

Abstract
In order to investigate the involvement of Ras and/or Rho proteins in the induction of the inducible isoform of nitric oxide synthase (NOS II) we used HMG-CoA reductase inhibitors (statins) and Clostridium difficile toxin B (TcdB) as pharmacological tools. Statins indirectly inhibit small G proteins by preventing their essential farnesylation (Ras) and/or geranylgeranylation (Rho). In contrast, TcdB is a glucosyltransferase and inactivates Rho-proteins directly.
Human A549/8- and DLD-1 cells as well as murine 3T3 fibroblasts were preincubated for 18 h with statins (1–100 μM) or TcdB (0.01–10 ng ml−1). Then NOS II expression was induced by cytokines. NOS II mRNA was measured after 4–8 h by RNase protection assay, and NO production were measured by the Griess assay after 24 h.
Statins and TcdB markedly increased cytokine-induced NOS II mRNA expression and NO production. Statin-mediated enhancement of NOS II mRNA expression was reversed almost completely by cotreatment with mevalonate or geranylgeranylpyrophosphate. It was only slightly reduced by farnesylpyrophosphate. Therefore, small G proteins of the Rho family are likely to be involved in NOS II induction.
In A549/8 cells stably transfected with a luciferase reporter gene under the control of a 16 kb fragment of the human NOS II promoter (pNOS2(16)Luc), statins produced only a small increase in cytokine-induced NOS II promoter activity. In contrast, statins had a considerable superinducing effect in DLD-1 cells stably transfected with pNOS2(16)Luc.
In conclusion, our studies provide evidence that statins and TcdB potentiate cytokine-induced NOS II expression via inhibition of small G proteins of the Rho family. This in turn results in an enhanced NOS II promoter activity and/or a prolonged NOS II mRNA stability.
Keywords: Nitric oxide synthase II, HMG-CoA inhibitors, statins, Clostridium difficile toxin B, small G proteins, Rho family, promoter activity, RNA stability, post-transcriptional regulation
Introduction
NO synthase II (NOS II), the high output NOS, is normally absent from resting cells (Förstermann et al., 1998; Förstermann & Kleinert, 1995). Cells must be activated to express the enzyme, the activity of which is then independent of elevated Ca2+ concentrations. In different types of cells and tissues, NOS II expression can be induced following exposure to immunological and inflammatory stimuli such as cytokines or lipopolysaccharide (LPS). Once synthesized, the enzyme is active for hours to days and generates large amounts of NO that can have beneficial effects, such as antimicrobial, antiatherogenic or antiapoptotic actions. However, inappropriate NOS II induction can have detrimental consequences, such as cellular injury in arthritis, colitis, or septic shock (Geller & Billiar, 1998; Kröncke et al., 1998; MacMicking et al., 1997; Nathan & Xie, 1994).
The regulation of NOS II expression is cell- and species-specific, with a wide variety of signal transduction pathways involved. Murine cells express NOS II after incubation with bacterial lipopolysaccharide (LPS), stimulatory cytokines (interferon-γ [IFN-γ], interleukin-1β [IL-1β], interleukin-6 [IL-6], tumour necrosis factor-α [TNF-α]) and/or other compounds. In most human cells, a cytokine combination consisting of IFN-γ, IL-1β, and TNF-α is required to achieve a significant NOS II expression (Förstermann & Kleinert, 1995; Kleinert et al., 2000). Data generated over the last years have revealed a variety of physical stimuli (e.g. heat shock, hypoxia, oxidative stress) and compounds that are able to induce, enhance or inhibit LPS/cytokine-induced NOS II expression in different cell- and animal models (Förstermann & Kleinert, 1995; Kleinert et al., 2000). For some agents (e.g. platelet-derived growth factor or cyclic AMP-elevating compounds) opposing effects (induction, superinduction, inhibition of induction) on NOS II expression have been described depending on the cell model used (Eberhardt et al., 1998; Gilbert & Herschman, 1993; Kleinert et al., 1996b; Kunz et al., 1997; Messmer & Brüne, 1994).
