimate the area perfused by the occluded artery (ischaemic region). The left ventricle was then cut into six pieces perpendicular to the apex-base axis. These specimens were incubated with 1% triphenyltetrazolium chloride at 37°C to stain the non-infarcted region. The ischaemic, infarcted, and non-ischaemic areas of tissue were separated with scissors and weighted. The area at risk and the infarct size were defined as the ratios of the mass of the ischaemic region to the left ventricular mass and the mass of the infarct region to that of the ischaemic region, respectively, and were expressed as percentages.
Myocardial tissue sampling
To obtain tissue samples for the measurement of Mn-SOD content and activity, rats were killed by an overdose of sodium pentobarbitone 24 h after preconditioning or sham-operation as described in Figure 1 (on day 2). The myocardial tissue was rinsed in phosphate-buffered saline (PBS), and then blood in left and right coronary arteries was washed out with an adequate volume of PBS from ascending aorta retrogradely. Evans blue dye (2%) was introduced after reocclusion of LCA to estimate the area perfused by the occluded artery (LCA region), and the myocardium in the LCA region was cut out with scissors (Yamashita et al., 1998a). Only left ventricular myocardial samples of LCA region were rapidly frozen in liquid nitrogen and stored at −80°C until use.
Measurement of activity and content of Mn-SOD
Myocardial Mn-SOD activity and content were determined in rats sacrificed after recovery interval of 24 h and in control rats those did not receive ischaemic preconditioning treatment. Mn-SOD activity of the myocardial samples was determined by the nitroblue tetrazolium method (Yamashita et al., 1998b; 1999a). Myocardial Mn-SOD content in rats was measured by means of an enzyme-linked immunosorbent assay (ELISA) as reported previously (Yamashita et al., 1997b; 1998a; 1999a). The activity and content of Mn-SOD were expressed relative to the protein concentration in the supernatant determined using the method of Lowry.
Administration of the reagents
The phosphorothioated oligodeoxynucleotides were purchased from Bex (Tokyo, Japan). Twenty-two-mer phosphorothioated derivative of the antisense oligodeoxynucleotides (ASODN, CACGCCGCCCGACACAACATTG) to Mn-SOD (Yamashita et al., 1994; 1999a), sense oligodeoxynucleotides (SODN, CAATGTTGTGTCGGGCGGCGTG) to Mn-SOD, or scrambled oligodeoxynucleotide (TCTCAGTGAGAGCCCTCATTCTGT) was injected i.p. just after ischaemic preconditioning at a dose of 10 mg kg−1 (Yamashita et al., 1999a). To optimize the experimental conditions for the in vivo delivery of systemically injected oligodeoxyribonucleotides, we evaluated the time-course of their accumulation in the heart. In experiments with 5′-FITC labeled ASODN to Mn-SOD, we found that significant labelling of these tissues occurred at these times following the intraperitoneal injection; in endothelial cells at 2–4 h, in vascular smooth muscle at 4 h, and in cardiac myocytes at 8 h (Yamashita et al., 1999a). We could not determine the subcellular distribution of ASODN in the cardiac myocytes. Anti-murine TNF-α antibody (0.1–1 ml) and/or anti-murine IL-1β antibody (0.1–1 mg), were injected i.p. 20 min before ischaemic preconditioning. Anti-murine TNF-α antibody and anti-murine IL-1β antibody were obtained from Genzyme (Cambridge, MA, U.S.A.). According to instructions of products (Genzyme), the TNF-α antibody in the range of less than 10 μl can neutralize approximately 1000 units of mouse TNF-α bioactivity. The IL-1β antibody in the range of 1–20 μg can neutralize the bioactivity of approximately 1 ng of mouse IL-1β. Both antibodies cross-react with rat cytokines (Teti et al., 1993; Wu et al., 1994). In our previous study, simultaneous administration of the antibodies to TNF-α (0.5 ml) and IL-1β (0.5 mg) effectively suppressed the increased levels of cytokines in myocardium induced by treadmill exercise in rats (Yamashita et al., 1999a)
Materials
Chemicals were purchased from Sigma Immunochemicals (St. Louis, MO, U.S.A.), and Wako Fine Chemicals (Osaka, Japan).
Statistics
Data are expressed as the mean±s.e.mean. Differences in haemodynamic parameters during the ischaemia-reperfusion protocol were assessed with respect to time and treatment by repeated measures analysis of variance (ANOVA) followed by Fisher's protected least significant difference method. Comparisons in infarct size, area at risk, and Mn-SOD content and activity between groups were assessed by one-way ANOVA with the Bonferroni's post hoc test for multiple comparisons. A level of P<0.05 was defined as statistically significant.
Results
Exclusion because of VF or death
A total of five rats developed serious VF during sustained coronary occlusion (two in the control group, one in the sham-operated control group, one in the ischaemic preconditioning group with sense ODN treated, and one in the ischaemic preconditioning group with TNF-α and IL-1β antibodies pretreated) were excluded from the evaluation of myocardial infarct size. Nine rats died prematurely (probably because of arrhythmia or heart failure) during the 48-h reperfusion period (three in the control group, two in the sham-operated control group, one in the ischaemic preconditioning group, one in the ischaemic preconditioning group with scrambled ODN treated, one in the ischaemic preconditioning group with TNF-α antibody pretreated, one in the ischaemic preconditioning group with IL-1β antibody pretreated).
