

Abstract
The release of PGE2 and nitric oxide (NO) from the respiratory epithelium may act to dampen inflammation. In other tissues, oncostatin M (OSM), a potent inducer of epithelial antiproteases, has also been shown to interact with IL-1β to stimulate PGE2 release. However, whether OSM interacts with pro-inflammatory cytokines and proteases in the production of anti-inflammatory eicosanoids and NO from airway epithelium is unknown.
The effect of OSM and the related cytokine leukaemia inhibitory factor (LIF) on PGE2 and NO production by the respiratory epithelial cell line, A549 in response to pro-inflammatory cytokines as well as protease-rich house dust mite (HDM) fractions and a protease-deficient rye grass pollen extract was examined by immunohistochemistry, cell culture, ELISA and enzyme-immunoassay.
Cells treated with a mixture of IL-1β, IFNγ and LPS for 48 h produced a 9 fold increase in PGE2 and a 3 fold increase in NO levels (both P<0.05). Both OSM and LIF were without effect. However, OSM added together with the cytokine mixture synergistically enhanced PGE2 production (22 fold, P<0.05). OSM also synergistically enhanced PGE2 production in response to a cysteine protease-enriched, but not serine protease-enriched HDM fraction (P<0.05). Rye grass extract, neither alone nor in combination with OSM, induced PGE2 or NO production, although it did induce the release of GM-CSF.
These observations suggest that OSM is an important co-factor in the release of PGE2 and NO from respiratory epithelial cells and may play a role in defense against exogenous proteases such as those derived from HDM.
Keywords: Oncostatin M, cytokine, epithelium, PGE2, inflammation, dust mites
Introduction
The respiratory epithelium plays an important role in airway homeostasis since it is the tissue of first contact to insults from the external environment (Folkerts & Nijkamp, 1998; Knight et al., 1994). Rather than acting simply as a physical barrier, the epithelium is metabolically active and has been shown to regulate both the inflammatory and subsequent repair processes characteristic of respiratory diseases such as asthma. In this context, the epithelium releases a battery of pro-inflammatory cytokines and growth factors such as IL-1, IL-6, GM-CSF and TGFβ as well as putative anti-inflammatory mediators such as PGE2 and nitric oxide (NO) (Knight et al., 1994). PGE2 is the predominant cyclo-oxygenase metabolite produced by the epithelium and has been shown to exert a variety of anti-inflammatory or protective effects both in vitro and in vivo. They include protection against allergen-induced bronchoconstriction (Gauvreau et al., 1999), the relaxation of airway smooth muscle (Knight et al., 1995; Pavord & Tattersfield, 1995), inhibition of cholinergic neurotransmission (Ito et al., 1990), inhibition of mast cell mediator release (Peters et al., 1982) and the modulation of fibroblast proliferation (McAnulty et al., 1997).
The mechanisms underlying the release of PGE2 and NO from respiratory epithelium are not well understood, but a number of stimuli have been shown to modulate epithelial function. In this regard, proteases, released from a diverse array of exogenous sources such as house dust mites (Winton et al., 1998), fungi (Tomee et al., 1997) and grass pollens (Hassim et al., 1998; Tomee et al., 1998) have been shown to be particularly important. They have been shown to induce apoptosis (Winton et al., 1998), increase the flux of macromolecules across sheets of airway mucosa (Herbert et al., 1995) and induce the release of cytokines such as IL-6, IL-8 (King et al., 1998), and IL-1β (Calderon et al., 1997; Hastie et al., 1996; Yu et al., 1997). In other tissues, IL-1β has been shown to be a potent inducer of COX-2 expression and PGE2 release. However, whether allergenic proteases modulate PGE2 and NO release directly or through the release of intermediary cytokines is unknown.
More recently, it has been shown that PGE2 release induced by IL-1β may be augmented by concomitant exposure to the cytokine Oncostatin M (OSM) (Bernard et al., 1999). This cytokine, first described in cells associated with inflammation such as T-lymphocytes and monocyte/macrophages (Rose & Bruce, 1991; Zarling et al., 1986), belongs to the IL-6 family of cytokines, which also includes leukaemia inhibitory factor (LIF), IL-11, ciliary neurotrophic factor and cardiotrophin 1 (Taga, 1997). Like all members of this cytokine family, OSM exhibits a variety of biological activities. To this end, we have begun examining the localisation and role of these cytokines in airway inflammation (Knight et al., 1998; 1999a, 1999b).
In this study, we investigated the role of OSM and LIF on the release of PGE2 and NO from the respiratory epithelial cell line, A549. Given their association with allergic disease, we have also investigated the role of protease-enriched fractions isolated from the house dust mite Dermatophagoides pteronyssinus and a protease-deficient extract of rye grass pollen, on PGE2 and NO release. The data obtained show that OSM, but not LIF, enhanced the release of PGE2 from A549 cells exposed to either a stimulatory cytokine mix or mite proteases, in particular, the cysteine protease enriched fraction. In contrast, the rye grass pollen extract was without effect.
Methods
Cell culture
A549 cells, a human pulmonary epithelial type II cell line was obtained from American Type Culture Collection and cultured according to standard conditions as described previously (Watkins et al., 1997a). Cells were seeded into 24-well tissue culture plates (Nunc, Naperville, U.S.A.) and grown to 80% confluence in Hams F-12 media supplemented with 10% (v v−1) foetal calf serum (FCS), 100 μg ml−1 gentamicin and L-glutamine (2 mM). At this time, cells were then washed in phosphate buffered saline (PBS) and incubated in serum-free medium for 16 h prior to the commencement of each experiment. For all experiments, cells were exposed to cytokine/LPS mix or allergen extracts for 48 h, and each experiment was performed in quadruplicate. In the present study, the mite protease-enriched fractions and rye grass pollen extracts were titrated to obtain the maximum concentration that did not induce significant amounts of cell detachment. For the mite protease fractions this concentration was determined to be 0.5 Azocoll unit mg protein−1. In contrast, the rye grass extract did not induce significant amounts of cell detachment. At the conclusion of each experiment, supernatants were collected and cleared by centrifugation at 12,000×g for 5 min at 4°C and then stored at −20°C until required. Cells from each well were then trypsinized and counted using a Neubauer haemocytometer. Viability of cells was determined by trypan blue exclusion. For immunohistochemistry, cells were seeded into 8-chamber culture slides (Falcon, Perth, Australia) at a density of 3×104/chamber and grown in Ham's F-12 medium for 48 h. The cells were then washed in PBS before being fixed by immersion in ice-cold acetone for 20 min and processed for immunohistochemistry.
