

Abstract
In previous studies cannabinoid agonists have been found to inhibit and cannabinoid antagonists to enhance electrically-evoked [3H]-acetycholine (ACh) release in hippocampal slices. The present study was undertaken to determine if similar cannabinoid effects could be observed in synaptosomes.
[3H]-ACh release was evoked by two methods, both sensitive to presynaptic receptor effects. The first involved the addition of 1.3 mM calcium following perfusion with calcium-free Krebs and the second the addition of 11 mM potassium following perfusion with normal Krebs.
In hippocampal synaptosomes the 1.3 mM calcium-evoked release and the high potassium-evoked [3H]-ACh release were inhibited by the cannabinoid agonist, WIN 55212-2, by 59 and 39%, respectively, and with an EC50 of approximately 1 nM. WIN 55212-2 produced a similar, although less potent, inhibition of [3H]-ACh release in cortical synaptosomes. No inhibitory effect of WIN 55212-2 on [3H]-ACh release in striatal synaptosomes was observed, supporting previous data collected in this area with brain slices.
The cannabinoid antagonist, SR 141716A, produced a robust enhancement of 1.3 mM calcium-evoked [3H]-ACh release in hippocampal synaptosomes (EC50<0.3 nM) but had no effect on potassium-evoked release or on [3H]-ACh release in the cortex or striatum.
In conclusion our data demonstrates the inhibitory effects of WIN 55212-2 observed on ACh release in brain slices can be observed in hippocampal and cortex synaptosomes, suggesting a direct action of these compounds on the synaptic terminals. The SR 141716A-induced enhancement of ACh release can similarly be observed in hippocampal synaptosomes and is probably due to an inverse agonist action at constitutively active receptors.
Keywords: Cannabinoid receptors, acetylcholine, synaptosomes, hippocampus, cortex
Introduction
The CB1 cannabinoid receptor, the target site for the active ingredient of marijuana, delta-9-tetrahydrocannabinol, is abundant in the brain with high densities of receptors in the cerebellum, globus pallidus and substantia nigra and moderate densities in the hippocampus and cortex. A principal function of cannabinoid receptors in these regions of the brain is to presynaptically inhibit neurotransmitter release. Amongst the transmitters which have been found to be subject to cannabinoid regulation are acetylcholine (Gifford & Ashby, 1996; Gessa et al., 1997), glutamate (Shen et al., 1996; Sullivan, 1999; Levenes et al., 1998), noradrenaline (Schlicker et al., 1997) and GABA (Miller & Walker, 1995). Electrophysiological studies have indicated that inhibition of neurotransmitter release by cannabinoid receptors is a result of a reduction of N and P/Q type calcium currents (Mackie & Hille, 1992; Pan et al., 1996; Twitchell et al., 1997) and an enhancement of potassium A-currents (Deadwyler et al., 1995).
Two common preparations for examining transmitter release in vitro are brain slices and synaptosomes. In the case of brain slices electrical stimulation is usually used to evoke transmitter release whereas in synaptosomes potassium or veratridine stimulation must be used since the synaptosomes are too small to be stimulated electrically. However, a disadvantage of potassium and veratridine stimulation is that presynaptic receptor effects are less reliably observed with these modes of depolarization than with electrical stimulation (Raiteri et al., 1978). An alternative method which has been used in synaptosomes to evoke transmitter release is the addition of 1.3 mM calcium following perfusion with calcium-free buffer (Bowyer & Weiner, 1987; 1990a; Vickroy et al., 1993). The advantage of evoking transmitter release in synaptosomes in this way is that it appears to be readily inhibited by activation of presynaptic receptors. Thus in striatal synaptosomes the calcium-evoked dopamine release has been found to be inhibited by D2 receptor agonists (Bowyer & Weiner, 1987) and in hippocampal synaptosomes the calcium-evoked norephinephrine and acetylcholine release can be inhibited by adrenergic and muscarinic receptor agonists, respectively (Bowyer & Weiner, 1990b; Vickroy et al., 1993). Although the mechanism of transmitter release by this method has not yet been fully established, it can be blocked by N-type calcium channel blockers (Bowyer & Weiner, 1990a; Vickroy et al., 1992) and hence may involve calcium entry through tonically active channels of this type.
