

Abstract
The intermolecular cross-regulation mediated by the prostanoid IP-receptor (IP)/EP1 receptor (EP1) agonists PGI2 and 17 phenyl trinor PGE2 on TP receptor (TP) signalling within platelets was compared to that which occurs to the individual TPα and TPβ receptors over-expressed in human embryonic kidney (HEK) 293 cells. Ligand mediated TP receptor activation was monitored by analysing mobilization of intracellular calcium ([Ca2+]i) following stimulation with the selective thromboxane (TX) A2 mimetic U46619.
Consistent with previous studies, in platelets, PGI2 acting through endogenous IP receptors completely inhibited U46619-mediated TP receptor signalling in a protein kinase (PK) A-dependent, PKC-independent manner.
In HEK 293 cells, PGI2, acting through endogenous AH6809 sensitive EP1 rather than IP receptors, and the selective EP1 receptor agonist 17 phenyl trinor PGE2 antagonized U46619-mediated signalling by both TPα and TPβ receptors in a PKC-dependent, PKA-independent manner.
The maximum response induced by either ligand was significantly (P<0.005) greater for the TPα receptor than the TPβ receptor, pointing to possible physiologic differences between the TP isoforms, although the potency of each ligand was similar for both TP receptors.
TPΔ328, a truncated variant of TP receptor lacking the C-tail sequences unique to TPα or TPβ receptors, was not sensitive to EP1 receptor-mediated regulation by PGI2 or 17 phenyl trinor PGE2
In conclusion, these data confirm that TPα and TPβ receptors are subject to cross regulation by EP1 receptor signalling in HEK 293 cells mediated by PKC at sites unique to the individual TP receptors and that TPα receptor responses are significantly more reduced by EP1 receptor regulation than those of the TPβ receptor.
Keywords: Thromboxane A2, prostaglandin I2, prostaglandin E2, EP1 receptor, G protein-coupled receptor, calcium, protein kinase C, signalling, TP isoforms
Introduction
Prostaglandins (PGs) and thromboxanes (TX) regulate an array of functions under normal and pathophysiological conditions (Narumiya et al., 1999). The five primary prostanoids, PGD2, PGE2, PGF2α, PGI2 and TXA2 mediate their actions through specific G protein-coupled receptors (GPCRs) termed prostanoid-DP, EP, FP, IP and TP receptors, respectively, to signal activation or inhibition of adenylyl cyclase or activation of phospholipase (PL) C and elevation of intracellular calcium ([Ca2+]i). EP1 - EP4 receptors mediate the diverse actions of PGE2 and may couple to elevation of [Ca2+]i (EP1), activation (EP2 and EP4) or inhibition of adenylyl cyclase (EP3) (Funk et al., 1993; An et al., 1994; Foord et al., 1996).
Further diversity of prostanoid receptors is achieved by alternative mRNA splicing yielding, for example, receptors which differ in their carboxyl terminal tails (C-tails), such as the human TP receptor isoforms TPα and TPβ (Hirata et al., 1991; Raychowdhury et al., 1994) and the EP3 receptor isoforms (Namba et al., 1993; Negishi et al., 1993). TPα and TPβ receptors diverge subsequent to amino acid 328 and hence, differ exclusively in their C-tail sequences. They exhibit identical ligand binding and couple similarly to Gq/G11 (Kinsella et al., 1997; Walsh et al., 1998), G16 and G12 (Walsh et al., 2000a), but oppositely regulate adenylyl cyclase (Hirata et al., 1996).
Overlapping ligand specificities and the co-existence of more than one prostanoid receptor type, subtype or isoform within a given cell adds to the complexity of the downstream signalling resulting from receptor activation (Fennekohl et al., 1999; van der Vuurst et al., 1997; Kiriyama et al., 1997). Also, cross-regulation at the intracellular level between the prostanoid receptors and their respective effector systems has been observed. However, such cross-regulation is not readily predictable. For example, platelet aggregation mediated by TP, but not the ADP or thrombin receptors, is particularly sensitive to PGI2 (prostacyclin) inhibition in a cyclic AMP-dependent (PK) A-dependent manner (Manganello et al., 1999). Whereas some of the platelet targets have been identified (Murray et al., 1990; Manganello et al., 1999), such as PLC, myosin light chain kinase, thrombolamban and Gα13, the precise mechanism has not been fully elucidated. In human platelets, iloprost stimulation of cyclic AMP is enhanced by prior exposure to the TP receptor agonist U46619, but not by to platelet-activating factor or thrombin, and is independent of protein kinase (PK) C (Murray et al., 1990). In human MEG-01 cells, both thrombin and TP receptor agonists STA2 and U46619 augment cyclic AMP generation by both iloprost and forskolin and this augmentation is mediated by PKC (Watanabe et al., 1996). Despite these studies, the extent of cross-talk between other prostanoid receptor types has not been fully explored. Thus, in this study, we have examined the intermolecular cross-talk mediated by the IP and EP/EP1 receptor agonists PGI2, PGE2 and 17 phenyl trinor PGE2 on TP signalling within platelets and compared it to that which occurs to the individual TPα and TPβ receptors over expressed in human embryonic kidney (HEK) 293 cells. Our results demonstrate that TPα and TPβ receptors are subject to differential desensitization or functional antagonism in response to EP1 receptor activation and that these effects are mediated at PKC phosphorylation sites unique to the individual TP receptors.
Methods
Materials
U46619, SQ29,548, 17 phenyl trinor PGE2, PGE2 and PGI2 were obtained from Cayman Chemical Company. FURA2/AM was from Calbiochem. [3H]-cyclic AMP (15–30 Ci mmol−1) was from American Radiolabeled Chemicals Inc. AH6809 was from Tocris, U.K. Cicaprost was from Schering AG, Berlin.
Cell culture and transfections
HEK 293 cells were obtained from the American Type Culture Collection. HEK.α10, HEK.β3 and HEK.TPΔ328 stable cell lines, over-expressing TPα, TPβ and TPΔ328 receptors, respectively, have been previously described (Walsh et al., 1998). HEK 293 cells were transiently transfected with pCMV:Gα11 or pCMV5 using the calcium phosphate/DNA co-precipitation (Kinsella et al., 1997) and were harvested 48 h post-transfection.