PGE2 enzyme immunoassay
PGE2 was measured using a competitive enzyme immunoassay according to the manufacturer's instructions (Cayman Chemical, Ann Arbor, MI, U.S.A.). Plates were read using an ELISA plate reader (Spectramax 250, Molecular Devices Co., Sunnyvale, CA, U.S.A.) at 450 nm. The manufacturer's specifications for this assay include an intra-assay coefficient of variation of <10%, cross reactivity with PGD2 and PGF2α of less than 1% and linearity over the range of 10–1000 pg ml−1.
GM-CSF ELISA
GMCSF was quantified by specific capture ELISA using commercially available antibodies. Briefly, 96-well plates (Maxisorp, Nunc) were coated with 100 μl of the capture antibody (500 ng ml−1 in 0.1 M NaHCO3/NaCO3 buffer, pH 9.6) and incubated overnight at 4°C. After washing, samples or standards were added to the plate and allowed to incubate at room temperature overnight. After multiple washes in wash buffer (0.1% (v v−1) Tween 20 in PBS), the biotinylated 2° antibody was then added and the plate allowed to incubate at room temperature for 1 h, after which peroxidase-labelled streptavidin (Dako, Sydney, Australia) was added. After 30 min, substrate (K-Blue ELISA substrate, Elisa systems, Brisbane, Australia) was added to each well. The reaction was terminated by the addition of 1 M phosphoric acid and the plates read on a ELISA plate reader (Spectramax) at 450 nm.
NO analysis
The NO content of cell culture supernatants was measured as NO2− using a chemiluminescence NO analyser (Seivers model 280, Boulder, CO, U.S.A.).
Immunocytochemistry
Immunocytochemical expression of OSMR and LIFR was investigated using affinity purified polyclonal antibodies to OSMR and LIFR respectively. The OSMR antibody used was specific for the β-chain of OSMR and does not cross-react with the LIFR (Mosley et al., 1996). Similarly, according to the manufacturer's specifications, the LIFR antibody does not cross-react with OSM. At the completion of each experiment, cells were fixed in ice-cold acetone for 20 min. Non-specific binding was blocked by the addition of 0.25% (w v−1) casein. Cells were then incubated with either anti-OSMR or anti-LIFR antibodies for 1 h at room temperature at concentrations of 1 and 2.5 μg ml−1 respectively. All dilutions were prepared in Tris buffered saline (TBS) containing 0.1% (w v−1) bovine serum albumin (BSA). Negative controls included cells that were incubated in this buffer but without the primary antibody and use of irrelevant IgG ((NH4)2SO4 cut of a rabbit anti-house dust mite) instead of the appropriate primary antibody. Sections were then incubated for 1 h with a FITC-conjugated secondary antibody at a concentration of 15 μg ml−1 (Dako), and mounted under a glass coverslip. Staining was observed using a Zeiss epifluorescence microscope (Zeiss, Sydney, Australia).
Preparation of protease fractions from house dust mite (HDM) and rye grass pollen
Cysteine protease-enriched and serine protease-enriched fractions of whole mite media were prepared as described previously (Winton et al., 1998). Briefly, spent medium from cultures of D. pteronyssinus was extracted in five volumes of PBS and centrifuged at 48,000×g for 20 min at 4°C. Ammonium sulphate was gradually added to the supernatant to achieve a 50% saturated solution. After further centrifugation (48,000×g, 20 min, 4°C), the pellet (enriched for cysteine protease activity), was redissolved in a minimum volume of distilled H2O. Ammonium sulphate was then added to the supernatant from the first cut to achieve 80% saturation. This fraction (enriched for serine protease activity) was also resuspended in a minimum of distilled H2O. The cysteine and serine protease fractions were dialyzed against distilled H2O and lyophilized prior to reconstitution in Hams F-12 medium as required. Extracts were also assayed for endotoxin using the Limulus amebocyte lysis assay and found to be below the limit of detection (<0.06 ng ml−1). Rye grass pollen was extracted with PBS at 10% (w v−1) overnight at 4°C and the resulting diffusate was then dialyzed against PBS overnight at 4°C. The protein concentrations of the cysteine protease enriched fraction, serine protease enriched fraction and rye grass extract as determined by the method of Bradford (Bradford, 1976) were 255, 140 and 450 μg ml−1 respectively.
Determination of protease activity of the HDM fractions and Rye grass extracts
Protease activities of D. pteronyssinus protease fractions and Rye grass (Lolium perenne) pollen extract were determined spectrophotometrically using insoluble diazotized collagen (Azocoll) as described previously (Stewart et al., 1989) and expressed in Azocoll unit ml−1 (Table 1). To measure cysteine proteinase activity, fractions were activated with dithiothreitol (DTT, 1.5 mM) before assay.
Table 1.
Protease activity* and Der p 1 concentration of fractionated house dust mite fecal pellets and extracts of Rye grass
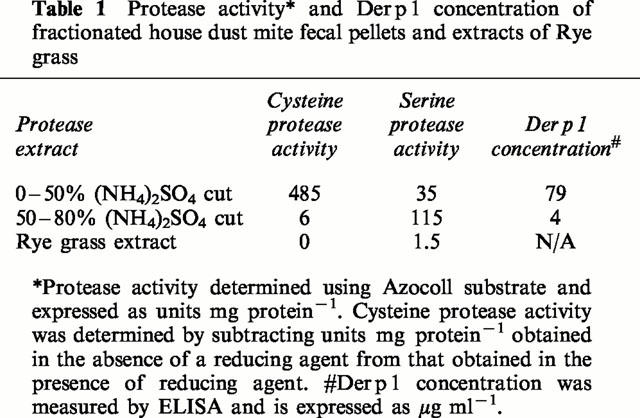
Determination of Der p 1 concentration in fractionated HDM fecal pellets
The concentrations of immunoreactive Der p 1 in the mite proteinase fractions were determined using a capture-ELISA as previously described (Hill et al., 1997), using commercially obtained monoclonal antibodies.