Haemodynamic data and rectal temperature
No significant differences were observed in the rate-pressure product or in the rectal temperature during the infarct protocol among the groups before ischaemia, at the end of the ischaemic period, or 0.5 h after reperfusion (data not shown).
Direct relationship between cardioprotection and induction of Mn-SOD
The size of the area at risk expressed as a percentage of left ventricular area did not differ significantly among the groups (Figures 2 and 4). There was no significant difference in the size of the myocardial infarction for recovery interval of 24 h between sham-operated control group and untreated control group (Figure 2). As we previously reported, the induction of a brief period of ischaemia reduced the size of myocardial infarction (Figure 2) and increased Mn-SOD activity and content in the LCA region of left ventricular myocardium in rats 24 h after ischaemic preconditioning (Figure 3). The activity of the cytosolic isoform of SOD (Cu, Zn-SOD) in the LCA region did not change 24 h after ischaemic preconditioning (data not shown). We examined the relationship between the acquisition of tolerance to ischaemia/reperfusion and the induction of Mn-SOD in the myocardium 24 h after preconditioning. We manipulated the level of expression of Mn-SOD, using ASODN that corresponded to the initiation site of Mn-SOD translation; this reagent was administered i.p. to rats immediately after preconditioning. The administration of ASODN completely inhibited the increases in Mn-SOD activity and content 24 h after preconditioning (Figure 3). However, SODN did not attenuate the increases in Mn-SOD activity and content induced by preconditioning (Figure 3). As shown in Figure 2, the expected decrease in infarct size induced by preconditioning was abolished in rats treated with ASODN to Mn-SOD, in which the induction of Mn-SOD was specifically inhibited. SODN or scrambled ODN, which did not attenuate the induction of Mn-SOD in myocardium following preconditioning, had no effect on infarct size or Mn-SOD activity.
Figure 2.
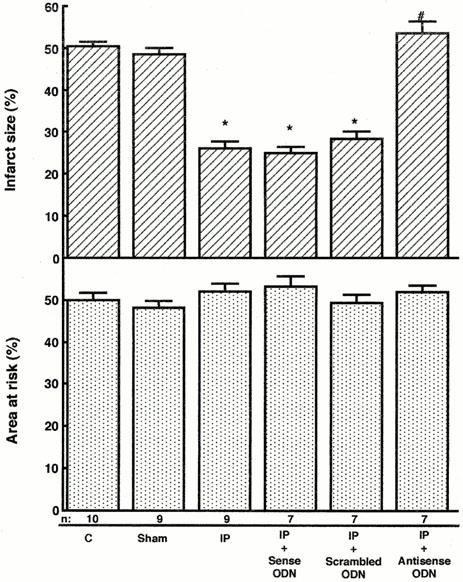
Effects of oligodeoxynucleotides (ODN) on the size of myocardial infarct in rats. Infarct size was calculated as the ratio of the mass of the infarct region to that of the ischaemic region in hearts from rats exposed to treatments as described in Figure 1. Rats in the control ‘C', sham-operated, and preconditioning groups received saline i.p. 24 h before sustained ischaemia. ‘Sham' indicates sham-operated control group, which was instrumented with a suture around the left coronary artery in the same manner as ischaemic preconditioning group with 24 h recovery; ‘IP' indicates rats from ischaemic preconditioning group with 24 h recovery; ‘IP+Sense ODN' indicates 24 h-recovery rats receiving sense ODN just after ischaemic preconditioning treatment; ‘IP+Scrambled ODN' indicates 24 h-recovery rats receiving scrambled ODN just after ischaemic preconditioning treatment; ‘IP+Antisense ODN' indicates 24 h-recovery rats receiving antisense ODN just after ischaemic preconditioning treatment. The number of rats used was indicated in each column. The infarct size in the sham-operated control rats was not significantly different from that in the control rats. Data are expressed as the mean±s.e.mean. *Indicates a value of P<0.05 versus sham-operated control rats and #indicates n.s. versus sham-operated rats by ANOVA with the Bonferroni's post hoc test for multiple comparisons.
Figure 4.