The cytokines used to induce NOS II expression in different cell models are known to activate small G proteins of the Ras/Rho family (Denhardt, 1996; Lim et al., 1996). Small G protein of the Ras/Rho family can be involved in the activation of the mitogen activated protein kinase (MAPK) pathways (Denhardt, 1996; Lim et al., 1996). Ras proteins seem to activate predominantly the extracellular regulated kinase pathway (ERK1/2-MAPK). Rho proteins, on the other hand, are believed to activate the Jun-N terminal kinase (JNK, also termed stress activated protein kinase SAPK) and the p38 MAPK (Denhardt, 1996; Lim et al., 1996; Mackay & Hall, 1998). As small G proteins of the Ras/Rho family can be activated only after prenylation (Gibbs & Oliff, 1997), inhibition of the prenyl biosynthesis by HMG-CoA reductase inhibitors (statins) leads to inactivation of these proteins (see Figure 1). Specific inhibitors of the enzymes transferring the prenyl moiety to proteins have been developed. These farnesyltransferase- and geranylgeranyltransferase inhibitors can be used to prevent specifically protein farnesylation and geranylgeranylation, respectively (Gibbs & Oliff, 1997). Several bacterial toxins directly modify and thereby inhibit the Ras/Rho family of small G proteins. Toxin B from Clostridium difficile (TcdB) specifically inactivates the Rho-family of small G proteins (Rho, Rac, Cdc42) by UDP-glycosylation (see Figure 1) (von Eichel-Streiber et al., 1996).
Figure 1.

Biosynthesis of farnesylpyrophosphate and geranylgeranyl-pyrophosphate: effect of statins and toxin B from Clostridium difficile. Farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP, synthesized from FPP by addition of a C5-moiety) are long lipophilic carbon chains that occur as intermediate products within the cholesterol biosynthesis pathway. Their formation depends on a sufficient supply of mevalonate by HMG-CoA reductase. Statins inhibit competitively this key enzyme of prenyl and cholesterol biosynthesis. Post-translational farnesylation or geranylgeranylation of small G proteins of the Ras/Rho family is an essential prerequisite for their anchoring in the cell membrane and thus for their activity. For Ras proteins farnesylation is the predominant mechanism, whereas Rho proteins are mainly geranylgeranylated. Toxin B from Clostridium difficile (TcdB) inactivates directly and specifically Rho proteins by UDP-glucosylation.
The involvement of small G proteins in NOS II induction is complex. Pahan et al. (1997) described an inhibitory effect of lovastatin on LPS-induced NOS II expression in rat primary astrocytes, which could be reversed by mevalonate and farnesylpyrophosphate. As the Ras family of small G protein needs to be farnesylated for activation (Gibbs & Oliff, 1997), the authors conclude, that Ras proteins are involved in NOS II induction in these cells. Using lovastatin and a specific inhibitor of geranylgeranyltransferase I, Finder et al. (1997) observed a superinduction of IL-1β-induced NOS II expression in rat pulmonary artery smooth muscle cells and rat hepatocytes. On the other hand, blockade of protein farnesylation by a specific farnesyltransferase inhibitor resulted in a decreased NOS II induction (Finder et al., 1997). Thus, Rho proteins, which need to be geranylgeranylated, inhibit NOS II induction in these cells, whereas activation of Ras proteins is required for NOS II induction.
In the current study, we sought to investigate the involvement of small G proteins of the Ras/Rho family in the induction of NOS II expression in human cells. Our data demonstrate, that NOS II induction seems to be under the negative control of Rho proteins.
Methods
Reagents
Trypsin-, glutamine-, and pyruvate-solutions, agarose, tRNA, BSA, lovastatin, mevalonate, farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP) were purchased from Sigma, Deisenhofen, Germany. Isotopes were obtained from NEN/Dupont, Köln, Germany. Restriction enzymes, Taq polymerase, Klenow DNA polymerase, T7-Sequencing Kit, dNTPs and NTPs were purchased from Amersham-Pharmacia, Freiburg, Germany. T3 and T7 RNA polymerase, RNase A, RNase T1, DNase I and DOTAP were obtained from Roche Diagnostics, Mannheim, Germany. Human IFN-γ, IL1-β, TNFα, FCS, DMEM and RPMI were purchased from PAN-Systems, Nürnberg, Germany. The Dual-Luciferase Reporter Assay System, Passive Lysis Buffer, pGL2-Basic and pRL-SV40 were purchased from Promega, Heidelberg, Germany. G 418 was purchased from Calbiochem, Bad Soden, Germany. pCR-Script was from Stratagene, Heidelberg, Germany. Clostridium difficile Toxin B (TcdB) was isolated as described (von Eichel-Streiber et al., 1987; 1995). Atorvastatin was a gift from Gödecke/Parke Davis, Freiburg, Germany.