Calcium measurements
Measurements of intracellular calcium ([Ca2+]i) in FURA2 preloaded cells and platelets were carried out as previously described (Kinsella et al., 1997). Cells were stimulated with 1 μM ligand (U46619, cicaprost, PGE2, PGI2 or 17-phenyl trinor PGE2) unless otherwise stated or, for dose response studies, with 10−12–10−5 M agent. The results, representative data from at least four independent experiments, are plotted as changes in intracellular Ca2+ mobilized (Δ[Ca2+]i (nM)) as a function of time (s) upon ligand stimulation. Alternatively changes in ligand-mediated intracellular Ca2+ mobilized (Δ[Ca2+]i±s.e.mean, nM; n=4) were calculated; those levels of [Ca2+]i mobilized following stimulation with U46619 only were set to represent 100% and thereafter, the level of U46619-mediated [Ca2+]i mobilized subsequent to prior stimulation with PGI2, PGE2 or 17 phenyl trinor PGE2 were calculated as a percentage of that value.
Measurement of cyclic AMP
Ligand mediated cyclic AMP measurements were carried out essentially as previously described (Hayes et al., 1999). HEK 293 cells were washed three times in ice-cold phosphate-buffered saline and approximately 1–2×106 cells were resuspended in 200 μl HEPES-buffered saline (HBS) (in mM; NaCl 140, KCl 4.7, CaCl2 2.2, KH2PO4 1.2, glucose 11, HEPES-NaOH 15, pH 7.4) containing 1 mM 3-isobutyl-1-methylxanthine and preincubated at 37°C for 10 min. Thereafter, cells were stimulated in the presence of 1 μM PGI2 (from a 5 μM PGI2 stock, 50 μl) or 1 μM PGE2 (from a 5 μM PGE2 stock, 50 μl) or in the presence of HBS (50 μl) at 37°C for 10 min. Reactions were terminated by heat inactivation (100°C, 5 min) and the level of cyclic AMP produced was quantified by radioimmunoassay using the cyclic AMP binding protein from bovine adrenal medulla essentially as described by Hayes et al. (1999). Levels of cyclic AMP produced by ligand-stimulated cells over basal stimulation, in the presence of HBS, were expressed in pmol cyclic AMP/mg cell protein±standard error of the mean (pmol mg−1±s.e.mean) and as fold stimulation over basal (fold increase±s.e.mean; n=3).
Data analyses
Statistical analyses were carried out using the unpaired Student's t-test using the Statworks Analysis Package. P-Values of less than or equal to 0.05 were considered to indicate a statistically significant difference.
Results
Effects of PGI2 on U46619-mediated [Ca2+]i mobilization in human platelets and in HEK 293 cells
The effect of PGI2 on TP receptor mediated signalling was investigated within the platelet and was compared to that which occurs within HEK 293 cells over expressing the individual α or β isoforms of the TP receptor. Stimulation of platelets with U46619 (1 μM) induced a significant transient rise in [Ca2+]i levels (Figure 1A; Δ[Ca2+]i=153±26.9 nM). While PGI2 at 1 μM (Figure 1B) or 10 μM (data not shown) did not induce an increase in [Ca2+]i mobilization, it completely blocked [Ca2+]i mobilization in response to subsequent stimulation with U46619 (Figure 1B).
Figure 1.
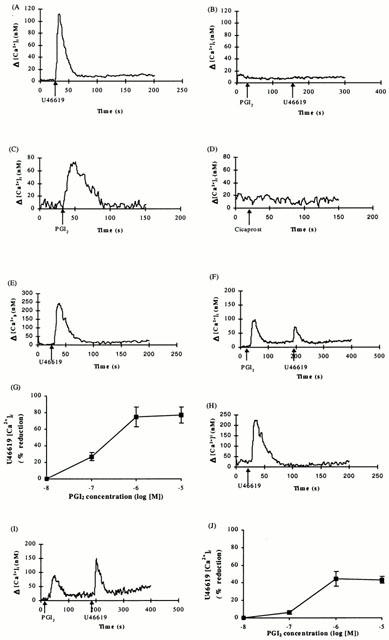
Analysis of ligand-induced [Ca2+]i mobilization in platelets and HEK 293 cells. Platelets (A,B), HEK 293 cells (C,D) or HEK.α10 cells (E–G) and HEK.β3 cells (H–J), transiently co-transfected with pCMV:Gα11, were stimulated with either U46619 (1 μM), PGI2 (1 μM), Cicaprost (1 μM), PGI2 (1 μM) or PGI2 (10−8 to 10−5 M) followed by U46619 (1 μM). In [Ca2+]i traces (A–F, H–I), ligands were added at the times indicated by the arrows. Dose response curves (G,J) indicate percentage (%) reduction in U46619 (1 μM) response induced by the indicated PGI2 concentration. Data presented are representative of four independent experiments.
In contrast to that observed in platelets, both HEK.α10 ([ΔCa2+]i=86.0±21.8 nM) and HEK.β3 ([ΔCa2+]i=75.0± 7.75 nM), or the control non-transfected HEK 293 cells (Figure 1C; [ΔCa2+]i=79.1±16.4 nM) exhibited mobilization of [Ca2+]i in response to PGI2 (1 μM) but not in response to the IP agonist cicaprost (1 μM; Figure 1D). Both TP receptors stably expressed in HEK 293 cells exhibited efficient mobilization of [Ca2+]i in response to stimulation with the TXA2 mimetic U46619 with EC50 values of 20±0.7 and 65±12 nM, respectively. At 1 μM, U46619 resulted in maximal [Ca2+]i mobilization (Figure 1E,H; Δ[Ca2+]i=248±17.5 nM for TPα receptor; Δ[Ca2+]i=184±23.2 nM for TPβ receptor). The control, non-transfected HEK 293 cells did not exhibit a measurable rise in [Ca2+]i (data not shown). Prior stimulation with PGI2 reduced subsequent U46619 mediated mobilization of [Ca2+]i by HEK.α10 cells, with an IC50 value of 0.3±0.05 μM; 1 μM PGI2 resulted in maximal reduction to 25.2±4.1% (P=0.0002; Δ[Ca2+]i=63.7±14.0 nM) of that originally observed in the absence of PGI2 (Figure 1F,G). In HEK.β3 cells, U46619-mediated changes in intracellular [Ca2+]i were also reduced by prior stimulation with PGI2. The IC50 was 0.4±0.08 μM whilst the maximal effect was again observed with 1 μM PGI2, with signalling reduced to 55.6±10.7% (P=0.04; Δ[Ca2+]i=105±23.4 nM) of that originally observed in the absence of PGI2 (Figure 1I,J). The TPα receptor U46619 response was significantly more reduced in the presence of PGI2 than that of the TPβ receptor (P<0.005).