In previous studies with brain slices we have found that [3H]-ACh release in the hippocampus is potently inhibited by the cannabinoid agonists such as WIN 55212-2 and CP 55,940 (Gifford & Ashby, 1996; Gifford et al., 1997a). Studies performed by Nakazi et al. (2000) have indicated that ACh release in the cortex may also be subject to cannabinoid regulation. In addition to being inhibited by cannabinoid agonists such as WIN 55212-2 and CP 55,940 [3H]-ACh release in the hippocampus can also be inhibited by the putative endogenous transmitter, anandamide, provided that breakdown of anandamide by fatty acid amide hydrolase is inhibited (Gifford et al., 1999). Interestingly, in the hippocampus the cannabinoid antagonist, SR 141716A, in addition to antagonizing the effects of cannabinoid agonists, will by itself produce a substantial enhancement of acetylcholine release (Gifford & Ashby, 1996; Gessa et al., 1997; Gifford et al., 1997b). This suggests that SR 141716A antagonizes actions of anandamide or other endogenous cannabinoids in the slice. Alternatively, it may be that cannabinoid receptors on the acetylcholine terminals are constitutively active and that SR 141716A acts as an inverse agonist at these receptors.
The objective of the present study were (1) to determine if the inhibitory effects of cannabinoid agonists and the potentiating effects of cannabinoid antagonists on hippocampal [3H]-ACh release observed in electrically stimulated brain slices could also be observed in a synaptosomal preparation; (2) to compare the 1.3 mM calcium and high potassium modes of stimulation of transmitter release in the latter preparation; and (3) to determine if cortical ACh release in synaptosomes is subjected to a similar cannabinoid regulation as hippocampal ACh release.
Methods
Synaptosomal experiments
For each of the synaptosomal experiments, a male Sprague-Dawley rat (200–350 g, Taconic, Germantown, NY, U.S.A) was killed by decapitation, its brain removed and the hippocampus, frontal cortex or striatum dissected out. Following dissection, the tissue was placed in approximately 2.5 ml of a cold 0.32 M sucrose solution then homogenized gently in a glass tube with a Teflon pestle using three up and down strokes.
The homogenate was centrifuged at 1000×g for 5 min at 4°C and the resulting supernatant removed and centrifuged at 14,000×g for 15 min. The pellet from the second centrifugation was resuspended in 3 ml of Krebs buffer (mM: NaCl 119.5, KCl 3.3, CaCl2 1.3, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, glucose 11 EDTA 0.03, pH 7.4, saturated with 95% O2/5 % CO2), containing 15 μCi [3H]-choline, and incubated at 35°C for approximately 20 min to allow [3H]-choline uptake into the synaptosomes.
The synaptosomal suspension was subsequently loaded into ten superfusion chambers that were constructed from Swinnex Millipore filter units. To retain the synaptosomes, glass fibre (GF/B) filters were placed inside the filter units. To minimize drug binding, teflon tubing was used for all the inlet tubes to the chambers and the peristaltic pump tubing, which has high drug binding, was moved to the outflow side of the chambers between the chambers and the fraction collector. The chambers were perfused with oxygenated Krebs medium at 35°C and at a superfusion rate of 1.6 ml min−1. At frequent intervals the chambers were briefly inverted to allow air bubbles trapped under the filters to escape. This was essential in order to ensure a uniform flow of medium over the entire filter area. To maintain consistency with our previous slice experiments, 1 μM physostigmine (to prevent hydrolysis of the released acetylcholine) and 0.3 μM quinuclidinyl benzilate (to prevent auto-inhibition of release via presynaptically located muscarinic receptors) were included in all superfusion buffers. After a period of 30 min in calcium containing Krebs, the superfusion medium was switched to a calcium-free Krebs medium which contained 2.6 μM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N′-tetraacetic acid). The synaptosomes were perfused with the calcium-free Krebs for 80 min before the addition 1.3 mM CaCl2 to the Krebs medium to evoke [3H]-ACh release. After the calcium addition, the synaptosomes were perfused for a further 30 min (in the presence of calcium) before evoking [3H]-ACh release a second time, in this case, by the addition of 6 mM KCl to the same Krebs medium to raise the total potassium concentration in the Krebs to 11 mM. The time-line for calcium and potassium additions is summarized in Figure 1.