Materials and reagents
Cell culture media and additives were purchased from Gibco BRL (Melbourne, Australia), trypsin-versene solution was purchased from Commonwealth Serum Laboratories (Perth, Australia), cell culture plastic ware and disposables from Falcon (Perth, Australia). ELISA plates were purchased from Nunc (Naperville, U.S.A.). OSM, LIF, LIFR, and GM-CSF antibodies were purchased from R&D systems (Minneapolis, MN, U.S.A.). The polyclonal antibody to OSMR was provided by Immunex Corporation (Seattle, WA, U.S.A.). Monoclonal antibodies against Der p 1 were obtained from Indoor Biotechnologies (Charlottesville, VA, U.S.A.). Rye grass pollen extracts were obtained from Greer Laboratories (Lenoir, NC, U.S.A.). All other cytokines were obtained from Boehringer-Mannheim (Sydney, Australia). Limulus amebocyte lysis assay was purchased from Endotect (ICN Biochemicals, Melbourne, Australia). Coomassie blue was purchased from BioRad (Sydney, Australia). K blue TMB substrate was obtained from Elisa Systems (Brisbane, Australia). Cell culture antibiotics and all other chemicals were purchased from Sigma (Sydney, Australia).
Statistical analyses
Data were expressed as mean±s.e.mean. Statistical comparisons of mean data were performed using one-way ANOVA with Bonferroni correction performed post-hoc to correct for multiple comparisons. A P value of <0.05 was considered significant.
Results
Immunohistochemical detection of LIFR and OSMR in A549 cells
Experiments were initially undertaken to determine the presence of both LIFR (type I receptor) and OSMR (type II receptor) on A549 cells. Figure 1 shows immunoreactive staining for both receptors in confluent cultures of these cells. Expression of OSMR was widespread, with essentially all cells showing some degree of staining. Expression of LIFR was also observed although the staining did not appear to be as intense as for OSMR. In contrast, staining in the negative control sections was not observed.
Figure 1.
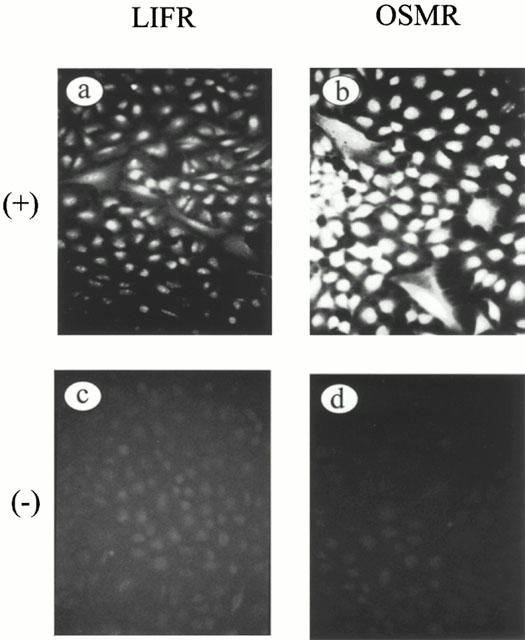
Immunofluorescent detection of OSMR and LIFR in A549 cells. Photomicrograph of positive staining for (a) LIFR and (b) OSMR. (c) and (d) represent corresponding negative controls for LIFR and OSMR respectively. (Final magnification:×100).
Release of OSM from A549 cells
A549 cells did not release detectable amounts of OSM either at rest or following exposure to both the cytokine/LPS mix and HDM protease-enriched fractions (data not shown).
Effect of cytokine mix, OSM and LIF on PGE2 release
The effect of OSM on the generation of PGE2 from A549 cells is shown in Figure 2a. Cells were exposed to either OSM (2, 20 or 200 ng ml−1) or a cytokine/LPS mix consisting of IL-1β (1 u ml−1), IFNγ (100 u ml−1) and LPS (0.1 μg ml−1) for 48 h. Unstimulated cells or cells exposed to OSM alone produced little PGE2 after 48 h in culture (Figure 2a). However, when cells were exposed to the cytokine/LPS mixture, a 9 fold increase in PGE2 production was detected (Figure 2a; P<0.05). When cells were exposed to a combination of OSM and cytokine/LPS mixture, a mean 22 fold increase in PGE2 production was observed when compared to unstimulated cells (P<0.01).
Figure 2.
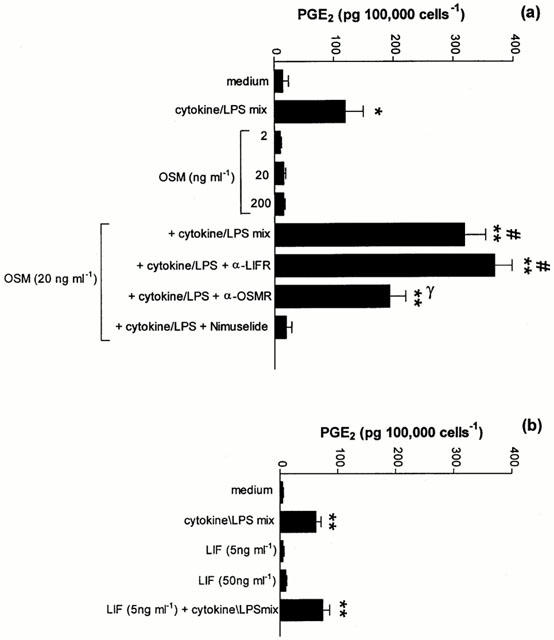
Effect of OSM, LIF and cytokine/LPS mix alone and in combination on PGE2 production from A549 cells. (a) Effect of OSM. A549 cells were treated with OSM or a mix of IL-1β, IFNγ and LPS for 48 h. Supernatants were collected and PGE2 quantified by EIA. Antibodies to OSMR and LIFR as well as the COX-2 specific inhibitor, nimesulide were added 30 min prior to addition of OSM and cytokine/LPS mix. Results are expressed as means±s.e.mean of four experiments performed in quadruplicate. (b) Effect of LIF. A549 cells were treated with LIF or a mix of IL-1β, IFNγ and LPS in the presence or absence of LIF for 48 h, before supernatants were assayed for PGE2 as described in (a). Results are expressed as mean±s.e.mean of four experiments performed in quadruplicate. *P<0.05 compared to medium control, **P<0.01 compared to medium control, #P<0.05 compared to cytokine/LPS mix, γP<0.05 compared to OSM+cytokine/LPS mix.