Effects of AH6809 on PGI2-induced inhibition of U46619-mediated [Ca2+]i mobilization by TPα and TPβ expressed in HEK 293 cells
Pre-treatment of HEK.α10 cells with the EP1 receptor antagonist AH6809 (1 μM) prior to the addition of PGI2 (1 μM) restored U46619-mediated [Ca2+]i mobilization to 74.5±6.4% whereas that of HEK.β3 cells was restored to 104.9±25.2%. AH6809 had no significant effect (P=0.97) on U46619-mediated [Ca2+]i mobilization in the absence of PGI2 pre-stimulation by either cell type. Neither HEK.α10 (1.5±0.2 fold increase in cyclic AMP), nor HEK.β3 (1.0±0.2 fold increase in cyclic AMP) cells led to significant increases in cyclic AMP (P=0.13) in response to PGI2 (1 μM). The presence of mRNA encoding EP1 receptor in HEK 293 cells was confirmed by selective RT-PCR (data not shown).
Effects of PGE2 and 17 phenyl trinor PGE2 on U46619-mediated [Ca2+]i mobilization by TPα and TPβ expressed in HEK 293 cells
The presence of EP type receptors coupled to both mobilization of [Ca2+]i and cyclic AMP generation in HEK 293 cells was further investigated using PGE2 as stimulating ligand. Stimulation of HEK 293 cells with 1 μM PGE2 generated a 17.2±1.4 fold increase in cyclic AMP. Treatment of HEK 293 cells with PGE2 (1 μM) lead to mobilization of [Ca2+]i (Δ[Ca2+]i=67.1±20.6 nM). Pre-stimulation of HEK.α10 cells with PGE2 (1 μM) reduced subsequent U46619-mediated mobilization of [Ca2+]i with an IC50 of 50±14 nM; 1 μM PGE2 resulted in maximal reduction to 24.9±2.8% (P=0.00015) of that originally observed in the absence of PGE2 (Figure 2A,C,E) whereas U46619-mediated changes in [Ca2+]i in HEK.β3 cells (Figure 2B,D,E) were maximally reduced only to 54.7±8.7% (P=0.028) by 1 μM PGE2 pre-stimulation (IC50 was 2±0.4×10−7 M). The TPα receptor U46619 response was significantly more reduced in the presence of PGE2 than was the TPβ receptor response (P<0.005).
Figure 2.
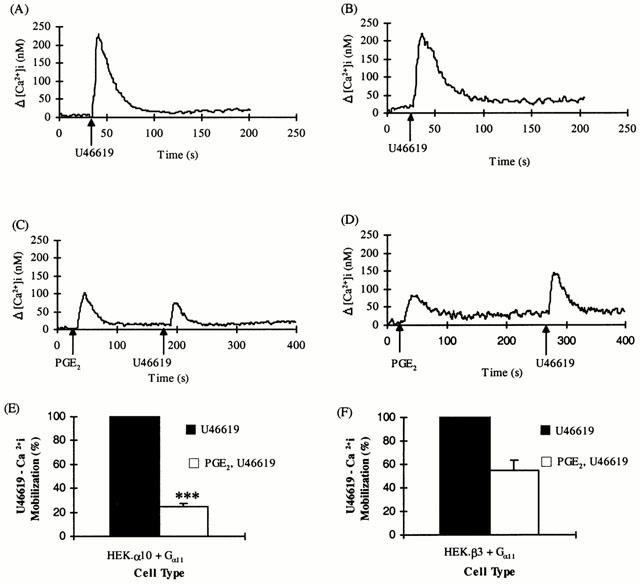
Effect of PGE2 on U46619-mediated [Ca2+]i mobilization by TPα and TPβ. HEK.α10 cells (A,C,E) and HEK.β3 cells (B,D,F), transiently co-transfected with pCMV:Gα11, were stimulated with either 1 μM U46619 (A,B) or 1 μM PGE2 followed by 1 μM U46619 (C–F), as indicated in the panels. (A–D) Data presented are representative of 5–6 independent experiments. (E,F) Levels of [Ca2+]i mobilized following stimulation with U46619 only were set to represent 100% and the level of U46619-mediated [Ca2+]i mobilized subsequent to prior stimulation with PGE2 were calculated as a percentage of that value (%±s.e.mean). ***Indicates HEK.α10 cells were significantly (P<0.005) more sensitive to PGE2 than HEK.β3 cells.
Stimulation of platelets with the selective EP1 agonist 17 phenyl trinor PGE2 (1 μM) did not generate a significant increase in intracellular Ca2+ (Figure 3A). However, 17 phenyl trinor PGE2 induced significant mobilization of [Ca2+]i in both HEK.α10 cells (Figure 3B; Δ[Ca2+]i=50.9±4.91 nM, n=5), HEK.β3 cells (Figure 3C; Δ[Ca2+]i=45.8±7.09 nM, n=5) and in the control non-transfected HEK 293 cell line (data not shown).
Figure 3.
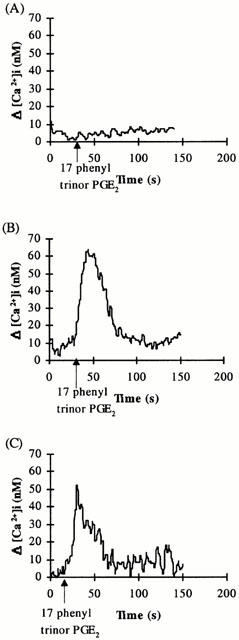
Mobilization of [Ca2+]i mediated by 17 phenyl trinor PGE2. Platelets (A) or HEK.α10 cells (B) and HEK.β3 cells (C), transiently co-transfected with pCMV:Gα11, were stimulated with 17 phenyl trinor PGE2 (1 μM), added at the times indicated by the arrows in the panels. Data presented are representative of at least four independent experiments.