Figure 1.
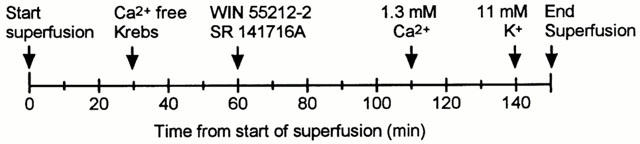
Times of calcium, potassium and drug additions to the synaptosomes.
To measure [3H]-ACh release, two 4 min fractions were collected prior to either the calcium or potassium addition and a third fraction was collected immediately following the calcium or potassium addition. To measure [3H]-ACh retained in the synaptosomes the filters were removed from the chambers after the final potassium stimulation and counted for radioactivity.
Drug treatments
WIN 55212-2, SR141716A and quinuclidinyl benzilate were dissolved in dimethylsulphoxide (DMSO) at a concentration of 1 mg ml−1. The final maximal concentration of DMSO, when dissolved in the Krebs medium, was 0.03%. Previously, we have not found any effects of this concentration of DMSO on [3H]-ACh release (Gifford & Ashby, 1996). Drugs were added 50 min prior to the first stimulation with calcium and kept in the medium for the remainer of the experiment. Two of the 10 chambers in each experiment were used as control (no drug) and were only given DMSO vehicle only.
Data analysis
Stimulation-evoked release was calculated by subtracting the mean level of counts in the two basal fractions collected immediately prior to calcium or potassium addition from that in the fraction collected immediately followed calcium or potassium addition. Results were expressed as fractional release, representing the ratio of [3H]-ACh evoked release relative to the total amount of [3H] radioactivity retained on the filters at the end of the experiment. To determine the effects of the drugs on stimulation-evoked release, the [3H]-ACh evoked release in the chambers exposed to drugs was expressed as a percentage of those obtained for the controls. Data was analysed using analysis of variance followed by Dunnett's test for comparing several drug-treated groups to a single control group.
In the case of the hippocampal synaptosomes, levels of [3H]-ACh released into the Krebs typically ranged from 1000–1200 c.p.m. per fraction prior to the calcium addition and rose to 1800–2500 c.p.m. following calcium addition (for chambers not exposed to WIN 55212-2 or SR 14116A). For potassium stimulation, basal levels of ACh release were typically 1500–1800 c.p.m. per fraction prior to the potassium addition and rose to 2500–3000 c.p.m. following potassium addition.
Drug sources
R (+) WIN 55212-2 mesylate and (±) quinuclidinyl benzilate were purchased from Research Biochemicals, (Natick, MA, U.S.A.). ω-Conotoxin was purchased from Alamone Labs (Jerusalem, Israel). Tetrodotoxin and physostigmine (eserine) was purchased from Sigma (St. Louis, MO, U.S.A.). SR141716A was obtained from Sanofi Research (Montpellier, France).
Results
The 1.3 mM calcium-evoked [3H]-ACh release was inhibited by both ω-conotoxin and by tetrodotoxin (Table 1). This suggests the involvement of N-type calcium channels and voltage-gated sodium channels, respectively, in this mode of transmitter release and confirms the observations of the effects of calcium channel and sodium channel blockade on calcium-evoked release made by Bowyer & Weiner (1990a) and Vickroy (1991). The 11 mM potassium-evoked release was also reduced by both the ω-conotoxin and by tetrodotoxin.
Table 1.
Effect of Ω-conotoxin and tetrodotoxin on the 1.3 mM calcium-evoked release and 11 mM potassium-evoked release of [3H]-ACH release in hippocampal synaptosomes

WIN55212-2 inhibited the 1.3 mM calcium-evoked release with an inhibition of 59% at 300 nM WIN 55212-2 and an EC50 of 0.6 nM (Figure 2a). The inhibition of calcium-evoked [3H]-ACh release by WIN 55212-2 was prevented by 300 nM SR141716A at all except the highest (3 μM) WIN 55212-2 concentration. Since at the 3 μM WIN 55212-2 plus 300 nM SR 141716A concentration the level of release was significantly below that of the WIN 55212-2 alone data points, a non-specific effect of the drug combination may have contributed to the inhibition of the release at this concentration.