Treatment of A549 cells with the COX-2 selective inhibitor nimesulide (1 μM) for 30 min prior to the addition of the cytokine mix completely suppressed the production of PGE2 in response to the cytokine/LPS mix (Figure 2a).
Whether OSM was acting via the type I or specific type II receptor was investigated by pretreating cells with neutralising antibodies to either LIFR or OSMR at a dilution of 1:100 for 30 min prior to the addition of the cytokine/LPS mix (Figure 2a). Blockade of the OSMR completely inhibited the synergistic interaction between OSM and cytokine/LPS mixture. In contrast, selective neutralisation of LIFR did not influence PGE2 production, confirming that OSM was interacting with the type II receptor. As was observed for OSM, exposure of A549 cells to LIF (5 or 50 ng ml−1) for 48 h did not induce PGE2 release (Figure 2b). In contrast to OSM, concomitant addition of LIF and cytokine/LPS mix did not result in any synergistic interaction (Figure 2b).
Effects of HDM protease-enriched fractions on PGE2 release
The cysteine protease and serine protease activities of the mite protease fractions were confirmed by the Azocoll degradation assay (Table 1). As expected, the protease activity of the cysteine-enriched fraction was significantly enhanced following exposure to the reducing agent DTT (1.5 mM). The Der p 1 concentration of both protease-enriched fractions is also shown in Table 1. The concentration of Der p 1 in the cysteine protease-enriched fraction was 15 fold higher than the serine protease-enriched fraction.
Exposure of A549 cells to the cysteine protease-enriched fraction induced a dose-dependent increase in PGE2 production (Figure 3a). The effect on PGE2 release was significantly enhanced following activation by DTT (P<0.05). A synergistic interaction between the cysteine protease enriched fraction and OSM was observed only in the presence of DTT (P<0.05; Figure 3a) and was abolished by heating allergen samples to 65°C for 10 min prior to adding them to the cell cultures. The addition of DTT alone did not significantly increase the release of PGE2 per se (Figure 3a). Exposure to the serine protease enriched fraction also dose-dependently stimulated significant amounts of PGE2 from A549 cells (P<0.05; Figure 3b). Heating allergen samples prior to adding them to the cell cultures also abolished this effect. However, when OSM was added in combination with the serine protease enriched fraction, a synergistic increase in PGE2 production was not observed (Figure 3b).
Figure 3.
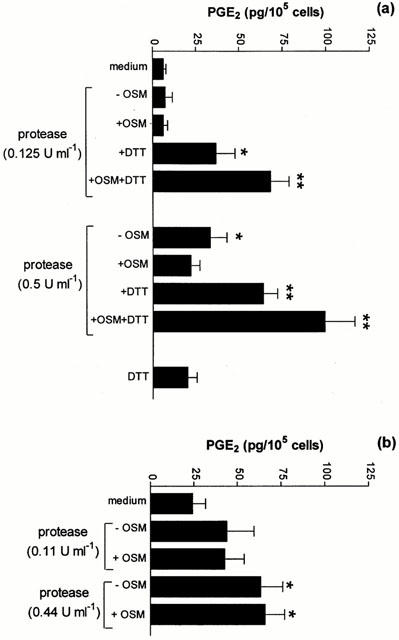
Effect of protease-enriched fractions of HDM fecal pellets on PGE2 production by A549 cells. (a) Effect of cysteine protease-enriched fraction. Cells were exposed to cysteine protease-enriched fractions in the absence or presence of DTT (1.5 mM) and/or OSM (20 ng ml−1). After 48 h, cell culture supernatants were taken and the amount of PGE2 released quantified by EIA. Results are expressed as mean±s.e.mean of four experiments performed in quadruplicate. (b) Effect of serine protease-enriched fraction. Cells were exposed to dilutions of serine protease-enriched fractions for 48 h. Supernatants were assayed for PGE2 as described in (a). Results are expressed as means±s.e.mean of four experiments performed in quadruplicate. *P<0.05 compared to medium control. **P<0.01 compared to medium control.
Effects of OSM and rye grass pollen extract on PGE2 release
Exposure of A549 cells to the rye grass pollen extract did not elicit measurable quantities of PGE2 either alone or in combination with OSM (Figure 4a). However, the extract induced the release of significant amounts of GM-CSF from these cells (P<0.05, Figure 4b). In the same experiment, OSM also induced the release of significant levels of GM-CSF (P<0.05, Figure 4b).
Figure 4.
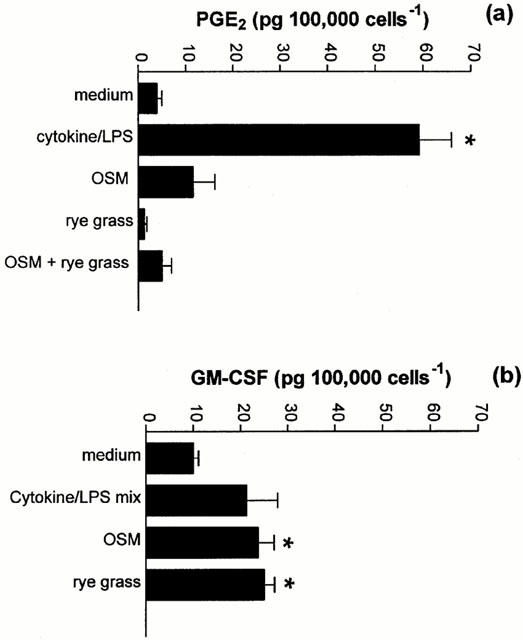
The effect of Rye grass pollen on PGE2 and GM-CSF release. Cells were exposed to extracts of rye grass pollen or OSM (20 ng ml−1) for 48 h. After this time, cell culture supernatants were taken and the amount of PGE2 released quantified by EIA (a), while the concentration of GM-CSF was quantified by specific ELISA (b). Results are expressed as means±s.e.mean of four experiments performed in quadruplicate. *P<0.05 compared to medium control.