Whereas pre-treatment of platelets with 17 phenyl trinor PGE2 (1 μM) did not affect subsequent mobilization of [Ca2+]i in response to U46619 (Figure 4A,B), it significantly reduced U46619-mediated TP signalling in HE 293 cells (Figure 4C–F), with an IC50 of approximately 0.4 μM for both cell types. One μM 17 phenyl trinor PGE2 was necessary to produce the maximal effect observed for this ligand; in HEK.α10 cells (Figure 4C,D), the initial U46619-induced [Ca2+]i mobilization was reduced to 50.5±6.7% (P=0.0025; Δ[Ca2+]i=89.5± 14.5 nM) while in HEK.β3 cells (Figure 4E,F), U46619-induced mobilization of [Ca2+]i was decreased to 78.5±4.9% (P=0.01; Δ[Ca2+]i=98.8±3.8 nM) of the original response following pre-stimulation with 17 phenyl trinor PGE2. The TPα receptor U46619 response was significantly more reduced (P<0.005) in the presence of the EP1 agonist 17 phenyl trinor PGE2 than was the TPβ receptor response.
Figure 4.
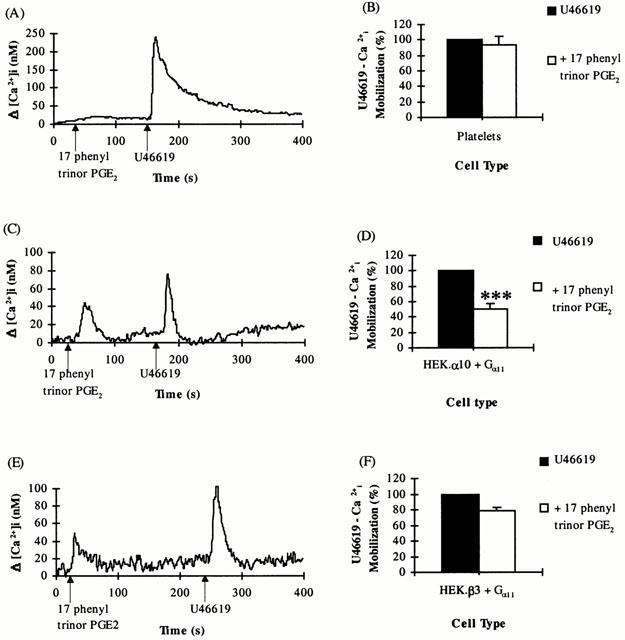
Effect of 17 phenyl trinor PGE2 on U46619-mediated [Ca2+]i mobilization. Platelets (A,B) or HEK.α10 cells (C,D) and HEK.β3 cells (E,F), transiently co-transfected with pCMV:Gα11, were stimulated with either 1 μM U46619 (B,D,F) or 1 μM 17 phenyl trinor PGE2 followed by 1 μM U46619 (A–F), as indicated in the panels. (A,C,E) Data presented are representative of at least four independent experiments. (B,D,F) Levels of [Ca2+]i mobilized following stimulation with U46619 only were set to represent 100% and the level of U46619-mediated [Ca2+]i mobilized subsequent to prior stimulation with 17 phenyl trinor PGE2 were calculated as a percentage of that value (%±s.e.mean). ***Indicates HEK.α10 cells were significantly (P<0.005) more sensitive to 17 phenyl trinor PGE2 than HEK.β3 cells.
Effects of H-89 and GF 109203X on PGI2 and 17 phenyl trinor PGE2-induced inhibition of U46619-mediated [Ca2+]i mobilization by TPα and TPβ expressed in HEK 293 cells
To investigate whether the second messenger protein kinases (PKs) may be involved in the PGI2 or 17 phenyl trinor PGE2 mediated functional antagonism of TP receptor signalling, the effects of H-89, a PKA inhibitor, and GF 109203X, a PKC inhibitor, on U46619-induced [Ca2+]i mobilization was investigated in HEK 293 cells and compared to that which occurs in platelets.
Pre-treatment of platelets with H-89 (10 μM, 1 min at 37°C) prior to subsequent stimulation with PGI2 (1 μM) followed by U46619 (1 μM) completely restored U46619-mediated [Ca2+]i responses to levels observed in the absence of pre-stimulation with PGI2 (Figure 5A). In contrast, pre-incubation of platelets with GF 109203X (50 nM, 2 min at 37°C) produced no significant effect on subsequent PGI2 mediated reduction of [Ca2+]i mobilization in response to U46619 (Figure 5A).
Figure 5.
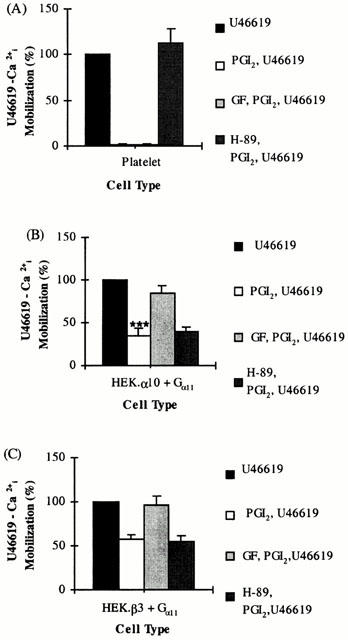
Effect of Kinase inhibitors on PGI2 mediated desensitization of TP signalling. Platelets (A) or HEK.α10 cells (B) and HEK.β3 cells (C), transiently co-transfected with pCMV:Gα11, were stimulated with either 1 μM U46619 (U46619) or 1 μM PGI2 followed by 1 μM U46619 (PGI2, U46619). Alternatively, cells were pre-incubated with 50 nM GF 109203X (GF, PGI2, U46619) or 10 μM H-89 (H-89, PGI2, U46619) and then stimulated with 1 μM PGI2 followed by 1 μM U46619. Levels of [Ca2+]i mobilized following stimulation with U46619 only were set to represent 100% and the level of U46619-mediated [Ca2+]i mobilized subsequent to prior stimulation with PGI2, in the absence or presence of GF 109203X or H-89 were calculated as a percentage of that value (%±s.e.mean, n=4). ***Indicates HEK.α10 cells were significantly (P<0.005) more sensitive to PGI2 than HEK.β3 cells.