Figure 2.
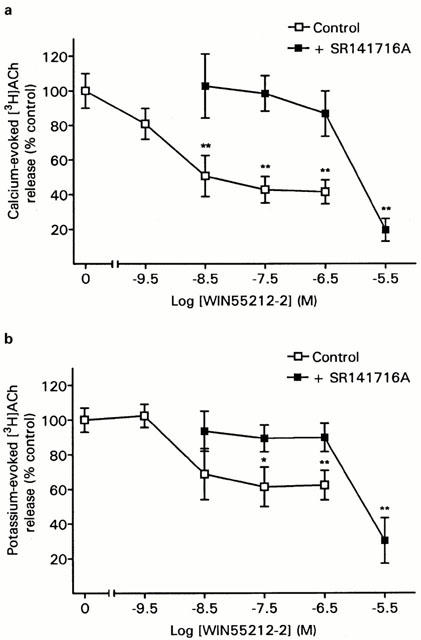
Inhibition [3H]-ACh release in hippocampal synaptosomes by WIN 55212. Synaptosomes were perfused with WIN 55212-2 either alone or in the presence of 300 nM 141716A. [3H]-ACh release was evoked by (a) 1.3 mM calcium or (b) 11 mM potassium. Data are means (±s.e.mean) of 8–10 determinations and are shown as a percentage of their respective controls (either vehicle or 300 nM SR 141716A, alone). ANOVA: 1.3 mM calcium with no SR 141716A, F(4,44)=16.8, P<0.01; 11 mM potassium with no SR 141716A, F(4,32)=4.1, P<0.01; 1.3 mM calcium+SR141716A, F(4,33)=6.9, P<0.01; 11 mM potassium+SR141716A, F(4,33)=9.8, P<0.01. *P<0.05, **P<0.01 versus control (Dunnett's test). Control values (fractional release): 1.3 mM calcium with no SR 1414716A, 0.0153±0.0012; 11 mM potasssium with no SR 14176A, 0.0215±0.0017; 1.3 mM calcium+SR 141716A, 0.0158±0.0017; 11 mM potassium+SR 141716A, 0.0167±0.0009.
In addition to inhibiting the 1.3 mM calcium-evoked release WIN 55212-2 also produced an inhibition of [3H]-ACh release evoked by 11 mM potassium, although the degree of inhibition was smaller and more variable than that obtained with the calcium-evoked release (Figure 2b). The maximal inhibition of potassium-evoked [3H]-ACh release of 39% was obtained at 30 nM WIN55212-2 and the EC50 was 1.6 nM.
SR 141716A, given alone, produced a significant enhancement in the calcium-evoked [3H]-ACh release from hippocampal synaptosomes at all concentrations tested (Figure 3a). The maximal increase was obtained at 3 nM SR 141716A and was 177% of control. In contrast to its effects on calcium-evoked release, SR 141716A failed to produce an enhancement of potassium-evoked release (Figure 3b).
Figure 3.
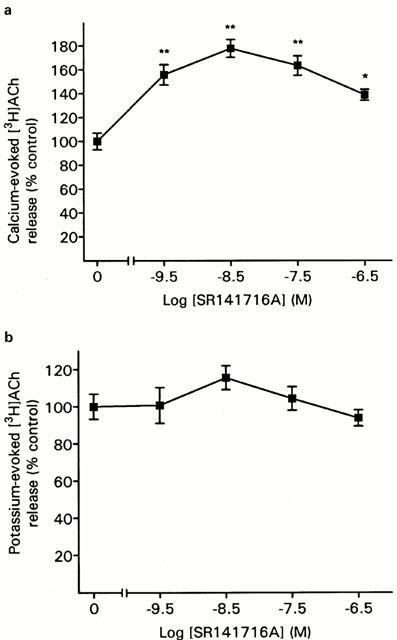
Effect of SR 141716A alone on [3H]-ACh release in hippocampal synaptosomes evoked by (a) 1.3 mM calcium or (b) 11 mM potassium. Data are means (±s.e.mean) of 12–14 determinations. ANOVA: 1.3 mM calcium, F(4,62)=5.2, P<0.01; 11 mM potassium, F(4,59)=0.98, P not significant. *P<0.05, **P<0.01 versus control (Dunnett's test). Control values (fractional release): 1.3 mM calcium, 0.0158±0.0013; 11 mM potassium, 0.0255±0.0018.