Effect of cytokine/LPS mix, OSM, LIF and proteases on NO production
Exposure of A549 cells to the cytokine/LPS mixture significantly increased NO production over the 48 h period (P<0.05; Figure 5a). This enhancement was completely inhibited by pre-incubation of the cells with the NO synthase inhibitor L-NAME, suggesting that inducible NO synthase (iNOS) was being activated. As was observed for PGE2, both OSM and LIF did not stimulate significant NO release (Figure 5a). The combined addition of either OSM or LIF and cytokine/LPS did not induce a synergistic effect on NO release. In the absence of DTT, exposure of A549 cells to the mite cysteine-protease-enriched fraction did not induce the release of NO above control values. In contrast, following exposure to DTT, this fraction dose-dependently enhanced NO production (P<0.05). However, synergism with OSM was not observed (Figure 5b). Exposure to either DTT alone or a combination of DTT+OSM did not induce NO release above control levels. Exposure of A549 cells to the serine protease-enriched fraction (Figure 5c) or the rye grass pollen extract (Figure 5d) did not induce NO production, either alone or in combination with OSM.
Figure 5.
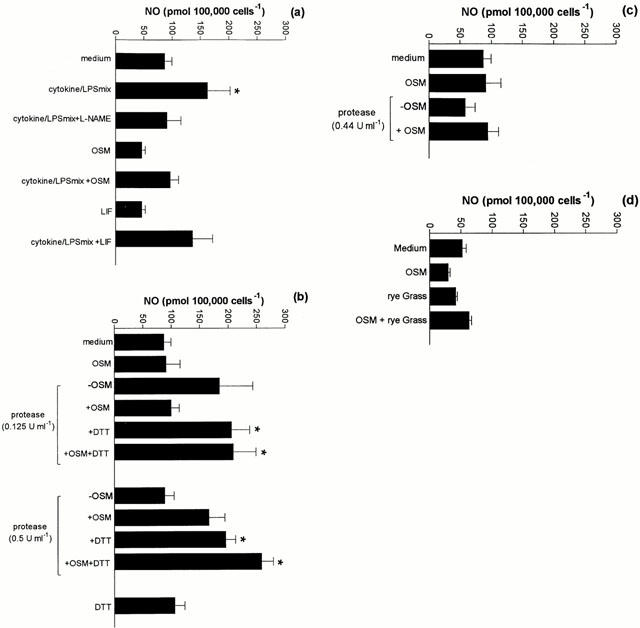
The effect of OSM, LIF, cytokine/LPS mix, protease-enriched fractions of HDM fecal pellets and protease-deficient rye grass pollen extracts on NO release from A549 cells. A549 cells were treated with either OSM (20 ng ml−1) or LIF (5 ng ml−1) and cytokine/LPS mix or a combination of both for 48 h. L-NAME was added 30 min prior to the addition of the cytokine/LPS cocktail. (b) The effect of cysteine protease-enriched fraction. A549 cells were exposed to cysteine protease-enriched fractions in the absence or presence of DTT and/or OSM. After 48 h supernatants were assayed for NO. (c) Effect of serine protease-enriched fraction. Cells were exposed to dilutions of serine protease-enriched fractions for 48 h. Supernatants were assayed for NO as described in (a). (d) Effect of Rye grass pollen extract. Cells were exposed to dilutions of protease-enriched fractions of rye grass pollen for 48 h. Supernatants were assayed for NO as described in (a). Results are expressed as mean±s.e.mean of four experiments performed in quadruplicate. *P<0.05 compared to medium control.
Discussion
This study has demonstrated that OSM synergistically enhanced the release of PGE2 in response to pro-inflammatory cytokines as well as the cysteine protease fraction-derived from the house dust mite, D. pteronyssinus. This enhancement appeared to involve an interaction between OSM and its specific type II receptor, rather than the type I (LIFR), despite the presence of both receptor subtypes on A549 cells. In contrast, LIF did not influence PGE2 production either alone or in combination with protease-enriched fractions of house dust mite fecal pellets. Neither OSM nor LIF influenced the release of NO from A549 cells per se. However, the cysteine protease-enriched fraction induced NO release following activation by DTT.
OSM is released by both resident structural cells as well as infiltrating inflammatory cells, suggesting that it may play an important role in modulating the inflammatory and repair processes. However, the biological effects of OSM in the context of lung inflammation are only just beginning to be explored. In other tissues, OSM appears to interact with different mediators to enhance cell activity. For example, OSM alone had a negligible effect on PGE2 release from synovial fibroblasts, but interacted additively with IL-1 to enhance the liberation of PGE2 (Hamilton et al., 1991). In these same cells, OSM and IL-1 were also shown to interact synergistically to increase the production of urokinase-type plasminogen activator. The results of the present study extend these observations in as much as OSM, while having no effect on PGE2 release per se, induced a synergistic increase in PGE2 production from epithelial cells when added in combination with a cytokine mixture containing IL-1β. The augmented PGE2 release was apparently due to induction of COX-2 activity, since pretreating cells with the COX-2 selective inhibitor nimesulide completely inhibited PGE2 release.
OSM has been shown to interact with both the LIFR (type I) and, more recently, a second specific OSMR (type II) (Heinrich et al., 1998; Mosley et al., 1996). Although both type I and II receptors are widely distributed on a variety of cultured cell types, assessing their distribution and role in human airways has only recently begun (Knight et al., 1999a). In the current study, we confirmed the presence of both type I and type II receptors on A549 cells. Pretreatment with a specific OSMR antibody completely negated the synergism between OSM and the cytokine/LPS mix, but neutralisation of the LIFR was without effect, confirming that in epithelial cells OSM was working specifically via the type II receptor.
The observation that mite proteases stimulated PGE2 release from respiratory epithelial cells is consistent with other data from our laboratory showing that they stimulate the release of a variety of cytokines such as IL-6, IL-8 and GM-CSF (King et al., 1998) from respiratory epithelial cells, as well as causing changes in mucosal permeability (Herbert et al., 1995) due to degradation of tight junction proteins (Wan et al., 1999). In the absence of exogenous reducing agents, the cysteine protease fraction still induced significant PGE2 release, suggesting that the epithelial cells may have the capacity to activate the latent cysteine protease activity, as has been described in canine kidney epithelial cells (Herbert et al., 1995). When OSM and cysteine protease-enriched fractions were added together in the presence of DTT, a synergistic increase in the generation of PGE2 was observed. This interaction appeared to be dependent on the biochemical activity of protease-enriched fractions since PGE2 liberation was not detected following heat inactivation. Exposure to the serine protease-enriched fraction also induced a dose-dependent production of PGE2 from A549 cells, although the magnitude was not as great as that for the cysteine protease-enriched fraction.