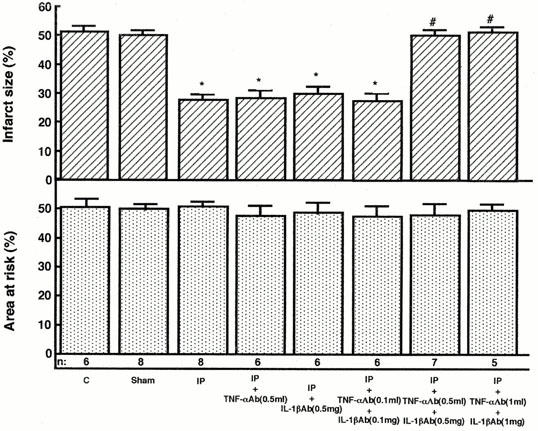
Effects of TNF-α and/or IL-1β antibodies on infarct size in rats. Infarct size was calculated as the ratio of the mass of the infarct region to that of the ischaemic region following reperfusion in rats exposed to treatments as described in Figure 1. Rats in the control group ‘C' received TNF-α (0.5 ml) and IL-1β (0.5 mg) antibodies i.p. 24 h before coronary occlusion. Rats in the sham-operated group received TNF-α (0.5 ml) and IL-1β (0.5 mg) antibodies i.p. prior to the sham-operation. ‘Sham' indicates sham-operated group, which was instrumented with a suture around the left coronary artery in the same manner as ischaemic preconditioning group with 24 h recovery; ‘IP' indicates rats from ischaemic preconditioning group; ‘IP+TNF-αAb' indicates rats receiving TNF-α antibody (0.5 ml) prior to ischaemic preconditioning treatment; ‘IP+IL-1βAb' indicates rats receiving IL-1β antibody (0.5 mg) prior to ischaemic preconditioning treatment; ‘IP+TNF-αAb+IL-1βAb' indicates rats receiving TNF-α(0.1, 0.5, 1 ml) and IL-1β (0.1, 0.5, 1 mg) antibodies prior to ischaemic preconditioning treatment. The number of rats used was indicated in each column. Data are expressed as the mean±s.e.mean. *Indicates a value of P<0.05 versus sham-operated rats and #indicates n.s. versus sham-operated rats by ANOVA with the Bonferroni's post hoc test for multiple comparisons.
Figure 3.
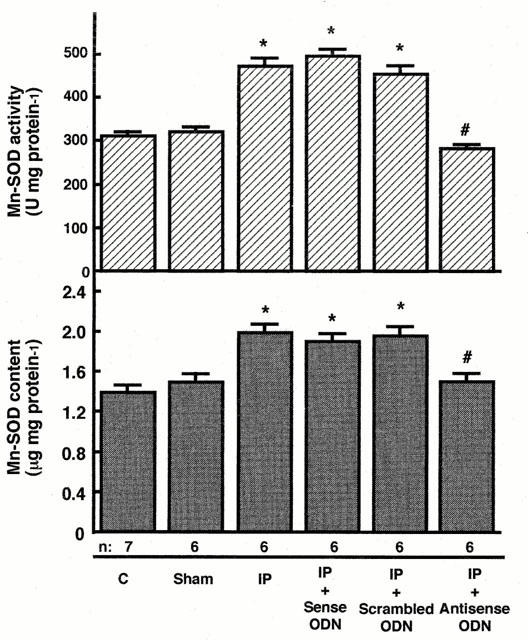
Effects of oligodeoxynucleotides (ODN) on manganese superoxide dismutase (Mn-SOD) activity and content in rat myocardium. Activity of Mn-SOD was determined by the nitroblue tetrazolium method and the content was determined by ELISA in cardiac tissue homogenates from rats 24 h after preconditioning or sham-operated as described in Figure 1. Groups are defined as in the legend to Figure 2. Data are expressed as the mean±s.e.mean of values from at least six rats. *Indicates a value of P<0.05 versus sham-operated control rats and #indicates n.s. versus sham-operated rats by ANOVA with the Bonferroni's post hoc test for multiple comparisons.
Involvement of cytokines in the ischaemia-induced cardioprotection
TNF-α and IL-1β are involved in the exercise-induced cardioprotection (Yamashita et al., 1999a). To investigate a possible role of these cytokines in delayed ischaemic preconditioning, we administered the neutralizing antibodies to these cytokines i.p. 20 min before ischaemic preconditioning. As shown in Figure 4, the administration of the antibody to TNF-α did not influence the size of infarct 24 h after ischaemic preconditioning. The administration of the antibody to IL-1β also did not alter the size of the myocardial infarct 24 h after ischaemic preconditioning. The simultaneous administration of the antibodies to TNF-α (0.1 ml) and IL-1β (0.1 mg) had no effect on the size of myocardial infarct 24 h after ischaemic preconditioning. However, the simultaneous administration of the antibodies to these cytokines in the range of 0.5–1 ml and 0.5–1 mg, respectively, abolished the protection against ischaemic damage 24 h after ischaemic preconditioning. Antibody to TNF-α (0.5 ml) or IL-1β (0.5 mg) had no effect on the increase in Mn-SOD activity induced by ischaemic preconditioning (Figure 5). However, the simultaneous administration of the antibodies to these cytokines eliminated the increase in Mn-SOD activity 24 h after ischaemic preconditioning (Figure 5).
Figure 5.