Cell culture, cytokine treatment, RNA isolation and nitrite measurement
The human alveolar epithelium-like A549/8 cells (Edgell et al., 1983), the human colon adenocarcinoma DLD-1 cells (ATCC) and the murine fibroblasts NIH-3T3 (ATCC) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) with 5–10% foetal bovine serum, 2 mM L-glutamine, penicillin and streptomycin. For RNA isolation and NO production studies, they were plated onto 10 cm-diameter (58 cm2/well) dishes, whereas those experiments involving luciferase activity determinations were performed with cells plated onto 6-well plates (9.6 cm2/well) or 24-well plates (1.75 cm2/well). Eighteen hours prior to cytokine induction, cells were washed with PBS solution and incubated with DMEM containing 2 mM L-glutamine in the absence of serum and phenol red. During this incubation time as well as the following induction period the cells were treated with or without different concentrations of atorvastatin, lovastatin or TcdB. A549/8 and DLD-1 cells were induced with a cytokine mixture composed of INF-γ (100 u ml−1), IL1-β (50 u ml−1) and TNF-α (10 ng ml−1) for the corresponding time periods depending on the experiment. NIH-3T3 cells were induced with TNF-α (10 ng ml−1) for 4 h. Afterwards, the supernatant of the cells (300 μl) was used to measure NO2− by the Griess reaction and cells were processed for RNA isolation by guanidinium thiocyanate/phenol/chloroform extraction as described (Chomczynski & Sacchi, 1987; Kleinert et al., 1998a).
RNase protection analysis
In order to generate a luciferase cDNA plasmid for in vitro transcription, a 230 bp HinfI-fragment (positions 306–535) of pGL2-Basic was cloned after blunting into the EcoRV site of pCR-Script, generating pCR-Luc. DNA sequences of the clones were determined using the dideoxy chain termination method with a sequencing kit from Amersham-Pharmacia. For the generation of radiolabelled human NOS II-, human β-actin-, luciferase-, murine NOS II- and murine β-actin-antisense probes for RNase protection assays, 0.5 μg of the linearized plasmids pCR_NOS II_human, pCR_β-actin_human (Kleinert et al., 1996a), pCR-Luc, pCR_NOS II_mouse and pCR_β-actin_mouse (Kleinert et al., 1996b) were in vitro transcribed using T3 or T7 RNA polymerase and α-32P-UTP. To quantify human and murine NOS II mRNA levels, RNase protection experiments were performed as described (Kleinert et al., 1998a). In all experiments, β-actin mRNA expression was determined for normalization purposes. Densitometric analyses were performed using a PhosphoImager (BioRad, Munich, Germany). The protected fragments of human NOS II-, human β-actin-, luciferase-, murine NOS II- and murine β-actin-mRNA were 386 nt or 195 nt, 108 nt, 230 nt, 184 nt and 108 nt, respectively.
Stable transfection of A549/8 and DLD-1 cells; analysis of the human NOS II promoter activity
To generate A549/8- or DLD-1 cells stably transfected with a construct containing a 16 kb fragment of the human NOS II promoter cloned in front of a luciferase reporter gene cells were transfected by lipofection with DOTAP according to the manufacturer's recommendations using 4.5 μg of pNOS2(16)Luc (de Vera et al., 1996) and 0.5 μg of pRc-CMV (Invitrogen; containing a neomycin resistance gene). The transfected cells were selected by G 418-treatment (1 mg ml−1). Different cell clones were analysed for luciferase activity and checked for integration of the transfected DNA by PCR.
In order to investigate the effect of the inhibition of small G proteins on cytokine-induced NOS II promoter activity, the stably transfected cells were incubated for 18 h with DMEM without FCS and without phenol red in the presence or absence of atorvastatin and lovastatin. Then the cells were induced with CM for 4–6 h in the presence or absence of these compounds. After CM incubation cells were lysed in 1× Passive Lysis Buffer (Promega). Firefly luciferase activity was determined using the Dual Luciferase Assay Kit (Promega). Protein concentrations of the extracts were determined by Bradford reagent using BSA as standard. Luciferase activity was normalized by protein content of the extracts.
Results
Atorvastatin and lovastatin enhance NOS II induction in human DLD-1- or A549/8 cells as well as in murine NIHH 3T3 cells
In human DLD-1 colon carcinoma cells, both atorvastatin and lovastatin enhanced NOS II mRNA expression in a concentration-dependent manner (Figure 2a,b). Similar results were generated with human A549/8 aveolar carcinoma cells (Figure 2c). The same concentration-dependent enhancement of NOS II expression was seen in TNF-α-induced murine 3T3 fibroblasts (n=6; data not shown). Neither of these statins alone induced NOS II mRNA expression in any cell line (n=5; data not shown). As shown in Figure 3. both statins enhanced cytokine-induced NO production in DLD-1 and A549/8 cells.
Figure 2.