In cortical synaptosomes WIN 55212-2 inhibited calcium-evoked release with an inhibition of up to 52% at 300 nM WIN 55212-2 (Figure 4a). The EC50 for the inhibition by WIN 55212-2, assuming the inhibition at 300 nM WIN 55212-2 represents the maximal effect, was 10 nM WIN 55212-2 thus appeared to be less potent in this area than in the hippocampus. Similar data was obtained for the potassium-evoked release (Figure 4b). No enhancement of either the calcium or potassium-evoked release by SR 141716A was observed in this region.
Figure 4.
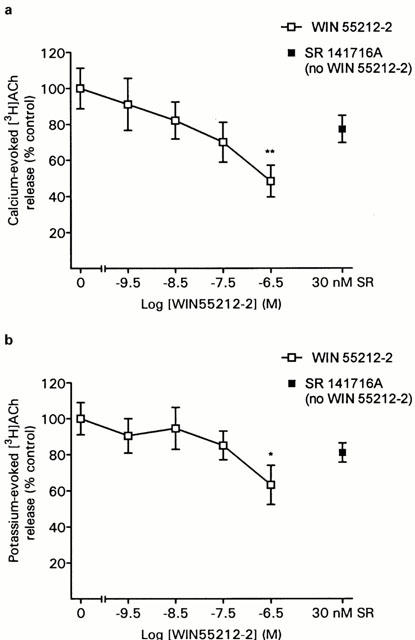
Effect of WIN 55212-2 on [3H]-ACh release from cortical synaptosomes evoked by (a) 1.3 mM calcium or (b) 11 mM potassium. Data are means (±s.e.mean) of eight determinations. ANOVA: 1.3 mM calcium, F(5,35)=3.7, P<0.01; 11 mM potassium, F(5,35)=2.4, P=0.057. *P<0.05, **P<0.01 versus control (Dunnett's test). Control values (fractional release): 1.3 mM calcium, 0.0184±0.0018; 11 mM potassium, 0.0303±0.0024.
In striatal synaptosomes the calcium and potassium-evoked [3H]-ACh were unaffected by either WIN 55212-2 or SR 141716A (Table 2).
Table 2.
Effect of WIN 55212-2 or SR 141716A on 1.3 mM calcium-evoked or 11 mM potassium-evoked release of [3H]-ACh release in striatal synaptosomes

Discussion
In both hippocampal and cortical synatosomes the 1.3 mM calcium and the 11 mM potassium-evoked acetylcholine release were inhibited by WIN 55212-2. The maximum inhibition of the evoked release by WIN 55212-2 was obtained using 1.3 mM calcium-evoked release in the hippocampus. The degree of inhibition was similar to that obtained using electrically-evoked [3H]-ACh release in hippocampal brain slices (Gifford & Ashby, 1996; Gifford et al., 1998). The EC50 for the inhibition by WIN 55212-2, however, was approximately 5–10 times lower in the synaptosomes than in our previous experiments in brain slices. This may be because in the synaptosomes the WIN 55212-2 has direct access to the receptors whereas in the brain slices the WIN 55212-2 must first diffuse into the slice to reach the receptors. Surprisingly, in the case of the cortex synaptosomes in the present study, WIN 55212-2 was found to be significantly less potent in inhibiting [3H]-ACh release than in the hippocampal synaptosomes. One possible explanation for this may be that there is a difference in the size of the receptor reserve between the two areas. Thus whereas the receptor reserve is very large in the case of the hippocampus (Gifford et al., 1998), it is possible that it may not be as large in the cortex, which would result in a reduced agonist potency in this area.