Whereas H-89 had no significant effect on inhibition of PGI2-mediated desensitization of U46619 responses in either HEK.α10 (P=0.553; Figure 5B) or HEK.β3 (P=0.776; Figure 5C) cells, GF 109203X significantly impaired PGI2-mediated functional antagonism of U46619 responses in both cell types (Figure 5B,C). In HEK.α10 cells, the levels of [Ca2+]i mobilization in the presence of GF 109203X were restored to 84.7±9.1% (P=0.02) of original U46619 responses in the absence of PGI2 treatment (Figure 5B) whilst in HEKβ3 cells, intracellular Ca2+ mobilization was restored to 95.7±10.9% (P=0.03) of original U46619 responses (Figure 5C).
Similarly, whereas H-89 did not prevent 17 phenyl trinor PGE2 mediated functional antagonism of TP receptor signalling in HEK.α10 (P=0.852) or HEK.β3 cells (P=0.168; Figure 6); GF 109203X significantly increased the U46619 responses observed subsequent to 17 phenyl trinor PGE2 pre-treatment in both cell types (Figure 6). In HEK.α10 cells, [Ca2+]i mobilization was restored to 72.4±8.2% (P=0.003) of original U46619 responses in the absence of 17 phenyl trinor PGE2 treatment (Figure 6A) whilst in HEK.β3 cells, [Ca2+]i mobilization was restored to 104±4.58% (P=0.03) of the original U46619 responses (Figure 6B).
Figure 6.
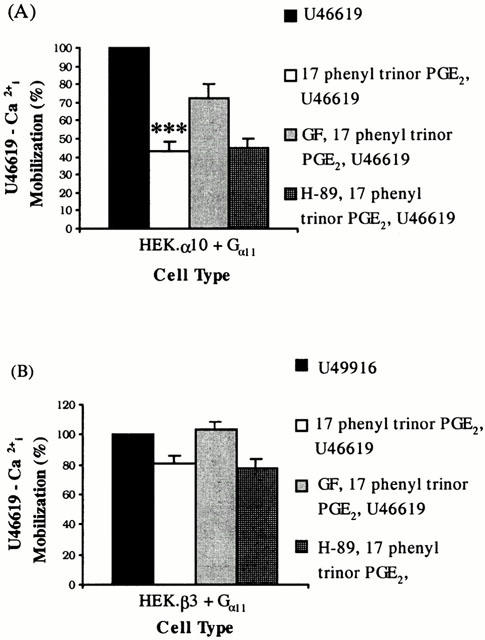
Effect of Kinase inhibitors on 17 phenyl trinor PGE2-mediated desensitization of TP signalling. HEK.α10 cells (A) or HEK.β3 cells (B), transiently co-transfected with pCMV:Gα11, were stimulated with either 1 μM U46619 (U46619) or 1 μM 17 phenyl trinor PGE2 followed by 1 μM U46619 (17 phenyl trinor PGE2, U46619). Alternatively, cells were pre-incubated with 50 nM GF 109203X (GF, 17 phenyl trinor PGE2, U46619) or 10 μM H-89 (H-89, 17 phenyl trinor PGE2, U46619) and then stimulated with 1 μM 17 phenyl trinor PGE2 followed by 1 μM U46619. Levels of [Ca2+]i mobilized following stimulation with U46619 only were set to represent 100% and the level of U46619-mediated [Ca2+]i mobilized subsequent to prior stimulation with 17 phenyl trinor PGE2, in the absence or presence of GF 109203X or H-89 were calculated as a percentage of that value (%±s.e.mean), n=4). ***Indicates HEK.α10 cells were significantly (P<0.005) more sensitive to 17 phenyl trinor PGE2 than HEK.β3 cells.
Effect of 17 phenyl trinor PGE2 on U46619-mediated [Ca2+]i mobilization by TPΔ328 expressed in HEK 293 cells
Stimulation of HEK.TPΔ328, which lack the C-tail sequences distal to the point of divergence of TPα and TPβ receptor, with U46619 (1 μM) resulted in a significant transient rise in [Ca2+]i mobilization (Figure 7A; Δ[Ca2+]i=56.0±2.1 nM, n=3). Whereas 17 phenyl trinor PGE2 mediated [Ca2+]i mobilization in HEK.TPΔ328 receptor (Figure 7B; Δ[Ca2+]i=40.1±2.9 nM, n=3), pre-stimulation of those cells with 17 phenyl trinor PGE2 did not significantly reduce (P=0.61) subsequent U46619-induced [Ca2+]i mobilization (Figure 7B; Δ[Ca2+]i=57.1±3.82 nM, n=5). Similarly, pre-stimulation of HEK.TPΔ328 cells with PGI2 did not significantly reduce (P=0.54) subsequent U46619-induced [Ca2+]i mobilization (data not shown).
Figure 7.
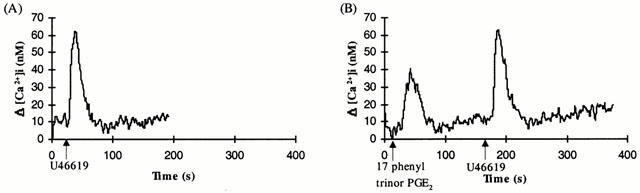
Effect of 17 phenyl trinor PGE2 on U46619-mediated [Ca2+]i mobilization by TPΔ328. HEK.TPΔ328 cells, transiently co-transfected with pCMV:Gα11, were stimulated with either 1 μM U46619 (A) or with 1 μM 17 phenyl trinor PGE2 followed by 1 μM U46619 (B) as indicated in the panels. The ligands were added at the times indicated by the arrows. Data presented are representative of four independent experiments.