Enhancement of cytokine-induced NOS II mRNA expression by atorvastatin and lovastatin in human DLD-1- and A549/8 cells. (A) Representative RNase protection assay performed with total RNA from human DLD-1 cells incubated for 8 h with serum-free medium alone (Co) or with serum-free medium containing a mixture of cytokines (CM; 100 u ml−1 IFN-γ and 10 ng ml−1 TNF-α and 50 u ml−1 IL-1β) in the presence or absence of atorvastatin (atorva; 3–100 μM) and lovastatin (lova; 3–100 μM), respectively. Cells were preincubated for 18 h with serum-free medium with or without statins. Experiments were performed using antisense RNA probes for human NOS II and β-actin (for normalization). The positions of the protected NOS II- and β-actin fragments are indicated. (T: tRNA lane, negative control; A: β-actin antisense probe; N: NOS II antisense probe; M: molecular weight standard, φX174 restricted with HinfI). (B) Densitometric analyses of ten different gels similar to the one shown in (A). Columns (means±s.e.mean) represent relative NOS II mRNA levels at the different concentrations of the respective statin (*=P<0.05; **=P<0.01; ***=P<0.001; ns=not significant vs CM). (C) Data similar to (B) generated with A549/8 cells. Columns (means±s.e.mean) represent the relative NOS II mRNA levels at the different concentration of the respective statin (**=P<0.01; ***=P<0.001; ns=not significant vs CM).
Figure 3.

Enhancement of cytokine-induced NO production by atorvastatin and lovastatin in human DLD-1 and A549/8 cells. (A) The figure shows the statistical analysis of eight different Griess assays. Supernatants form untreated DLD-1 cells (Co) or cells stimulated for 24 h with CM in the presence or absence of atorvastatin (atorva; 3–100 μM) or lovastatin (lova; 3–100 μM). Cells were preincubated for 18 h with serum-free medium with or without statins. Columns (means±s.e.mean) represent the relative nitrite levels at different concentration of the respective statin (***=P<0.001; ns=not significant vs CM). (B) Data similar to (A) generated with A549/8 cells. Columns (means±s.e.mean) represent the relative nitrite levels at different concentration of the respective statin (*=P<0.05; **=P<0.01; ***=P<0.001; ns=not significant vs CM).
Mevalonate reverses the statin effect on NOS II expression
As statins are inhibitors of the HMG-CoA reductase, incubation of cells with these compounds results in a depletion of mevalonate (Figure 1). In order to test whether the statin-mediated superinduction of NOS II expression was specific and dependent on mevalonate depletion, DLD-1 cells were incubated with lovastatin (30 μM) in the presence or absence of mevalonate (100 and 300 μM). As shown in Figure 4a,b, substitution with mevalonate blocked statin-dependent superinduction of NOS II mRNA expression.
Figure 4.

Reversal of the statin effect on cytokine-induced NOS II mRNA expression by mevalonate and geranylgeranylpyrophosphate. (A) Representative RNase protection assay performed with total RNA from human DLD-1 cells incubated with medium alone (Co) or with a cytokine mixture (CM) in the presence or absence of lovastatin (lova; 30 μM) for 8 h. Mevalonate (meval, 100 and 300 μM) was included in some experiments. Cells were preincubated for 18 h with serum-free medium with or without lovastatin and mevalonate. Experiments were performed using antisense RNA probes for human NOS II and β-actin (for normalization). The positions of protected NOS II and β-actin fragments are indicated. (T: tRNA lane, negative control; A: β-actin antisense probe; N: NOS II antisense probe). (B) Densitometric analyses of four different gels similar to the one shown in (A). Columns (means±s.e.mean) represent the relative NOS II mRNA levels at the different concentrations of the respective compound (***=P<0.001 vs CM; ###=P<0.001 vs CM and 30 μM lovastatin). (C) Same experiments as in (A) performed with geranylgeranylpyrophosphate (GGPP, 5 and 10 μM) or farnesylpyrophosphate (FPP, 5 and 10 μM) instead of mevalonate. (D) Densitometric analyses of five different gels similar to the one shown in (C). Columns (means±s.e.mean) represent the relative NOS II mRNA levels at the different concentrations of the respective compound (***=P<0.001 vs CM; ###=P<0.001 vs CM and 30 μM lovastatin).
Geranylgeranylpyrophosphate inhibits statin-dependent superinduction of NOS II expression
Farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP) are important for the post-translational modification of small G proteins of the Ras/Rho family (Figure 1). Prenylation is prerequisite for the activation of these proteins. Ras proteins are predominantly farnesylated, whereas the Rho proteins are mainly geranylgeranylated. To test whether Ras or Rho proteins are important for the statin-dependent superinduction of NOS II mRNA expression, DLD-1 cells were incubated with lovastatin (30 μM) in the presence or absence of FPP (5 or 10 μM) or GGPP (5 or 10 μM). As shown in Figures 4c,d, GGPP reversed the lovastatin-induced NOS II superinduction almost completely. In contrast, FPP diminished the statin-induced superinduction only slightly.
Clostridium difficile toxin B (TcdB) enhances cytokine-induced NOS II expression in human cells
TcdB is a glucosyltransferase that specifically inactivates the Rho proteins without affecting small G proteins of the Ras family. As shown in Figure 5a,b, TcdB enhanced concentration-dependently cytokine-induced NOS II mRNA expression in A549/8 cells. The same effect of TcdB on iNOS expression was seen in murine 3T3 cells (n=6; data not shown).