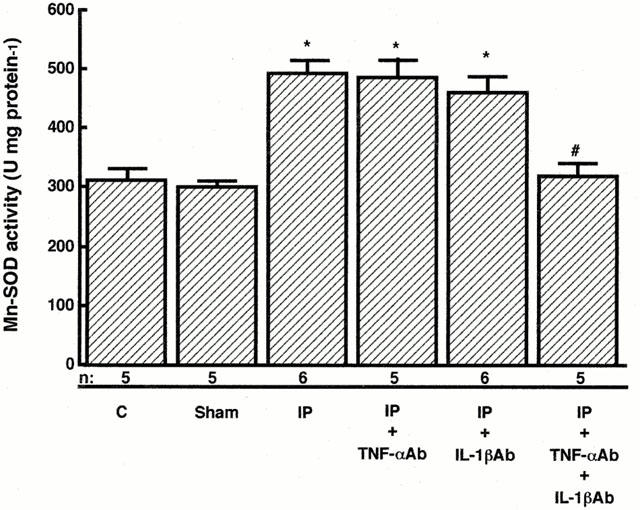
Effects of TNF-α and/or IL-1β antibodies on manganese superoxide dismutase (Mn-SOD) activity in rat myocardium. Activity of Mn-SOD was determined by the nitroblue tetrazolium method in cardiac tissue homogenates from rats 24 h after preconditioning or sham-operated as described in Figure 1. Groups are defined as in the legend to Figure 4. ‘IP+TNF-αAb+IL-1βAb' indicates rats receiving TNF-α (0.5 ml) and IL-1β (0.5 mg) antibodies prior to ischaemic preconditioning treatment. Data are expressed as the mean±s.e.mean of values from at least five rats. *Indicates a value of P<0.05 versus sham-operated rats and #indicates n.s. versus sham-operated rats by ANOVA with the Bonferroni's post hoc test for multiple comparisons.
Discussion
We report for the first time evidence that Mn-SOD antisense abrogates delayed ischaemic preconditioning in vivo. In this study, we found that manipulations including the administration of ASODN to Mn-SOD and the neutralizing antibodies to TNF-α and IL-1β, which inhibited the induction of Mn-SOD at the late phase, abolished the delayed protection against ischaemia/reperfusion injury induced by ischaemic preconditioning. The time course for the cardioprotection paralleled the change in Mn-SOD activity in rat (Yamashita et al., 1998b) and canine (Hoshida et al., 1993) models. Brief sublethal ischaemic or anoxic insult also has been shown to increase Mn-SOD activity or mRNA and to induce cardioprotection or cytoprotection, in a biphasic manner (Das et al., 1993; Hoshida et al., 1993; Zhou et al., 1996). Mn-SOD is directly associated with the delayed protection of the myocyte against hypoxia-reoxygenation injury in an in vitro model (Yamashita et al., 1994; 1996; 1997b). Taken together, these results indicated that Mn-SOD plays a central role in the protective effect in delayed preconditioning in rats. On the other hand, several lines of evidence suggest that HSPs confer protection against ischaemic injury. It has been reported that HSPs are induced after hypoxic/ischaemic preconditioning (Dillmann et al., 1986; Currie et al., 1987; Das et al., 1993; Iwaki et al., 1993; Marber et al., 1993; Hutter et al., 1994). Moreover, transgenic mice overexpressing human HSP70 increased the resistance of the heart to ischaemia/reperfusion injury (Marber et al., 1995; Plumier et al., 1995). Although we did not examine whether HSPs were induced by ischaemic preconditioning in our experimental model, the induction of HSPs also may play an important role in overall cellular defence mechanisms against oxidant stress. Myocardial ischaemia has been shown to induce HSPs possibly via accumulation of reactive oxygen species (Currie et al., 1987; Karmazyn et al., 1990; Maulik et al., 1993; Yamashita et al., 1998b). It remains to be determined if HSP70 and Mn-SOD are independent or interacting factors in ischaemic preconditioning. Das et al. (1993) reported that mRNA of catalase, which is other endogenous antioxidant enzyme, in myocardium is also increased after ischaemic preconditioning and could protect the heart against ischaemia/reperfusion injury. Although we did not examine myocardial catalase activity in the present study, it is conceivable that ischaemic preconditioning-induced increase in endogenous catalase activity in myocardial tissue could facilitate or enhance the detoxification of the superoxide in mitochondria and thus result in reduced tissue injury during the process of ischaemia/reperfusion.
The neutralizing antibodies to TNF-α and IL-1β, which inhibited the activation of Mn-SOD at the late phase, abolished the delayed cardioprotection against ischaemia/reperfusion injury induced by ischaemic preconditioning. We recently reported that TNF-α or IL-1β can mimic the biphasic pattern of cardioprotection and activation of Mn-SOD (Yamashita et al., 1999a). Although either of these cytokines is sufficient for both Mn-SOD activation and cardioprotection, the blockade of both cytokines is necessary for the inhibition of Mn-SOD activation and cardioprotection induced by ischaemic preconditioning because there is some redundancy of the effects of TNF-α and IL-1β (Dinarello, 1989; Aggarwal & Natarajan, 1996). These results suggest that both TNF-α and IL-1β are involved in the ischaemia-induced delayed cardioprotection via the induction and activation of Mn-SOD. TNF-α and IL-1β cause rapid activation and nuclear translocation of the transcription factor NF-κB in Jurkat T-lymphoma cells or HeLa cells (Osborn et al., 1989; Beg et al., 1993; Beg & Baldwin, 1994), and strongly correlates with the induction of Mn-SOD in adenocarcinoma cells (Das et al., 1995), indicating that TNF-α and IL-1β may regulate Mn-SOD expression through NF-κB.