In both the hippocampus experiments the effects of WIN 55212-2 on 11 mM potassium-evoked release appeared slightly smaller than its effects on 1.3 mM calcium-evoked release. This was probably not a consequence of possible receptor desensitization as a result of the longer exposure time to the WIN 55212-2 for the potassium stimulation since in our previous study (Gifford et al., 1998) we did not observe any reduction in the effect of WIN 55212-2 on electrically-evoked [3H]-ACh release in the hippocampus, even after 100 min of exposure of the tissue to this drug.
In contrast to the inhibitory effects of WIN 55212-2 on [3H]-ACh release in the hippocampus and cortex, WIN 55212-2 did not inhibit [3H]-ACh release in the striatum. This supports previous experiments in brain slices in which we also found no effect of cannabinoid agonists on [3H]-ACh release in this brain area (Gifford et al., 1997a). The cholinergic innervation in the striatum differs from that in the hippocampus and cortex in that whereas in the latter areas it is provided by cholinergic cell groups originating in the basal forebrain in the striatum it is provided instead by local interneurons. Recent data indicates that these striatal cholinergic interneurones do not contain mRNA for CB1 receptors (Hohmann & Herkenham, 2000).
Electrophysiological studies on cultured hippocampal and neuroblastoma cells have suggested that activation of cannabinoid receptors produce approximately 50% inhibition of the opening of membrane voltage activated calcium channels of the N and P/Q classes (Mackie & Hille, 1992; Pan et al., 1996; Twitchell et al., 1997). This action of cannabinoid receptors is similar to that of other presynaptic neurotransmitter receptors coupled to inhibition of cyclic AMP, such as mu opioid, dopamine D2, muscarinic and serotonin receptors. Since calcium entry via these calcium channels appears to mediate the synaptosomal 1.3 mM calcium-evoked release, as indicated by the effects of ω-conotoxin, this can explain the inhibitory effect of WIN 55212-2 observed in the present study. In addition to being blocked by calcium channel antagonists the 1.3 mM calcium-evoked acetylcholine release can also be largely inhibited by tetrodotoxin. This suggests that the calcium addition may depolarize the synaptosomes sufficiently to produce opening of voltage activated sodium channels. If this is the case then it is possible that cannabinoid receptor induced stimulation of the opening of A-type potassium channels may also contribute to the inhibition of acetylcholine release by WIN 55212-2 since the opening of these channels will counteract the membrane depolarization from the calcium addition.
In the present study the cannabinoid antagonist, SR 141716A, in addition to antagonizing the effects of WIN 55212-2, produced on its own a potent enhancement of 1.3 mM calcium-evoked release in the hippocampal synaptosomes. The degree of enhancement of the [3H]-ACh release by SR 141716A was similar to that we observed in experiments on electrically stimulated hippocampal slices (Gifford & Ashby, 1996). In the hippocampal slices one explanation of the effect of SR 141716A is an antagonism of tonic inhibition of the release by endogenously released endocannabinoids. However, a tonic endocannabinoid tone is less likely in synaptosomes than in slices because in the synaptosomes any endocannabinoids released into the extracellular medium would be rapidly washed away by the flow of medium past the synaptosomal bed. Only if the endocannabinoids were able to activate the cannabinoid receptors without leaving the synaptosomes would an endocannabinoid tone be a plausible explanation for the SR 141716A effect. It is thus more likely that the SR 141716A effect in the synaptosomes is a result of an inhibition of constitutive activity in the cannabinoid receptors by an inverse agonist action of SR 141716A. This explanation has some support from results obtained in cloned CB1 receptors in cell expression systems. In at least some of these systems constitutive activity of CB1 receptors has been observed, and has been found to be inhibited by the presence of SR 141716A (Landsman et al., 1997; Pan et al., 1998).
In contrast to its effects in the hippocampus SR 141716A did not enhance [3H]-ACh release in cortical synaptosomes. A similar lack of effect of SR 141716A in the cortex was observed in our previous study in brain slices (Gifford et al., 1997b) and may indicate that the receptors in this region are not constitutively active. This could result from either a smaller ratio of membrane receptors to G-proteins than in the hippocampus or from a coupling of the receptors to different G-proteins with lower affinity for the receptor.