Figure 5.
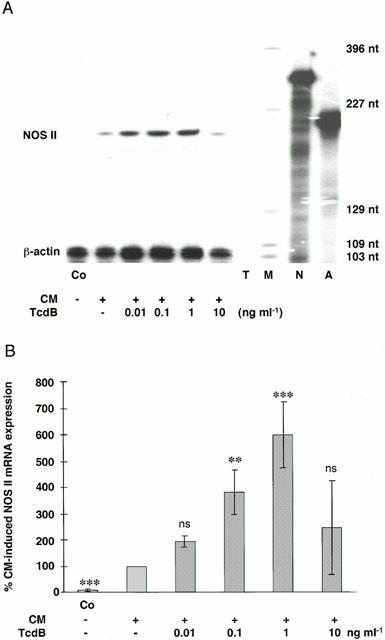
Toxin B from Clostridium difficile (TcdB) enhanced cytokine-induced NOS II mRNA expression in human A549/8 cells. (A) Representative RNase protection assay performed with total RNA from human A549/8 cells incubated with medium alone (Co) or with a cytokine mixture (CM) in the presence or absence different concentrations of toxin B from Clostridium difficile (TcdB) for 8 h. Cells were preincubated for 18 h with serum-free medium with or without TcdB. Experiments were performed using antisense RNA probes for human NOS II and β-actin (for normalization). The positions of the protected NOS II and β-actin fragments are indicated. (T: tRNA lane, negative control; A: β-actin antisense probe; N: NOS II antisense probe; M: molecular weight standard, pGL2-Basic restricted with HinfI). (B) Densitometric analyses of five different gels as shown in (A). Columns (means±s.e.mean) represent the relative NOS II mRNA levels at the different concentrations of TcdB (**=P<0.01; ***=P<0.001; ns=not significant vs CM). At the highest concentration of TcdB (10 ng ml−1) the cells showed morphological changes indicating cell toxicity.
Effects of atorvastatin and lovastatin on NOS II promoter activity in DLD-1- or A549/8 cells
In DLD-1 cells stably transfected with pNOS2(16)Luc (16 kb human NOS II promoter fragment cloned in front of a luciferase reporter gene), both statins produced an increase in CM-induced NOS II promoter activity (up to 3 fold, Figure 6a). In contrast, in A549/8 cells stably transfected with pNOS2(16)Luc, both statins only produced a small increase in CM-induced NOS II promoter activity (up to 1.5 fold, Figure 6b). Neither of the statins, when given alone, induced NOS II promoter activity in these two cell lines (n=4; data not shown). We confirmed these results by determining luciferase mRNA expression with RNase protection assays (n=5; data not shown).
Figure 6.

Effect of atorvastatin and lovastatin on the activity of a human 16 kb NOS II promoter fragment. (A) DLD-1 cells stably transfected with a construct containing a 16 kb human NOS II promoter fragment in front of a luciferase reporter gene (pNOS2(16)Luc, (de Vera et al., 1996)) were preincubated with or without atorvastatin (atorva, 3–30 μM) or lovastatin (lova; 3–30 μM) for 18 h in serum-free medium. Then cells were incubated with CM for 4 h in the presence or absence of the respective statin. Cell extracts were prepared and luciferase activity and protein content of the extracts determined. The columns (means±s.e.mean) represent relative luciferase activities (per cent of the CM effect) at different concentration of the statins (**=P<0.01; ***=P<0.001; ns=not significant vs CM). (B) The same experiments as in (A) with A549/8 cells stably transfected with pNOS2(16)Luc. The columns (means±s.e.mean) represent relative luciferase activities (per cent of the CM effect) at different concentrations of the statins (**=P<0.01; ***=P<0.001; ns=not significant vs CM).
Discussion
The current study provides evidence for an inhibitory role of small G proteins in the induction of NOS II expression in human (and murine) cells. Inhibition of protein-prenylation by the statins atorvastatin or lovastatin resulted in a superinduction of cytokine-mediated NOS II expression (Figures 2 and 3). This superinduction was abrogated by mevalonate and GGPP, but only modestly affected by FPP (Figure 4). As Rho-proteins are mainly geranylgeranylated, whereas Ras proteins are predominantly farnesylated (Figure 1), these data point to a negative involvement of Rho proteins in NOS II induction. This was corroborated by the TcdB-induced enhancement of cytokine-induced NOS II expression (Figure 5), because TcdB directly inactivates Rho proteins.