Sublethal ischaemia increases the generation of reactive oxygen species in canine heart muscle (Bolli et al., 1988). Ischaemic preconditioning may be mediated by reactive oxygen species produced during preconditioning in in vivo rabbit (Tanaka et al., 1994; Baines et al., 1997) and rat (Yamashita et al., 1998b), and in vitro rat (Osada et al., 1994; Chen et al., 1995; Zhou et al., 1996) models. We reported that the production of reactive oxygen species during ischaemic preconditioning resulted in the induction of Mn-SOD in the late phase, and also a biphasic cardioprotection following brief ischaemia (Yamashita et al., 1998a). TNF-α and IL-1β are induced following the generation of reactive oxygen species in heart tissue (Yamashita et al., 1999a). These data indicate that the production of TNF-α and IL-1β probably via the generation of reactive oxygen species during ischaemic preconditioning leads to the induction of Mn-SOD, which plays major roles in the acquisition of delayed cardioprotection against ischaemia/reperfusion injury in rats.
Several factors other than reactive oxygen species have been proposed to play a role in triggering the second window of ischaemic preconditioning. Baxter et al. (1994) reported a potential role for adenosine in triggering the second window of preconditioning and activation of adenosine receptor increased Mn-SOD activity in myocardium 24 h after treatment (Dana et al., 1998). Recent evidence from Bolli's laboratory has implicated roles for nitric oxide (NO) as both a trigger and mediator of the second window of the preconditioning (Bolli et al., 1997). In this study, we did not examine the contribution of adenosine and NO in the delayed preconditioning. Adenosine and NO may have a role in the signal transduction pathway. It is well known that NO can upregulate gene transcription including NF-κB (Gross & Wolin, 1995). Takano et al. (1998) reported that NO-donor induced late preconditioning was abrogated by an antioxidant, indicating that NO might be one possible source of reactive oxygen species for triggering the cardioprotective response. Further studies are needed to clarify the role of adenosine and NO in the induction of Mn-SOD and cytokines.
We reported that a single session of exercise (Yamashita et al., 1999a), whole-body hyperthermia (Yamashita et al., 1998b), and pretreatment with monophosphoryl lipid A (Yamashita et al., 1999b) induced a biphasic cardioprotection, which corresponded to the activation of Mn-SOD. The reactive oxygen species, produced during exercise, increased the levels of TNF-α and IL-1β in myocardium, and induced both biphasic activation and/or induction of Mn-SOD in myocardium and biphasic cardioprotection (Yamashita et al., 1999a). Neta et al. (1991) reported that radioresistance induced by lipopolysaccharide (LPS) depended on the induction of IL-1 and TNF. The delayed cardioprotection against ischaemia/reperfusion injury following sublethal stress such as brief ischaemia, exercise, ionizing radiation and LPS may involve a common signal transduction mechanism that mediate the induction and activation of Mn-SOD via the production of reactive oxygen species and cytokines.
Conclusions
Transient sublethal ischaemia resulted in increases in activity and protein level of Mn-SOD and the acquisition of tolerance to subsequent lethal ischaemia/reperfusion injury 24 h later in rats. The manipulations including the administration of ASODN to Mn-SOD and of the neutralizing antibodies to TNF-α and IL-1β, both of which inhibited the increases in activity and protein level of Mn-SOD at the late phase, abolished the delayed protection against ischaemia/reperfusion injury induced by ischaemic preconditioning. These results indicate that TNF-α and IL-1β may be involved in the brief ischaemia-induced delayed cardioprotection via the activation of Mn-SOD.
Acknowledgments
This work was supported in part by a research grant from the Ministries of Education, Science, and Culture of Japan (S. Hoshida, and M. Hori). We thank Dr Keiichiro Suzuki for providing polyclonal antibody to rat Mn-SOD.