The mechanism(s) by which house dust mite-derived proteases augmented PGE2 production is unknown, but recent studies have demonstrated the presence of protease-activated receptors (PAR) in a variety of tissues, including the lung. To date four PARs have been described (Dery et al., 1998) and activation of PAR-2 was shown to induce the release of PGE2 from airway epithelium, which consequently inhibited contractile responses of the airway smooth muscle (Cocks et al., 1999; Lan et al., 2000). However, whether OSM influences the expression and/or function of PAR-2 is unknown. It is also possible that activation of PARs by mite proteases leads to the release of specific cytokines that in turn induce the release of PGE2. In this regard, we have shown that specific PAR agonist peptides induce the release of this prostanoid (Thompson et al., 1999). Interestingly, there was no such interaction between OSM and the serine protease-enriched fraction. The reasons for this are currently under investigation, but it is known that OSM is a potent inducer of the serine protease inhibitors α1-antichymotrypsin (Cichy et al., 1995) and α1-protease inhibitor (Cichy et al., 1998; Sallenave et al., 1997). In this regard, the latter is known to inhibit all of the known mite serine proteases (Stewart et al., 1994). Thus, OSM may play an important role in restricting damage to human airways due to certain classes of exogenous proteases from sources such as HDM. To this end, studies using individual mite proteases are currently underway.
In addition to examining the role of OSM in PGE2 production, we also examined its effect on NO release. This mediator is thought to be important in the process of airway inflammation, although its precise role is unknown (Watkins et al., 1997b). In this study, we confirmed earlier findings demonstrating that a mixture of IL-1β, IFNγ and LPS induced NO production (Watkins et al., 1997b) and that its release was inhibited by L-NAME suggesting the involvement of iNOS. In contrast, neither OSM nor LIF induced NO production from A549 cells. Whilst our data indicate that neither cytokine appears to play a role in NO production in respiratory epithelium, LIF has been shown to superinduce NO production in vascular tissue (Moran et al., 1997). The reasons for these differences are not known, although it may relate to a greater contribution of endothelial NOS in the vasculature as compared to iNOS in the airways. Interestingly, co-induction of NO with prostaglandin production has been demonstrated in several animal models of inflammation, both in vivo and in vitro (Swierkosz et al., 1995; Vane et al., 1994) and our laboratory has demonstrated that NO interacts with COX-2 in human cultured epithelial cells (Watkins et al., 1997a). However, the results from this study suggest that the two pathways may also be independently stimulated.
Exposure of A549 cells to cysteine protease-enriched fractions resulted in a concentration-dependent increase in NO production that was enhanced further by activation with DTT. However, there was no observable synergistic increase in NO production following the addition of OSM. The reasons for this are not understood, although it is possible that the cells were already releasing a maximum amount of NO in response to the allergen fraction. Exposure of cells to the serine protease-enriched fraction at the concentrations chosen did not evoke NO release and this was not influenced by the further addition of OSM.
In addition to examining dust mite allergenic protease fractions, we also studied a protease-free (as judged by the Azocoll assay) rye grass pollen extract and showed that pollen extracts had little effect on PGE2 and NO release, suggesting that protease activity per se is important in these activities. However, the rye grass pollen extract was biologically active since we were able to demonstrate that airway epithelial cells release GM-CSF following exposure to this pollen extract. In other studies (Tomee et al., 1998), the release of IL-6 and IL-8 from A549 cells in response to allergens has been documented. Taken together, these data indicate differential control of the release of cytokines and mediators such as PGE2 and NO. We also showed that OSM alone induced the release of GM-CSF from epithelial cells, suggesting an interaction between these cytokines. Indeed, GM-CSF has been shown to induce the release of OSM from neutrophils (Grenier et al., 1999), suggesting that a positive feedback reaction may occur within the context of allergic inflammation.
In conclusion, we have demonstrated that OSM acts as a co-factor in the induction of PGE2 release from cultured human airway epithelial cells in response to cytokine and protease stimuli. These results suggest OSM plays a critical role in regulating the enzyme/mediator profile of airway epithelial cells and indicates a potentially important role of OSM in the regulation of the airway response to allergic inflammation.
Acknowledgments
The authors would like to acknowledge the financial support of the National Health and Medical Research Council of Australia, the Raine Medical Foundation and the Asthma Foundation of Western Australia. The authors would also like to thank Immunex Corp (Seattle, Washington) for the generous gift of the human OSMR antibody.