As these effects were also seen in murine 3T3 cells, the inhibitory role of Rho proteins seems to be a general principle in NOS II induction. Also in rat pulmonary artery smooth muscle cells and rat hepatocytes, Finder et al. (1997) demonstrated a superinduction of IL-1β-induced NOS II expression by lovastatin and by a specific inhibitor of the geranlygeranyltransferase. Specific blockade of protein farnesylation decreased IL1β-induced NOS II expression. Thus, also in these rat cells, Rho proteins seem to inhibit NOS II induction, whereas Ras proteins are required for NOS II expression.
In rat primary astrocytes Pahan et al. (1997) described an inhibitory effect of lovastatin on LPS-induced NOS II expression, which was reversed by mevalonate and farnesylpyrophosphate. These authors concluded that Ras proteins are positively involved in NOS II induction, because Ras proteins require farnesylation for activation (Gibbs & Oliff, 1997).
In our human cell models, we found no evidence for a major involvement of Ras proteins in NOS II induction. Ras proteins are believed to signal mainly through the ERK-1/2-MAPK-pathway (Denhardt, 1996). However, we have shown previously that cytokine-induction of DLD-1 cells did not result in ERK activation and that the specific inhibitor of ERK activation, PD 98059, did not interfere with the NOS II induction process (Kleinert et al., 1998a). Also, in A549/8 cells, PD98059 did not modify cytokine-induced NOS II expression (data not shown).
In order to analyse the molecular mechanism of Rho protein-mediated regulation of NOS II expression, we tested the effect of statins on human A549/8- and DLD-1 cells stably transfected with pNOS2(16)Luc (16 kb human NOS II promoter fragment cloned in front of a luciferase reporter gene (de Vera et al., 1996)). In DLD-1 cells, statin treatment resulted in a marked enhancement of NOS II promoter activity (Figure 6a) which explains the observed statin-dependent upregulation of NOS II expression. In contrast, in A549/8 cells, statins enhanced cytokine-induced NOS II promoter activity only modestly (Figure 6b). Given the marked (3 fold) superinduction of NOS II expression by statins in these cells, the involvement of additional post-transcriptional mechanisms has to be postulated.
Small G proteins of the Rho family have been described to activate AP-1-dependent transcription (Chang et al., 1998; Naumann et al., 1998). In addition we have shown in previous studies that AP-1 negatively regulates NOS II expression in DLD-1 cells by inhibiting cytokine-induced NOS II promoter activity (Kleinert et al., 1998b). Therefore, Rho proteins may decrease NOS II expression via activation of AP-1.
Run-on analyses (de Vera et al., 1996; Linn et al., 1997; Rodriguez-Pascual et al., 2000) and transfection studies (see Figure 6) have demonstrated a basal activity of the human NOS II promoter in untreated cells, which, however, did not result in a measurable NOS II expression. Cytokine stimulation of human cells produced a marked expression of NOS II mRNA (and protein) accompanied by only a moderate (5–10 fold) increase in transcriptional activity of the human NOS II promoter (c.f. Figures 1 and 6). The 3′-untranslated region of the NOS II mRNA contains AU motifs, known to destabilize the mRNAs of proto-oncogenes, nuclear transcription factors and cytokines. Luciferase reporter gene constructs containing the NOS II 3′-untranslated region showed a significantly reduced luciferase activity (Rodriguez-Pascual et al., 2000). This provides evidence that NOS II expression is regulated at the transcriptional and post-transcriptional level. Based on our findings with A549/8 cells (see above), it seems likely that Rho proteins regulate NOS II expression by regulating NOS II mRNA stability.
Also, other isoforms of NOS seem to be under the expressional control of small G proteins. According to Laufs & Liao (1998), inhibition of small G proteins of the Rho family resulted in a stabilization of endothelial NOS III mRNA.
Interestingly, the same cytokines that induce NOS II expression also enhance Rho activity (Denhardt, 1996; Lim et al., 1996). Thus, the negative regulation of NOS II induction by Rho proteins could represent a negative feedback pathway controlling excessive NOS II expression.
In conclusion, our data demonstrate that statins and TcdB potentiate cytokine-induced NOS II expression via inhibition of small G proteins of the Rho family. The inhibition of Rho proteins results in an enhanced NOS II promoter activity and–depending on the cell type studied–in additional stabilization of the NOS II mRNA.
Acknowledgments
The expert technical assistance of K. Masch is gratefully acknowledged. F. Rodriguez-Pascual was the recipient of a FEBS Long-Term Fellowship. The plasmids pNOS(16)Luc and the full-length human NOS II cDNA were generous gifts of Dr D.A. Geller, Department of Surgery, Pittsburgh, PA, USA. This work was supported by Grant 8312-38 62 61 from the Innovation Foundation of the State of Rhineland-Palatinate (to H. Kleinert and U. Förstermann), Grant Kl 1020/4-1 from the Deutsche Forschungsgemeinschaft (to H. Kleinert), and by the Collaborative Research Center SFB553 (Project A1 to U. Förstermann).