Abbreviations
- ASODN
antisense oligodeoxynucleotides
- ELISA
enzyme-linked immunosorbent assay
- IL-1β
interleukin-1β
- IP
ischaemic preconditioning
- LCA
left coronary artery
- Mn-SOD
manganese superoxide dismutase
- NBT
nitroblue tetrazolium
- PBS
phosphate-buffered saline
- TNF-α
tumour necrosis factor- α
- VF
ventricular fibrillation
References
- AGGARWAL B.B., NATARAJAN K. Tumor necrosis factor: developments during the last decade. Eur. Cytokine Netw. 1996;7:93–124. [PubMed] [Google Scholar]
- AKASHI M., HACHYA M., PAQUETTS R.L., OSAWA Y., SHIMIZU S., SUZUKI G. Irradiation increases manganese superoxide dismutase mRNA levels in human fibroblasts: possible mechanisms for its accumulation. J. Biol. Chem. 1995;270:15864–15869. doi: 10.1074/jbc.270.26.15864. [DOI] [PubMed] [Google Scholar]
- BAINES C.P., GOTO M., DOWNEY J.M. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J. Mol. Cell. Cardiol. 1997;29:207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- BAXTER G.F., GOMA F.M., YELLON D.M. Involvement of protein kinase C in the delayed cytoprotection following sublethal ischaemia in rabbit myocardium. Br. J. Pharmacol. 1995;90:2993–3000. doi: 10.1111/j.1476-5381.1995.tb15866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAXTER G.F., GOMA F.M., YELLON D.M. Characterization of the infarctlimiting effect of delayed preconditioning: timecourse and dose-dependency studies in rabbit myocardium. Basic Res. Cardiol. 1997;92:159–167. doi: 10.1007/BF00788633. [DOI] [PubMed] [Google Scholar]
- BAXTER G.F., MARBER M.S., PATEL V.C., YELLON D.M. Adenosine receptor involvement in a delayed phase of protection 24 hours following ischemic preconditioning. Circulation. 1994;117:1685–1692. doi: 10.1161/01.cir.90.6.2993. [DOI] [PubMed] [Google Scholar]
- BEG A.A., BALDWIN A.S. Activation of multiple NF-κB/Rel DNA-binding complexes by tumor necrosis factor. Oncogene. 1994;9:1487–1492. [PubMed] [Google Scholar]
- BEG A.A., FINCO T.S., NANTERMET P.V., BALDWIN A.S. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of. IκBα: a mechanism for NF-κB activation. Mol. Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLLI R., MANCHIKALAPUDI S., TANG X.-L., TAKANO H., QIU Y., GUO Y., ZHANG Q., JADOON A.K. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase: evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ. Res. 1997;81:1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- BOLLI R., PATEL B.S., JEROUDI M.O., LAI E.K., McCAY P.B. Demonstration of free radical generation in ‘‘stunned'' myocardium of intact dogs with the use of the spin trap alpha-phenyl N-tert-butyl nitrone. J. Clin. Invest. 1988;82:476–485. doi: 10.1172/JCI113621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROWN J.M., WHITE C.W., TERADA L.S., GROSSO M.A., SHANLEY P.F., MULVIN D.W., BANERJEE A., WHITMAN G.J., HARKEN A.H., REPINE J.E. Interleukin 1 pretreatment decreases ischemia/reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5026–5030. doi: 10.1073/pnas.87.13.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN W., GABEL S., STEENBERGEN C., MURPHY E. A redox-based mechanism for cardioprotection induced by ischemic preconditioning in perfused rat heart. Circ. Res. 1995;77:424–429. doi: 10.1161/01.res.77.2.424. [DOI] [PubMed] [Google Scholar]
- CURRIE R.W. Effects of ischemia and perfusion temperature on the synthesis of stress-induced (heat shock) proteins in isolated and perfused rat hearts. J. Mol. Cell. Cardiol. 1987;19:795–808. doi: 10.1016/s0022-2828(87)80390-5. [DOI] [PubMed] [Google Scholar]
- DANA A., YAMASHITA N., BAXTER G.F., YELLON D.M.Involvement of protein kinase and manganese-superoxide dismutase in adenosine induced delayed preconditioning J. Mol. Cell. Cardiol. 199830A75(abstract) [Google Scholar]
- DAS D.K., ENGELMAN R.M., KIMURA Y. Molecular adaptation of cellular defences following preconditioning of the heart by repeated ischaemia. Cardiovasc. Res. 1993;27:578–584. doi: 10.1093/cvr/27.4.578. [DOI] [PubMed] [Google Scholar]
- DAS K.C., LEWIS-MOLOCK Y., WHITE C.W. Activation of NF-κB and elevation of Mn-SOD gene expression by thiol reducing agents in lung adenocarcinoma (A594) cells. Am. J. Physiol. 1995;269:L588–L602. doi: 10.1152/ajplung.1995.269.5.L588. [DOI] [PubMed] [Google Scholar]
- DILLMANN W.H., MEHTA H.B., BARRIEUX A., GUTH B.D., NEELEY W.E., ROSS J., Jr Ischemia of the dog heart induces the appearance of a cardiac mRNA coding for a protein with migration characteristics similar to heat-shock/stress protein 71. Circ. Res. 1986;59:110–114. doi: 10.1161/01.res.59.1.110. [DOI] [PubMed] [Google Scholar]