Discussion
In this study we examined the cross-talk mediated by PGI2 (IP receptor agonist), PGE2 (EP receptor agonist) and 17 phenyl trinor PGE2 (EP1 receptor agonist) on U46619-mediated TP receptor signalling in HEK 293 cells stably over-expressing TPα or TPβ receptors and compared it to that which occurs within platelets. Consistent with previous studies, PGI2 failed to mobilize [Ca2+]i in platelets but abolished TP receptor mediated mobilization of [Ca2+]i in a PKA-dependent manner. In contrast, stimulation of HEK 293 cells with PGI2 demonstrated mobilization of [Ca2+]i through activation of endogenous AH6809 sensitive EP1 rather than IP receptors but failed to stimulate significant increases in cyclic AMP, despite the presence of low levels of endogenous IPs in HEK 293 cells (Hayes et al., 1999). Thus, PGI2 activates alternative receptors in human platelets and in kidney fibroblast HEK 293 cells.
Activation of EP1 receptors with the selective EP1 agonist 17 phenyl trinor PGE2 functionally antagonized U46619-activation of both TPα and TPβ receptors expressed in HEK 293 cells. Whereas 17 phenyl trinor PGE2 is a selective ligand that discriminates EP1 from related receptors, such as EP2, EP3, EP4 and IP receptors, it was less potent than PGI2 at mediating [Ca2+]i mobilization in HEK 293 cells, possibly accounting for why 17 phenyl trinor PGE2 promotes less functional antagonism of TP receptors than PGI2. Stimulation with PGE2 also confirmed the presence of EP type receptors which couple to [Ca2+]i mobilization in HEK 293 cells. Whereas both TP isoforms were regulated by PGE2, in a partially PKC-dependent manner (data not shown), TPα receptor signalling was significantly more reduced than was TPβ receptor. However, as HEK 293 cells produce a 17-fold increase in cyclic AMP in response to PGE2, pending the availability of selective ligands, further investigations are required to establish which EP subtype (EP1, EP2, EP4) or isoform (EP3 isoforms) is actually mediating TP receptor regulation in response to PGE2.
From dose response studies, there were no significant differences in the potency of PGI2, PGE2 or 17 phenyl trinor PGE2 in mediating functional antagonism of TPα or TPβ receptors, as assessed by measurement of the IC50 value for each respective ligand. Thus, in order to bring about a maximal effect on signalling by the TP isoforms, each agent was used at 1 μM throughout the studies. Despite the similar potency in functional antagonism of TPα or TPβ receptors observed for each respective ligand, TPα receptor responses were significantly more reduced than TPβ receptor responses for all ligands.
Structure function analyses of other receptors imply that many of the target desensitization/phosphorylation sites on GPCRs are mainly located within the C-tail regions (Lefkowitz, 1998). The PKC inhibitor GF 109203X, but not the PKA inhibitor H-89, significantly impaired PGI2 and 17 phenyl trinor PGE2 induced functional antagonism of both TP isoforms. TPΔ328, a truncated variant lacking the C-tail sequences distal to the point of divergence of TPα and TPβ receptor, was not sensitive to regulation by PGI2 or 17 phenyl trinor PGE2. These data confirm that the differential regulation of TPα and TPβ due to EP1 receptor signalling is due to unique elements in the C-tails of the TP isoforms. Moreover, as GF 109203X significantly alleviated 17 phenyl trinor PGE2 and PGI2 induced antagonism of TPα receptor and completely blocked antagonism of TPβ receptor signalling, these effects are most likely mediated at PKC phosphorylation sites unique to the individual TP receptors. The fact that GF 109203X did not fully block TPα receptor regulation may indicate a minor role for other kinases, or may reflect the relatively high TP receptor density in the stably transfected HEK 293 cells. Similar observations on the effect of receptor number on U46619-mediated desensitization of the mouse TP receptor have been reported (Spurney, 1998). To rule out the possibility that the differential effects of PGI2 or 17 phenyl trinor PGE2 in modifying the responses between TPα and TPβ receptors are not accounted for due to differences in relative receptor isoform density, we have confirmed these effects in a number of independent HEK 293 cell isolates that stably over express TPα and TPβ receptors at different levels and have found no difference in the behaviour patterns of the individual TP isoforms with the various antagonistic agents irrespective of receptor density – i.e. TPα receptor was significantly more sensitive to PGI2, PGE2 or 17 phenyl trinor PGE2 mediated antagonism than was TPβ receptor. Moreover, we have recently generated TP isoform specific antibodies (based on the unique C-tail sequences of TPα, TPβ) and confirmed by immuno-localization studies that the TPα, TPβ receptor are expressed to similar levels in the cells (HEK.α10 cells and HEK.β3 cells) used for the current study (data not shown).
In keeping with the involvement of PKC in EP1 receptor-mediated functional antagonism of the TP isoforms, we have established that the C-tail of TPα receptor may be phosphorylated in vitro by both PKC and PKA (Kinsella et al., 1994). Differences in the complement of serines or threonines within their unique C-tails, indicate that TPα and TPβ receptor may indeed be subject to differential phosphorylation. Consistent with this, we have recently established that the TPα but not the TPβ isoform, is subject to cicaprost induced desensitization mediated through direct cyclic AMP-dependent PKA phosphorylation of TPα at serine 329 (Walsh et al., 2000b). Thus, the TP isoforms are subject to differential counter regulation by IP (PKA-dependent) and EP1 (PKC dependent) receptors through their differential activation of alternate signal transduction cascades. Moreover, given that PGI2 a physiologic ligand, mediates activation of IP in platelets but AH6809 sensitive EP1 type receptors in kidney fibroblasts, the individual TP isoforms may be differentially regulated by PGI2 in cell/tissue-dependent manner.
Thus, in the current study, we report that both TPα and TPβ receptors are subject to functional antagonism by EP1 receptor, albeit at different levels of sensitivity, and point to additional differences in their signalling behaviour and responses to other signalling pathways. The PKC-dependent, EP1 receptor-mediated regulation of the TP isoforms is not predictable simply due to coincident activation of PKC associated with EP1 receptor/PLC coupling. For example, we and others (Kinsella et al., 1997; Thomas et al., 1995, Habib et al., 1997; 1999) have established that signalling by the TP receptors expressed in platelets or HEK 293 cells is not subject to desensitization due to thrombin activation of the PLC/PKC system and vice versa.