Abbreviations
- ERK
extracellular regulated kinase
- FPP
farnesylpyrophosphate
- GGPP
geranylgeranylpyrophosphate
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- IFN-γ
interferon-γ
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- JNK
Jun N-terminal kinase
- MAPK
mitogen activated protein kinase
- NO
nitric oxide
- NOS
NO synthase
- NOS II
NO synthase II
- iNOS
inducible NOS
- SAPK
stress-activated protein kinase
- TcdB
Toxin B from Clostridium difficile
- TNF-α
tumour necrosis factor-α
References
- CHANG J.H., PRATT J.C., SAWASDIKOSOL S., KAPELLER R., BURAKOFF S.J. The small GTP-binding protein Rho potentiates AP-1 transcription in T cells. Mol. Cell Biol. 1998;18:4986–4993. doi: 10.1128/mcb.18.9.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- DE VERA M.E., SHAPIRO R.A., NÜSSLER A.K., MUDGETT J.S., SIMMONS R.L., MORRIS S.M., BILLIAR T.R., GELLER D.A. Transcriptional regulation of human inducible nitric oxide synthase (NOS2) gene by cytokines: initial analysis of the human NOS2 promoter. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1054–1059. doi: 10.1073/pnas.93.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENHARDT D.T. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem. J. 1996;318:729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EBERHARDT W., PLUSS C., HUMMEL R., PFEILSCHIFTER J. Molecular mechanisms of inducible nitric oxide synthase gene expression by IL-1β and cAMP in rat mesangial cells. J. Immunol. 1998;160:4961–4969. [PubMed] [Google Scholar]
- EDGELL C.J., MCDONALD C.C., GRAHAM J.B. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc. Natl. Acad. Sci. U.S.A. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINDER J.D., LITZ J.L., BLASKOVICH M.A., MCGUIRE T.F., QIAN Y.M., HAMILTON A.D., DAVIES P., SEBTI S.M. Inhibition of protein geranylgeranylation causes a superinduction of nitric-oxide synthase-2 by interleukin-1β in vascular smooth muscle cells. J. Biol. Chem. 1997;272:13484–13488. doi: 10.1074/jbc.272.21.13484. [DOI] [PubMed] [Google Scholar]
- FÖRSTERMANN U., BOISSEL J.P., KLEINERT H. Expressional control of the ‘constitutive' isoforms of nitric oxide synthase (NOS I and NOS III) FASEB. J. 1998;12:773–790. [PubMed] [Google Scholar]
- FÖRSTERMANN U., KLEINERT H. Nitric oxide synthase: expression and expressional control of the three isoforms. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:351–364. doi: 10.1007/BF00172772. [DOI] [PubMed] [Google Scholar]
- GELLER D.A., BILLIAR T.R. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998;17:7–23. doi: 10.1023/a:1005940202801. [DOI] [PubMed] [Google Scholar]
- GIBBS J.B., OLIFF A. The potential of farnesyltransferase inhibitors as cancer chemotherapeutics. Annu. Rev. Pharmacol. Toxicol. 1997;37:143–166. doi: 10.1146/annurev.pharmtox.37.1.143. [DOI] [PubMed] [Google Scholar]
- GILBERT R.S., HERSCHMAN H.R. ‘Macrophage' nitric oxide synthase is a glucocorticoid-inhibitable primary response gene in 3T3 cells. J. Cell. Physiol. 1993;157:128–132. doi: 10.1002/jcp.1041570117. [DOI] [PubMed] [Google Scholar]
- KLEINERT H., BOISSEL J.P., SCHWARZ P.M., FÖRSTERMANN U.Regulation of the expression of nitric oxide synthase isoforms Nitric Oxide: Biology and Pathobiology 2000New York: Academic Press; 105–128.ed. Ignarro, L.J. pp [Google Scholar]
- KLEINERT H., EUCHENHOFER C., FRITZ G., IHRIG-BIEDERT I., FÖRSTERMANN U. Involvement of protein kinases in the induction of NO synthase II in human DLD-1 cells. Br. J. Pharmacol. 1998a;123:1716–1722. doi: 10.1038/sj.bjp.0701782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLEINERT H., EUCHENHOFER C., IHRIG-BIEDERT I., FÖRSTERMANN U. Glucocorticoids inhibit the induction of nitric oxide synthase II by down-regulating cytokine-induced activity of transcription factor nuclear factor-kB. Mol. Pharmacol. 1996a;49:15–21. [PubMed] [Google Scholar]