- DINARELLO C.A. Interleukin-1 and its biologically related cytokines. Adv. Immunol. 1989;44:153. doi: 10.1016/s0065-2776(08)60642-2. [DOI] [PubMed] [Google Scholar]
- EDDY L.J., GOEDDEL D.V., WONG G.H. Tumor necrosis factor-alpha pretreatment is protective in a rat model of myocardial ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 1992;184:1056–1059. doi: 10.1016/0006-291x(92)90698-k. [DOI] [PubMed] [Google Scholar]
- FUJII J., TANIGUCHI N. Phorbol ester induces manganese-superoxide dismutase in tumor necrosis factor-resistant cells. J. Biol. Chem. 1991;266:23142–23146. [PubMed] [Google Scholar]
- GROSS S.S., WOLIN M.S. Nitric oxide: pathophysiological mechanisms. Annu. Rev. Physiol. 1995;57:737–769. doi: 10.1146/annurev.ph.57.030195.003513. [DOI] [PubMed] [Google Scholar]
- HOSHIDA S., KUZUYA T., FUJI H., YAMASHITA N., OE H., HORI M., SUZUKI K., TANIGUCHI N., TADA M. Sublethal ischemia alters myocardial antioxidant activity in canine heart. Am. J. Physiol. 1993;264:H33–H39. doi: 10.1152/ajpheart.1993.264.1.H33. [DOI] [PubMed] [Google Scholar]
- HUTTER M.M., SIEVERS R.E., BARBOSA V., WOLFE C.L. Heat-shock protein induction in rat hearts.: a direct correlation between the amount of heat-shock protein induced and the degree of myocardial protection. Circulation. 1994;89:355–360. doi: 10.1161/01.cir.89.1.355. [DOI] [PubMed] [Google Scholar]
- IWAKI K., CHI S.-H., DILLMANN W.H., MESTRIL R. Induction of HSP70 in cultured rat neonatal cardiomyocytes by hypoxia and metabolic stress. Circulation. 1993;87:2023–2032. doi: 10.1161/01.cir.87.6.2023. [DOI] [PubMed] [Google Scholar]
- KARMAZYN M., MAILER K., CURRIE R.W. Acquisition and decay of heat-shock-enhanced postischemic ventricular recovery. Am. J. Physiol. 1990;259:H424–H431. doi: 10.1152/ajpheart.1990.259.2.H424. [DOI] [PubMed] [Google Scholar]
- KUZUYA T., HOSHIDA S., YAMASHITA N., FUJI H., OE H., HORI M., KAMADA T., TADA M. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ. Res. 1993;72:1293–1299. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- MARBER M.S., LATCHMAN D.S., WALKER J.M., YELLON D.M. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264. [DOI] [PubMed] [Google Scholar]
- MARBER M.S., MESTRIL R., CHI S.-H., SAYEN R., YELLON D.M., DILLMANN W.H. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J. Clin. Invest. 1995;95:1446–1456. doi: 10.1172/JCI117815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASUDA A., LONGO D.L., KOBAYASHI Y., APPELLA E., OPPENHEIM J.J., MATSUSHIMA K. Induction of mitochondrial manganese superoxide dismutase by interleukin 1. FASEB J. 1988;2:3087–3091. doi: 10.1096/fasebj.2.15.3263930. [DOI] [PubMed] [Google Scholar]
- MAULIK N., ENGELMAN R.M., WEI Z., LU D., ROUSOU J.A., DAS D.K. Interleukin 1α preconditioning reduces myocardial ischemia reperfusion injury. Circulation. 1993;88:II387–II394. [PubMed] [Google Scholar]
- MURRY C.E., JENNINGS R.B., REIMER K.A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- MURRY C.E., JENNINGS R.B., REIMER K.A. New insights into potential mechanisms of ischemic preconditioning. Circulation. 1991;84:442–445. doi: 10.1161/01.cir.84.1.442. [DOI] [PubMed] [Google Scholar]
- NELSON S.K., WONG G.H., MCCORD J.M. Leukemia inhibitory factor and tumor necrosis factor induce manganese superoxide dismutase and protect rabbit hearts from reperfusion injury. J. Mol. Cell. Cardiol. 1995;27:223–229. doi: 10.1016/s0022-2828(08)80021-1. [DOI] [PubMed] [Google Scholar]
- NETA R., OPPENHEIM J.J., SCHREIBER R.D., CHIZZONITE R., LEDNEY G.D., MACVITTIE T.J. Role of cytokines (interleukin 1, tumor necrosis factor, and transforming growth factor β) in natural and lipopolysaccharide-enhanced radioresistance. J. Exp. Med. 1991;173:1177–1182. doi: 10.1084/jem.173.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OBERLEY L.W., CLAIR D.K., ST, AUTOR A.P., OBERLEY T.D. Increase in manganese superoxide dismutase activity in the mouse heart after x-irradiation. Arch. Biochem. Biophys. 1987;254:69–80. doi: 10.1016/0003-9861(87)90082-8. [DOI] [PubMed] [Google Scholar]
- OSADA M., TAKADA S., SATO T., KOMORI S., TAMURA K. The protective effect of preconditioning on reperfusion-induced arrhythmias is lost by treatment with superoxide dismutase. Jpn. Circ. J. 1994;58:259–268. doi: 10.1253/jcj.58.259. [DOI] [PubMed] [Google Scholar]