EP1 receptors mediate contraction of smooth muscle in tissues such as the gastrointestinal tract (Lawrence & Jones, 1992), respiratory tract (McKenniff et al., 1988), myometrium (Coleman et al., 1990) and the iris sphincter muscle (Lawrence & Jones, 1992). Their exact role, however, may vary between species. Whereas EP1 receptors are expressed in HEL cells (Funk et al., 1993) and in other megakaryoblastic cell lines (van der Vuurst et al., 1997), there is little evidence to indicate their existence on platelets (Coleman et al., 1990). Our studies failed to demonstrate any evidence for EP1 receptors in platelets and hence, any involvement for molecular interplay between TP and EP1 receptor ligands in platelets. Consistent with this, AH6809 produced no significant effect on U46619 induced [Ca2+]i mobilization or on PGI2 (or cicaprost) mediated cross regulation of U46619 induced [Ca2+]i responses in human platelets (data not shown). Both TP and EP1 receptors are however abundantly co-expressed in kidney, lung, spleen and uterus where they bring about contraction of smooth muscle (Watabe et al., 1993; Hirata et al., 1991; Namba et al., 1992; Miggin & Kinsella., 1998). Thus, the current finding that TP isoforms are subject to functional antagonism by EP1 receptors may shed some light on how their activities are counter regulated in tissues such as kidney, lung, spleen and uterus.
Acknowledgments
This research was supported by grants from The Wellcome Trust, The Irish Heart Foundation, The Health Research Board of Ireland and Enterprise Ireland. We are very grateful to Leanne Kelley for assistance with platelet and cyclic AMP studies.
Abbreviations
- [Ca2+]i
intracellular calcium
- cyclic AMP
adenosine 3′,5′ -cyclic monophosphate
- EP
prostaglandin E receptor
- FBS
foetal bovine serum
- HEK
human embryonic kidney
- PG
prostaglandin
- PLC
phospholipase C
- TP
prostanoid TP receptor
- TXA2
thromboxane A2
References
- AN S., YANG J., SO S.W., ZENG I., GOETZL E.J. Isoforms of the EP3 subtype of human prostaglandin E2 receptor transduce both intracellular calcium and cAMP signals. Biochemistry. 1994;33:14496–14502. doi: 10.1021/bi00252a016. [DOI] [PubMed] [Google Scholar]
- COLEMAN R.A., KENNEDY I., HUMPREY P.P.A., BUNCE K., LUMLEY P. Comprehensive Medicinal Chemistry 1990Vol. 3New York: Pergamon Press Inc; 643–714.eds Hansch, C. Sammes, P.G., Taylor, J.B., Emmett, J.C. pp [Google Scholar]
- FENNEKOHL A., SCHIEFERDECKER H.L., JUNGERMANN K., PUSCHEL G.P. Differential expression of prostanoid receptors in hepatocytes, Kupffer cells, sinusoidal endothelial cells and stellate cells of rat liver. J. Hepatol. 1999;30:38–47. doi: 10.1016/s0168-8278(99)80006-3. [DOI] [PubMed] [Google Scholar]
- FOORD S.M., MARKS B., STOLZ M., BUFLIER E., FRASER N.J., LEE M.G. The structure of the prostaglandin EP4 receptor gene and related pseudogenes. Genomics. 1996;35:182–188. doi: 10.1006/geno.1996.0337. [DOI] [PubMed] [Google Scholar]
- FUNK C.D., FURCI L., FITZGERALD G.A., GRYGORCZYK R., ROCHETTE C., BAYNE M.A., ABRAMOVITZ M., ADAM M., METTERS K.M. Cloning and expression of a cDNA for the human prostaglandin E receptor EP1 subtype. J. Biol. Chem. 1993;268:26767–26772. [PubMed] [Google Scholar]
- HABIB A., FITZGERALD G.A., MACLOUF J. Phosphorylation of the thromboxane receptor alpha, the predominant isoform expressed in human platelets. J. Biol. Chem. 1999;274:2645–2651. doi: 10.1074/jbc.274.5.2645. [DOI] [PubMed] [Google Scholar]
- HABIB A., VEZZA R., CREMINON C., MACLOUF J., FITZGERALD G.A. Rapid, agonist-dependent phosphorylation in vivo of human thromboxane receptor isoforms. Minimal involvement of protein kinase C. J. Biol. Chem. 1997;272:7191–7200. doi: 10.1074/jbc.272.11.7191. [DOI] [PubMed] [Google Scholar]
- HAYES J.S., LAWLER O.A., WALSH M.-T., KINSELLA B.T. The prostacyclin receptor is isoprenylated. Isoprenylation is required for efficient receptor-effector coupling. J. Biol. Chem. 1999;274:23707–23718. doi: 10.1074/jbc.274.34.23707. [DOI] [PubMed] [Google Scholar]
- HIRATA M., HAYASHI Y., USHIKUBI F., YOKOTA Y., KAGEYAMA R., NAKANISHI S., NARUMIYA S. Cloning and expression of cDNA for a human thromboxane A2 receptor. Nature. 1991;349:617–620. doi: 10.1038/349617a0. [DOI] [PubMed] [Google Scholar]
- HIRATA T., USHIKUBI F., KAKIZUKA A., OKUMA M., NARUMIYA S. Two thromboxane A2 receptor isoforms in human platelets. J. Clin. Invest. 1996;97:949–956. doi: 10.1172/JCI118518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KINSELLA B.T., O'MAHONY D.J., FITZGERALD G.A. Phosphorylation and regulated expression of the human thromboxane A2 receptor. J. Biol. Chem. 1994;269:29914–29919. [PubMed] [Google Scholar]
- KINSELLA B.T., O'MAHONY D.J., FITZGERALD G.A. The human thromboxane A2 receptor alpha isoform (TPalpha) functionally couples to the G proteins Gq and G11in vivo and is activated by the isoprostane 8-epi prostaglandin F2 alpha. J. Pharmacol. Exp. Ther. 1997;281:957–964. [PubMed] [Google Scholar]