- KLEINERT H., EUCHENHOFER C., IHRIG-BIEDERT I., FÖRSTERMANN U. In murine 3T3 fibroblasts, different second messenger pathways resulting in the induction of NO synthase II (iNOS) converge in the activation of transcription factor NF-κB. J. Biol. Chem. 1996b;271:6039–6045. doi: 10.1074/jbc.271.11.6039. [DOI] [PubMed] [Google Scholar]
- KLEINERT H., WALLERATH T., FRITZ G., IHRIG-BIEDERT I., RODRIGUEZ-PASCUAL F., GELLER D.A., FÖRSTERMANN U. Cytokine induction of NO Synthase II in human DLD-1 cells: roles of the JAK-STAT, AP-1 and NF-κB-signaling pathways. Br. J. Pharmacol. 1998b;125:193–201. doi: 10.1038/sj.bjp.0702039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRÖNCKE K.D., FEHSEL K., KOLB-BACHOFEN V. Inducible nitric oxide synthase in human diseases. Clin. Exp. Immunol. 1998;113:147–156. doi: 10.1046/j.1365-2249.1998.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUNZ D., WALKER G., EBERHARDT W., MESSMER U.K., HUWILER A., PFEILSCHIFTER J. Platelet-derived growth factor and fibroblast growth factor differentially regulate interleukin 1β- and cAMP-induced nitric oxide synthase expression in rat renal mesangial cells. J. Clin. Invest. 1997;100:2800–2809. doi: 10.1172/JCI119827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAUFS U., LIAO J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J. Biol. Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- LIM L., MANSER E., LEUNG T., HALL C. Regulation of phosphorylation pathways by p21 GTPases. The p21 Ras- related Rho subfamily and its role in phosphorylation signalling pathways. Eur. J. Biochem. 1996;242:171–185. doi: 10.1111/j.1432-1033.1996.0171r.x. [DOI] [PubMed] [Google Scholar]
- LINN S.C., MORELLI P.J., EDRY I., COTTONGIM S.E., SZABO C., SALZMAN A.L. Transcriptional regulation of human inducible nitric oxide synthase gene in an intestinal epithelial cell line. Am J Physiol. 1997;272:G1499–G1508. doi: 10.1152/ajpgi.1997.272.6.G1499. [DOI] [PubMed] [Google Scholar]
- MACKAY D.J., HALL A. Rho GTPases. J. Biol. Chem. 1998;273:20685–20688. doi: 10.1074/jbc.273.33.20685. [DOI] [PubMed] [Google Scholar]
- MACMICKING J., XIE Q.W., NATHAN C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997;15:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- MESSMER U.K., BRÜNE B. Modulation of inducible nitric oxide synthase in Rinm5F Cells. Cell Signal. 1994;6:17–24. doi: 10.1016/0898-6568(94)90057-4. [DOI] [PubMed] [Google Scholar]
- NATHAN C., XIE Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- NAUMANN M., RUDEL T., WIELAND B., BARTSCH C., MEYER T.F. Coordinate activation of activator protein 1 and inflammatory cytokines in response to Neisseria gonorrhoeae epithelial cell contact involves stress response kinases. J. Exp. Med. 1998;188:1277–1286. doi: 10.1084/jem.188.7.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAHAN K., SHEIKH F.G., NAMBOODIRI A.M., SINGH I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J. Clin. Invest. 1997;100:2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGUEZ-PASCUAL F., HAUSDING M., IHRIG-BIEDERT I., FURNEAUX H., LEVY A.P., FORSTERMANN U., KLEINERT H.Complex contribution of the 3′-untranslated region to the expressional regulation of the human inducible nitric oxide synthase gene: Involvement of the RNA-binding protein HuR J Biol Chem 2000. published June 19, 2000 as 10.1074/jbc.M910460199 [DOI] [PubMed]
- VON EICHEL-STREIBER C., BOQUET P., SAUERBORN M., THELESTAM M. Large clostridial cytotoxins–a family of glycosyltransferases modifying small GTP-binding proteins. Trends Microbiol. 1996;4:375–382. doi: 10.1016/0966-842X(96)10061-5. [DOI] [PubMed] [Google Scholar]
- VON EICHEL-STREIBER C., HARPERATH U., BOSSE D., HADDING U. Purification of two high molecular weight toxins of Clostridium difficile which are antigenically related. Microb. Pathog. 1987;2:307–318. doi: 10.1016/0882-4010(87)90073-8. [DOI] [PubMed] [Google Scholar]
- VON EICHEL-STREIBER C., MEYER ZU HERINGDORF D., HABERMANN E., SARTINGEN S. Closing in on the toxic domain through analysis of a variant Clostridium difficile cytotoxin B. Mol. Microbiol. 1995;17:313–321. doi: 10.1111/j.1365-2958.1995.mmi_17020313.x. [DOI] [PubMed] [Google Scholar]