Interestingly, although the 1.3 mM calcium-evoked release was increased by SR 141716A the high potassium-evoked release was unaffected by this antagonist. One possible explanation is that with the potassium-evoked release one or more steps in the release pathway, such as calcium entry for example, were close to reaching saturation and thus any enhancing effect of SR 141716A on this step would not further increase release. A second possibility for the lack of effect of SR 141716A in enhancing the potassium-evoked release may be that the potassium-evoked release involves different membrane calcium currents than the calcium-evoked release and that these currents are not subjected to tonic cannabinoid inhibition.
To summarize, the results obtained in the present study indicate that in hippocampal synaptosomes both a cannabinoid agonist-induced inhibition and antagonist-induced stimulation of ACh release can be observed, with calcium-evoked release generally being more affected by the cannabinoid drugs than the potassium-evoked release. In synaptosomes prepared from the cortex only the agonist-induced inhibition was observed and in synaptosomes prepared from the striatum neither agonists or antagonist had an effect on the release. The results obtained in the synaptosome release experiments in the present study thus support those obtained in previous studies with brain slices. Since synaptosome experiments have the advantage over electrically-stimulated brain slice experiments of showing significantly less variability in the levels of transmitter release between individual chambers this preparation should be of value in future studies to assess the efficacy and potency of new cannabinoid compounds.
Acknowledgments
Supported by the National Institute on Drug Abuse Grant DA12412 and by the Department of Energy Office of Health and Environmental Research Contract DE-AC02-98CH10886.
Abbreviations
- ACh
Acetylcholine
- ANOVA
Analysis of Variance
- c.p.m.
Counts per minute
- DMSO
dimethylsulphoxide
References
- BOWYER J.F., WEINER N. Modulation of the Ca++-evoked release of [3H]dopamine from striatal synaptosomes by dopamine (D2) agonists and antagonists. J. Pharmacol. Exp. Ther. 1987;241:27–33. [PubMed] [Google Scholar]
- BOWYER J.F., WEINER N. Ca2(+)-evoked [3H]dopamine release from synaptosomes is dependent on neuronal type Ca2+ channels and is not mediated by acetylcholine, glutamate or aspartate release. J. Pharmacol. Exp. Ther. 1990a;254:664–670. [PubMed] [Google Scholar]
- BOWYER J.F., WEINER N. Alpha-2 adrenergic inhibition of Ca(++)-evoked [3H]norepinephrine release from synaptosomes is blocked by depolarization. J. Pharmacol. Exp. Ther. 1990b;253:1063–1069. [PubMed] [Google Scholar]
- DEADWYLER S.A., HAMPSON R.E., MU J., WHYTE A., CHILDERS S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J. Pharmacol. Exp. Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- GESSA G.L., MASCIA M.S., CASU M.A., CARTA G. Inhibition of hippocampal acetylcholine release by cannabinoids: reversal by SR 141716A. Eur. J. Pharmacol. 1997;327:R1–2. doi: 10.1016/s0014-2999(97)89683-5. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., ASHBY C.R. Electrically-evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55212-2, and is potentiated by the cannabinoid antagonist, SR-141716A. J. Pharmacol. Exp. Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- GIFFORD A.N., BRUNEUS M., GATLEY S.J., LAN R., MAKRIYANNIS A., VOLKOW N.D. Large receptor reserve for cannabinoid actions in the central nervous system. J. Pharmacol. Exp. Ther. 1998;288:478–483. [PubMed] [Google Scholar]