- OSBORN L., KUNKEL S., NABEL G.J. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. U.S.A. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PLUMIER J.-C.L., ROSS B.M., CURRIE R.W., ANGELIDIS C.E., KAZLARIS H., KOLLIAS G., PAGOULATORS G.N. Transgenic mice expressing the human heat shock protein 70 have improved postischemic myocardial recovery. J. Clin. Invest. 1995;95:1854–1860. doi: 10.1172/JCI117865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKANO H., TANG X.L., OUI Y., GUO Y., FRENCH B.A., BOLLI R. Nitric oxide donors induce late preconditioning against myocardial stunning and infarction in conscious rabbits via an antioxidant-sensitive mechanism. Circ. Res. 1998;83:73–84. doi: 10.1161/01.res.83.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA M., FUJIWARA H., YAMASAKI K., SASAYAMA S. Superoxide dismutase and N-2-mercaptopropionyl glycine attenuate infarct size limitation effect of ischaemic preconditioning in the rabbit. Cardiovasc. Res. 1994;28:980–986. doi: 10.1093/cvr/28.7.980. [DOI] [PubMed] [Google Scholar]
- TETI G., MANCUSO G., TOMASELLO F. Cytokine appearance and effects of anti-tumor necrosis factor alpha antibodies in a neonatal rat model of group B Streptococcal infection. Infection & Immunity. 1993;61:227–235. doi: 10.1128/iai.61.1.227-235.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALKER M.J., CURTIS M.J., HEARSE D.J., CAMBELL R.W., JANSE M.J., YELLON D.M., COBBE S.M., COKER S.J., HARNESS J.B., HARRON D.J., HIGGINS A.J., JULIAN D.G., LAB M.J., MANNING A.A., NORTHOVER B.J., PARRATT J.R., RIEMERSMA R.A., RIVA E., RUSSELL D.C., SHERIDAN D.J., WINSLOW E., WOODWARD B. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc. Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- WATANABE T., SUZUKI N., SHIMAMOTO N., FUJINO M., IMADA A. Contribution of endogenous endothelin to the extension of myocardial infarct size in rats. Circ. Res. 1991;69:370–377. doi: 10.1161/01.res.69.2.370. [DOI] [PubMed] [Google Scholar]
- WONG G.H.W., GOEDDEL D.V. Induction of manganese superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science. 1988;242:941–944. doi: 10.1126/science.3263703. [DOI] [PubMed] [Google Scholar]
- WU X., WITTER A.J., CARR L.S., CRIPPES B.A., DELARCO J.E., LEFKOWITH J.B. Cytokine-induced neutrophil chemoattractant mediates neutrophil influx in immune complex glomerulonephritis in rat. J. Clin. Invest. 1994;94:337–344. doi: 10.1172/JCI117326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., NISHIDA M., IGARASHI J., AOKI K., HORI M., KUZUYA T., TADA M. Time course of tolerance to ischemia-reperfusion injury and induction of heat shock protein 72 by heat stress in the rat heart. J. Mol. Cell. Cardiol. 1997a;29:1815–1821. doi: 10.1006/jmcc.1997.0416. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., NISHIDA M., IGARASHI J., TANIGUCHI N., TADA M., KUZUYA T., HORI M. Heat shock-induced manganese superoxide dismutase enhances the tolerance of cardiac myocytes to hypoxia-reoxygenation injury. J. Mol. Cell. Cardiol. 1997b;29:1805–1813. doi: 10.1006/jmcc.1997.0415. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., OTSU K., ASAHI M., KUZUYA T., HORI M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J. Exp. Med. 1999a;189:1699–1706. doi: 10.1084/jem.189.11.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., OTSU K., TANIGUCHI N., KUZUYA T., HORI M. Monophosphoryl lipid A provides biphasic cardioprotection against ischaemia-reperfusion injury in rat hearts. Br. J. Pharmacol. 1999b;128:412–418. doi: 10.1038/sj.bjp.0702809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., TANIGUCHI N., KUZUYA T., HORI M. A ‘‘second window of protection'' occurs 24 h after ischemic preconditioning in the rat heart. J. Mol. Cell. Cardiol. 1998a;30:1181–1189. doi: 10.1006/jmcc.1998.0682. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., HOSHIDA S., TANIGUCHI N., KUZUYA T., HORI M. Whole-body hyperthermia provides biphasic cardioprotection against ischemia/reperfusion injury in rat. Circulation. 1998b;98:1414–1421. doi: 10.1161/01.cir.98.14.1414. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., NISHIDA M., HOSHIDA S., IGARASHI J., HORI M., KUZUYA T., TADA M. α1-adrenergic stimulation induces tolerance of cardiac myocytes to hypoxia through induction and activation of Mn-SOD. Am. J. Physiol. 1996;271:H1356–H1362. doi: 10.1152/ajpheart.1996.271.4.H1356. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., NISHIDA M., HOSHIDA S., KUZUYA T., HORI M., TANIGUCHI N., KAMADA T., TADA M. Induction of manganese superoxide dismutase in rat cardiac myocytes increases tolerance to hypoxia 24 hours after preconditioning. J. Clin. Invest. 1994;94:2193–2199. doi: 10.1172/JCI117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU X., ZHAI X., ASHRAF M. Direct evidence that initial oxidative stress triggered by preconditioning contributes to second window of protection by endogenous antioxidant enzyme in myocytes. Circulation. 1996;93:1177–1184. doi: 10.1161/01.cir.93.6.1177. [DOI] [PubMed] [Google Scholar]