- KIRIYAMA M., USHIKUBI F., KOBAYASHI T., HIRATA M., SUGIMOTO Y., NARUMIYA S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWRENCE R.A., JONES R.L. Investigation of the prostaglandin E (EP-) receptor subtype mediating relaxation of the rabbit jugular vein. Br. J. Pharmacol. 1992;105:817–824. doi: 10.1111/j.1476-5381.1992.tb09063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFKOWITZ R.J. G protein-coupled receptors. III New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- MANGANELLO J.M., DJELLAS Y., BORG C., ANTONAKIS K., LEBRETON G.C. Cyclic AMP-dependent Phosphorylation of Thromboxane A2 receptor-associated Gα13. J. Biol. Chem. 1999;274:28003–28010. doi: 10.1074/jbc.274.39.28003. [DOI] [PubMed] [Google Scholar]
- MCKENNIFF M., RODGER I.W., NORMAN P., GARDINER P.J. Characterisation of receptors mediating the contractile effects of prostanoids in guinea-pig and human airways. Eur. J. Pharmacol. 1988;153:149–159. doi: 10.1016/0014-2999(88)90601-2. [DOI] [PubMed] [Google Scholar]
- MIGGIN S.M., KINSELLA B.T. Expression and tissue distribution of the mRNAs encoding the human thromboxane A2 receptor (TP) alpha and beta isoforms. Biochim. Biophys. Acta. 1998;1425:543–559. doi: 10.1016/s0304-4165(98)00109-3. [DOI] [PubMed] [Google Scholar]
- MURRAY R., SHIPP E., FITZGERALD G.A. Prostaglandin endoperoxide/thromboxane A2 receptor desensitization. J. Biol. Chem. 1990;265:21670–21675. [PubMed] [Google Scholar]
- NAMBA T., SUGIMOTO Y., HIRATA M., HAYASHI Y., HONDA A., WATABE A., NEGISHI M., ICHIKAWA A., NARUMIYA S. Mouse thromboxane A2 receptor: cDNA cloning, expression and northern blot analysis. Biochem. Biophys. Res. Commun. 1992;184:1197–1203. doi: 10.1016/s0006-291x(05)80009-9. [DOI] [PubMed] [Google Scholar]
- NAMBA T., SUGIMOTO Y., NEGISHI M., IRIE A., USHIKUBI F., KAKIZUKA A., ITO S., ICHIKAWA A., NARUMIYA S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993;365:166–170. doi: 10.1038/365166a0. [DOI] [PubMed] [Google Scholar]
- NARUMIYA S., SUGIMOTO Y., FUMITAKA U. Prostanoid receptors: Structures, Properties, and Functions. Physiol. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- NEGISHI M., SUGIMOTO Y., ICHIKAWA A. Prostanoid receptors and their biological actions. Prog. Lipid. Res. 1993;32:417–434. doi: 10.1016/0163-7827(93)90017-q. [DOI] [PubMed] [Google Scholar]
- RAYCHOWDHURY M.K., YUKAWA M., COLLINS L.J., MCGRAIL S.H., KENT K.C., WARE J.A.Alternative splicing produces a divergent cytoplasmiC-tail in the human endothelial thromboxane A2 receptor J. Biol. Chem. 199426919256–19261.Published erratum appears in J. Biol. Chem., (1995), 270, 7011 [PubMed] [Google Scholar]
- SPURNEY R.F. Effect of receptor number on desensitization of the mouse thromboxane receptor. Biochem. Pharmacol. 1998;55:1271–1281. doi: 10.1016/s0006-2952(97)00633-3. [DOI] [PubMed] [Google Scholar]
- THOMAS C.P., DUNN M.J., MATTERA R. Ca2+ signalling in K562 human erythroleukaemia cells: effect of dimethyl sulphoxide and role of G-proteins in thrombin- and thromboxane A2-activated pathways. Biochemistry. 1995;312:151–158. doi: 10.1042/bj3120151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAN DER VUURST H., VAN WILLIGEN G., VAN SPRONSEN A., HENDRIKS M., DONATH J., AKKERMAN J.W. Signal transduction through trimeric G proteins in megakaryoblastic cell lines. Arterioscler. Thromb. Vasc. Biol. 1997;17:1830–1836. doi: 10.1161/01.atv.17.9.1830. [DOI] [PubMed] [Google Scholar]
- WALSH M.-T., FOLEY J.F., KINSELLA B.T. Characterization of the role of N-linked glycosylation on the cell signaling and expression of the human thromboxane A2 receptor alpha and beta isoforms. J. Pharmacol. Exp. Ther. 1998;286:1026–1036. [PubMed] [Google Scholar]
- WALSH M.-T., FOLEY J.F., KINSELLA B.T. Investigation of the role of the carboxyl-terminal tails of the human thromboxane A2 receptor (TP) in mediating receptor: effector coupling. Biochim. Biophys. Acta. 2000a;1496:164–182. doi: 10.1016/s0167-4889(00)00031-8. [DOI] [PubMed] [Google Scholar]
- WALSH M.-T., FOLEY J.F., KINSELLA B.T.The α, but not the β, isoform of the human thromboxane A2 receptor (TP) is a target for prostacyclin mediated desensitisation J. Biol. Chem. 2000b275in press [DOI] [PubMed] [Google Scholar]
- WATABE A., SUGIMOTO Y., HONDA A., IRIE A., NAMBA T., NEGISHI M., ITO S., NARUMIYA S., ICHIKAWA A. Cloning and expression of cDNA for a mouse EP1 subtype of prostaglandin E receptor. J. Biol. Chem. 1993;268:20175–20178. [PubMed] [Google Scholar]
- WATANABE T., SUNAGA S., TOGO M., SATOH H., HIGASHIHARA M., HASHIMOTO Y., KUROKAWA K. Protein kinase C plays a key role in the cross talk between intracellular signalings via prostanoid receptors in a megakaryoblastic cell line, MEG-01 s. Biochim. Biophys. Acta. 1996;1304:161–169. doi: 10.1016/s0005-2760(96)00111-7. [DOI] [PubMed] [Google Scholar]