Abbreviations
- EIA
enzyme immunoassay
- GM-CSF
granulocyte macrophage-colony stimulating factor
- HDM
house dust mite
- LIF
leukemia inhibitory factor
- LIFR
leukaemia inhibitory factor receptor
- L-NAME
L-nitro-arginine methyl ester
- OSM
Oncostatin M
- OSMR
Oncostatin M receptor
- PAR
protease activated receptor
References
- BERNARD C., MERVAL R., LEBRET M., DELERIVE P., DUSANTER-FOURT I., LEHOUX S., CREMINON C., STAELS B., MACLOUF J., TEDGUI A. Oncostatin M induces interleukin-6 and cyclooxygenase-2 expression in human vascular smooth muscle cells: synergy with interleukin-1beta. Circ. Res. 1999;85:1124–1131. doi: 10.1161/01.res.85.12.1124. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- CALDERON M.A., DEVALIA J.L., PRIOR A.J., SAPSFORD R.J., DAVIES R.J. A comparison of cytokine release from epithelial cells cultured from nasal biopsy specimens of atopic patients with and without rhinitis and nonatopic subjects without rhinitis. J. Allergy Clin. Immunol. 1997;99:65–76. doi: 10.1016/s0091-6749(97)70302-6. [DOI] [PubMed] [Google Scholar]
- CICHY J., POTEMPA J., CHAWLA R.K., TRAVIS J. Stimulatory effects of inflammatory cytokines on a1-antichymotrypsin expression in human lung derived epithelial cells. J. Clin. Invest. 1995;95:2729–2733. doi: 10.1172/JCI117975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CICHY J., ROSE-JOHN S., TRAVIS J. Oncostatin M, leukemia inhibitory factor and interleukin 6 trigger different effects on a1-proteinase inhibitor synthesis in human lung derived epithelial cells. Biochem. J. 1998;329:335–339. doi: 10.1042/bj3290335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COCKS T.M., FONG B., CHOW J.M., ANDERSON G.P., FRAUMAN A.G., GOLDIE R.G., HENRY P.J., CARR M.J., HAMILTON J.R., MOFFATT J.D. A protective role for protease-activated receptors in the airways. Nature. 1999;398:156–160. doi: 10.1038/18223. [DOI] [PubMed] [Google Scholar]
- DERY O., CORVERA C.U., STEINHOFF M., BUNNETT N.W. Proteinase-activated receptors:novel mechanisms of signaling by serine proteases. Am. J. Physiol. (Lung Cell Mol. Biol.) 1998;43:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- FOLKERTS G., NIJKAMP F.P. Airway epithelium: more than just a barrier. Trends Pharmacol. Sci. 1998;19:334–341. doi: 10.1016/s0165-6147(98)01232-2. [DOI] [PubMed] [Google Scholar]
- GAUVREAU G.M., WATSON R.M., O'BYRNE P.M. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am. J. Respir. Crit. Care. Med. 1999;159:31–36. doi: 10.1164/ajrccm.159.1.9804030. [DOI] [PubMed] [Google Scholar]
- GRENIER A., DEHOUX M., BOUTTEN A., ARCE-VICIOSO M., DURAND G., GOUGEROT-POCIDALO M.A., CHOLLET-MARTIN S. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood. 1999;93:1413–1423. [PubMed] [Google Scholar]
- HAMILTON J.A., LEIZER T., PICCOLI D.S., ROYSTON K.M., BUTLER D.M., CROATTO M. Oncostatin M stimulates urokinase-type plasminogen activator activity in human synovial fibroblasts. Biochem. Biophys. Res. Commun. 1991;180:652–659. doi: 10.1016/s0006-291x(05)81115-5. [DOI] [PubMed] [Google Scholar]
- HASSIM Z., MARONESE S.E., KUMAR R.K. Injury to murine airway epithelial cells by pollen enzymes. Thorax. 1998;53:368–371. doi: 10.1136/thx.53.5.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASTIE A.T., EVERTS K.B., CHO S.K., ZANGRILLI J., SHAVER J.R., POLLICE M.B., FISH J.E., PETERS S.P. IL-1 beta release from cultured bronchial epithelial cells and bronchoalveolar lavage cells from allergic and normal humans following segmental challenge with ragweed. Cytokine. 1996;8:730–738. doi: 10.1006/cyto.1996.0097. [DOI] [PubMed] [Google Scholar]
- HEINRICH P.C., BEHRMANN I., MULLER-NEWEN G., SCHAPER F., GRAEVE L. Interleukin 6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERBERT C.A., KING C.M., RING P.C., HOLGATE S.T., STEWART G.A., THOMPSON P.J., ROBINSON C. Augmentation of permeability in the bronchial epithelium by the house dust mite allergen Der p 1. Am. J. Respir. Cell Mol. Biol. 1995;12:369–378. doi: 10.1165/ajrcmb.12.4.7695916. [DOI] [PubMed] [Google Scholar]
- HILL D.J., THOMPSON P.J., STEWART G.A., CARLIN J.B., NOLAN T.M., KEMP A.S., HOSKING C.S. The Melbourne house dust mite study: eliminating house dust mites in the domestic environment. J. Allergy Clin. Immunol. 1997;99:323–329. doi: 10.1016/s0091-6749(97)70049-6. [DOI] [PubMed] [Google Scholar]
- ITO I., SUZUKI H., AIZAWA H., HIROSE T., HAKODA H. Pre-junctional inhibitory action of prostaglandin E2 on excitatory neuro-effector transmission in the human bronchus. Prostaglandins. 1990;39:639–655. doi: 10.1016/0090-6980(90)90024-p. [DOI] [PubMed] [Google Scholar]
- KING C., BRENNAN S., THOMPSON P.J., STEWART G.A. Dust mite proteolytic allergens induce cytokine release from cultured airway epithelium. J. Immunol. 1998;161:3645–3651. [PubMed] [Google Scholar]
- KNIGHT D.A., CARROLL N.G., LENZO J.C., MCKAY K.O., JAMES A.L., THOMPSON P.J. Immunohistochemical localisation of oncostatin M receptors in human airways: altered pattern of expression in asthma. Eur. Respir. J. 1999a;14:528s. [Google Scholar]
- KNIGHT D.A., LYDELL C.P., ZHOU D., WEIR T.D., SCHELLENBERG R.R., BAI T.R. Leukemia inhibitory factor (LIF) and LIF receptor in human lung. Distribution and regulation of LIF release. Am. J. Respir. Cell Mol. Biol. 1999b;20:834–841. doi: 10.1165/ajrcmb.20.4.3429. [DOI] [PubMed] [Google Scholar]
- KNIGHT D.A., STEWART G.A., THOMPSON P.J. The respiratory epithelium and airway smooth muscle homeostasis: its relevance to asthma. Clin. Exp. Allergy. 1994;24:698–706. doi: 10.1111/j.1365-2222.1994.tb00980.x. [DOI] [PubMed] [Google Scholar]
- KNIGHT D.A., STEWART G.A., THOMPSON P.J. PGE2 but not prostacyclin inhibits histamine-induced contraction of human bronchial smooth muscle. Eur. J. Pharmacol. 1995;272:13–19. doi: 10.1016/0014-2999(94)00602-4. [DOI] [PubMed] [Google Scholar]