- GIFFORD A.N., BRUNEUS M., LIN S., GOUTOPOULOS A., MAKRIYANNIS A., VOLKOW N.D., GATLEY S.J. Potentiation of the action of anandamide on hippocampal slices by the fatty acid amide hydrolase inhibitor, palmitylsulphonyl fluoride (AM 374) Eur. J. Pharmacol. 1999;383:9–14. doi: 10.1016/s0014-2999(99)00609-3. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., SAMIIAN L., GATLEY S.J., ASHBY C.R. Examination of the effect of the cannabinoid receptor agonist, CP-55,940, on neurotransmitter release from brain slices. Eur. J. Pharmacol. 1997a;324:187–192. doi: 10.1016/s0014-2999(97)00082-4. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., TANG Y., GATLEY S.J., VOLKOW N.D., LAN R., MAKRIYANNIS A. Effect of the cannabinoid receptor SPECT agent, AM 281, on hippocampal acetylcholine release from rat brain slices. Neurosci. Lett. 1997b;238:84–86. doi: 10.1016/s0304-3940(97)00851-3. [DOI] [PubMed] [Google Scholar]
- HOHMANN A.G., HERKENHAM M. Localization of cannabinoid CB(1) receptor mRNA in neuronal subpopulations of rat striatum: A double-label in situ hybridization study. Synapse. 2000;37:71–80. doi: 10.1002/(SICI)1098-2396(200007)37:1<71::AID-SYN8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- LANDSMAN R.S., BURKEY T.H., CONSROE P., ROESKE W.R., YAMAMURA H.I. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur. J. Pharmacol. 1997;334:R1–R2. doi: 10.1016/s0014-2999(97)01160-6. [DOI] [PubMed] [Google Scholar]
- LEVENES C., DANIEL H., SOUBRIE P., CREPEL F. Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J. Physiol. 1998;510:867–879. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACKIE K., HILLE B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER A.S., WALKER J.M. Effects of a cannabinoid on spontaneous and evoked neuronal activity in the substantia nigra pars reticulata. Eur. J. Pharmacol. 1995;279:179–185. doi: 10.1016/0014-2999(95)00151-a. [DOI] [PubMed] [Google Scholar]
- NAKAZI M., BAUER U., NICKEL T., KATHMANN M., SCHLICKER E. Inhibition of serotonin release in the mouse brain via presynaptic cannabinoid CB1 receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2000;361:19–24. doi: 10.1007/s002109900147. [DOI] [PubMed] [Google Scholar]
- PAN X., IKEDA S.R., LEWIS D.L. Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol. Pharmacol. 1996;49:707–714. [PubMed] [Google Scholar]
- PAN X., IKEDA S.R., LEWIS D.L. SR 141716A acts as an inverse agonist to increase neuronal voltage-dependent Ca2+ currents by reversal of tonic CB1 cannabinoid receptor activity. Mol. Pharmacol. 1998;54:1064–1072. doi: 10.1124/mol.54.6.1064. [DOI] [PubMed] [Google Scholar]
- RAITERI M., CERVONI A.M., DEL CARMINE R., LEVI G. Do presynaptic autoreceptors control dopamine release. Nature. 1978;274:706–708. doi: 10.1038/274706a0. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., TIMM J., ZENTNER J., GOTHERT M. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn Schmiedebergs Arch. Pharmacol. 1997;356:583–589. doi: 10.1007/pl00005093. [DOI] [PubMed] [Google Scholar]
- SHEN M., PISER T.M., SEYBOLD V.S., THAYER S.A. Cannabinoid receptor agonists inhibit glutaminergic synaptic transmission in rat hippocampal cultures. J. Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SULLIVAN J.M. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J. Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- TWITCHELL W., BROWN S., MACKIE K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- VICKROY T.W. Neurobiochemical evidence for calcium-induced depolarization of EGTA-pretreated hippocampal synaptosomes. Brain Res. 1991;540:335–339. doi: 10.1016/0006-8993(91)90532-z. [DOI] [PubMed] [Google Scholar]
- VICKROY T.W., MALPHURS W.L., DEFIEBRE N.C. Absence of receptor reserve at hippocampal muscarinic autoreceptors which inhibit stimulus-dependent acetylcholine release. J. Pharmacol. Exp. Ther. 1993;267:1198–1204. [PubMed] [Google Scholar]
- VICKROY T.W., SCHNEIDER C.J., HILDRETH J.M. Pharmacological heterogeneity among calcium channels that subserve acetylcholine release in vertebrate forebrain. Neuropharmacology. 1992;31:307–309. doi: 10.1016/0028-3908(92)90181-n. [DOI] [PubMed] [Google Scholar]