- KNIGHT D.A., ZHOU D., BAI T.R. Leukemia Inhibitory Factor (LIF) induces substance P (SP) release and increases NK-1 receptor (NK-1R) gene expression in tracheal explants. Am. J. Respir. Crit. Care Med. 1998;157:A490. [Google Scholar]
- LAN R.S., STEWART G.A., HENRY P.J. Modulation of airway smooth muscle tone by protease activated receptor-1,-2,-3 and -4 in trachea isolated from influenza A virus-infected mice. Br. J. Pharmacol. 2000;129:63–70. doi: 10.1038/sj.bjp.0703007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCANULTY R.J., HERNANDEZ-RODRIGUEZ N.A., MUTSAERS S.E., COKER R.K., LAURENT G.J. Indomethacin suppresses the anti-proliferative effects of transforming growth factor-beta isoforms on fibroblast cell cultures. Biochem. J. 1997;321:639–643. doi: 10.1042/bj3210639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORAN C.S., CAMPBELL J.H., CAMPBELL G.R. Induction of smooth muscle cell nitric oxide synthase by human leukaemia inhibitory factor: effects in vitro and in vivo. J. Vasc. Res. 1997;34:378–385. doi: 10.1159/000159246. [DOI] [PubMed] [Google Scholar]
- MOSLEY B., IMUS C.D., FRIEND D., BOIANI N., THOMA B., PARK L.S., COSMAN D. Dual Oncostatin M receptors. J. Biol. Chem. 1996;70:32636–32643. doi: 10.1074/jbc.271.51.32635. [DOI] [PubMed] [Google Scholar]
- PAVORD I.D., TATTERSFIELD A.E. Bronchoprotective role for endogenous prostaglandin E2. Lancet. 1995;345:436–438. doi: 10.1016/s0140-6736(95)90409-3. [DOI] [PubMed] [Google Scholar]
- PETERS S.P., SCHULMAN E.S., SCHLEIMER R.P., MACGLASHAN D.W., JR, NEWBALL H.H., LICHTENSTEIN L.M. Dispersed human lung mast cells. Pharmacologic aspects and comparison with human lung tissue fragments. Am. Rev. Respir. Dis. 1982;126:1034–1039. doi: 10.1164/arrd.1982.126.6.1034. [DOI] [PubMed] [Google Scholar]
- ROSE T.M., BRUCE A.G. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc. Natl. Acad. Sci. 1991;88:8641–8645. doi: 10.1073/pnas.88.19.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALLENAVE J.-M., TREMBLAY G.M., GAULDIE J., RICHARDS C.D. Oncostatin M, but not Interleukin 6 or Leukemia Inhibitory Factor, stimulates expression of Alpha1-proteinase inhibitor in A549 alveolar epithelial cells. J. Interfer. Cytokine Res. 1997;17:337–346. doi: 10.1089/jir.1997.17.337. [DOI] [PubMed] [Google Scholar]
- STEWART G.A., KOLLINGER M.R., KING C.M., THOMPSON P.J. A comparative study of three serine proteases from Dermatophagoides pteronyssinus and D. farinae. Allergy. 1994;49:553–560. doi: 10.1111/j.1398-9995.1994.tb01128.x. [DOI] [PubMed] [Google Scholar]
- STEWART G.A., THOMPSON P.J., SIMPSON R.J.Protease antigens from house dust mite Lancet 19892154–155.(correction, 462) [DOI] [PubMed] [Google Scholar]
- SWIERKOSZ T.A., MITCHELL J.A., WARNER T.D., BOTTING R.M., VANE J.R. Co-induction of nitric oxide synthase and cyclo-oxygenase: interactions between nitric oxide and prostanoids. Br. J. Pharmacol. 1995;114:1335–1342. doi: 10.1111/j.1476-5381.1995.tb13353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAGA T. gp130 and the interleukin-6 family of cytokines. Ann. Rev. Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- THOMPSON P.J., KNIGHT D.A., NITHIANANTHAN A., STEWART G.A. The presence of Proteinase Activated Receptors (PAR) in cultured human airway epithelial cells: Differential effects on PGE2 release. Am. J. Respir. Crit. Care Med. 1999;159:A98. [Google Scholar]
- TOMEE J.F., VAN WEISSENBRUCH R., DE MONCHY J.G., KAUFFMAN H.F. Interactions between inhalant allergen extracts and airway epithelial cells: effect on cytokine production and cell detachment. J. Allergy Clin. Immunol. 1998;102:75–85. doi: 10.1016/s0091-6749(98)70057-0. [DOI] [PubMed] [Google Scholar]
- TOMEE J.F.C., WIERENGA A.T.J., HIEMSTRA P.S., KAUFFMAN H.F. Proteases from Aspergillus fumigatus induce release of proinflammatory cytokines and cell detachment in airway epithelial cell lines. J. Infectious Dis. 1997;176:300–303. doi: 10.1086/517272. [DOI] [PubMed] [Google Scholar]
- VANE J.R., MITCHELL J.A., APPLETON I., TOMLINSON A., BISHOP-BAILEY D., CROXTALL J., WILLOUGHBY D.A. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc. Natl. Acad. Sci. 1994;91:2046–2050. doi: 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAN H., WINTON H.L., SOELLER C., TOVEY E.R., GRUENERT D.C., THOMPSON P.J., STEWART G.A., TAYLOR G.W., GARROD D.R., CANNELL M.B., ROBINSON C. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J. Clin. Invest. 1999;104:123–133. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATKINS D.N., GARLEPP M.J., THOMPSON P.J. Regulation of the inducible cyclo-oxygenase pathway in human cultured epithelial (A549) cells by nitric oxide. Br. J. Pharmacol. 1997a;121:1482–1488. doi: 10.1038/sj.bjp.0701283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATKINS D.N., PERONI D.J., BASCLAIN K.E., GARLEPP M.J., THOMPSON P.J. Expression and activity of nitric oxide synthases in human airway epithelium. Am. J. Respir. Cell Mol. Biol. 1997b;16:629–639. doi: 10.1165/ajrcmb.16.6.9191464. [DOI] [PubMed] [Google Scholar]
- WINTON H.L., WAN H., CANNELL M.B., THOMPSON P.J., GARROD D.R., STEWART G.A., ROBINSON C. Class specific inhibition of house dust mite proteinases which cleave cell adhesion, induce cell death and which increase the permeability of lung epithelium. Br. J. Pharmacol. 1998;124:1048–1059. doi: 10.1038/sj.bjp.0701905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU C.K., YANG B.C., LEE S.C., WANG J.Y., HSIUE T.R., LEI H.Y. Dermatophagoides-farinae-induced pulmonary eosinophilic inflammation in mice. Int. Arch. Allergy Immunol. 1997;112:73–82. doi: 10.1159/000237434. [DOI] [PubMed] [Google Scholar]
- ZARLING J.M., SHOYAB M., MARQUARDT H., HANSON B., LIOUBIN N., TODARO G.J. Oncostatin M: A growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. 1986;83:9739–9743. doi: 10.1073/pnas.83.24.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]