

Abstract
The physiological role of the angiotensin II AT2 receptor subtype is not fully characterized. We studied whether AT2 receptor could antagonize AT1 mediated superoxide formation in endothelial cells.
In quiescent human umbilical vein endothelial cells (HUVEC) superoxide formation was measured after long-term incubation (6 h) with angiotensin II in the presence or absence of its receptor blocker candesartan (AT1) or PD123319 (AT2) using the cytochrome c assay. In separate experiments, the effects of AT2 mediated effects on activities of cellular phosphates including the src homology 2 domain containing phosphatases (SHP-1) was studied.
The basal superoxide formation (0.19±0.03 nmol superoxide mg protein−1 min−1) in HUVEC was increased by 37.1% after exposure to angiotensin II (100 nM,) which was due to an activation of a NAD(P)H oxidase. This was abolished by candesartan (1 μM) as well as the tyrosine kinase inhibitor genistein. In contrast, blockade of AT2 receptors by PD123319 enhanced the superoxide formation by 73.7% in intact cells. Stimulation of AT2 went along with an increased activity of tyrosine phosphatases in total cell lysates (29.8%) and, in particular, a marked stimulation of src homology 2 domain containing phosphatases (SHP-1, by 293.4%). The tyrosine phosphatase inhibitor vanadate, in turn, prevented the AT2 mediated effects on superoxide formation. The expression of both angiotensin II receptor subtypes AT1 and AT2 was confirmed by RT–PCR analysis.
It is concluded that AT2 functionally antagonizes the AT1 induced endothelial superoxide formation by a pathway involving tyrosine phosphatases.
Keywords: Angiotensin II receptor subtypes, superoxide anion, endothelial cell physiology, tyrosine phosphatases, Src homology 2 domain containing phosphatases
Introduction
Angiotensin II (AngII) plays an important role in the pathogenesis of cardiovascular disorders such as development of myocardial and smooth muscle hypertrophy, arteriosclerosis and neointima formation after balloon injury (Helin et al., 1997). This is, at least in part, due to an AngII-induced formation of superoxide anions in vascular tissues (Griendling et al., 1994; Pagano et al., 1998). Superoxide scavenges NO and therefore abolishes its vasoprotective effects resulting in an impairment of endothelium dependent dilation, enhanced platelet aggregation and intimal proliferation (Mügge et al., 1991; Ohara et al., 1993). The inactivation of NO by superoxide may be particularly important in endothelial cells (Ohara et al., 1993; Warnholtz et al., 1999) where this autacoid is synthesized.
Virtually all stimulating effects of AngII on cardiovascular cells are mediated by the AT1 receptor subtype. The physiological effects of AT2 receptor stimulation are, however, less clear (Horiuchi et al., 1999). Experimental data suggest that the AT2 receptor opposes some AT1 receptor mediated effects on cell growth as well as on water and electrolyte balance. (Munzenmaier & Greene, 1996; Hein et al., 1995). The AT1 in vascular cells is coupled via G-proteins to the inositol 1,4,5-triphosphate pathway, and additionally, to cytoplasmatic tyrosine kinases such as the JAK/STAT pathway (Schieffer et al., 1996), while the signal pathway of the AT2 mediated effects is not fully understood (Horiuchi et al., 1999). It was proposed that an activation of cellular phosphotyrosine phosphatase (PTP) activity plays a key role in AT2 signaling (Tsuzuki et al., 1996; Bedecs et al., 1997; Marrero et al., 1998). Recently, the vanadate sensitive src homology 2 domain containing PTP (SHP-1, previously termed PTP1C, SHPTP-1 and SHP) which is known to inhibit growth related signalling predominantly in hemopoietic cells (Klingmuller et al., 1995), has been shown to be activated by AT2 stimulation in N1E-115 cells (Bedecs et al., 1997). In vascular smooth muscle cells SHP-1 was responsible for JAK2 dephosphorylation and thus termination of AngII induced JAK/STAT pathway (Marrero et al., 1998).
With respect to endothelial superoxide formation, it has been demonstrated recently that AngII serves as an effective stimulus, acting via an AT1 dependent mechanism (Warnholtz et al., 1999). Nothing is known, however, about a potential antagonistic role of the AT2 receptor in this process. Here, we studied whether AT2 stimulation counteracts the AT1 mediated superoxide production in cultured endothelial cells and whether activation of PTP is involved in these AT2-dependent signaling pathways. An AT2 mediated reduction of endothelial superoxide might play an important role in the beneficial therapeutic effect of AT1 blockers.
Methods
Cell culture
Endothelial cells (HUVEC) were isolated from freshly obtained human umbilical veins and grown to confluence in Medium 199 supplemented with 16% foetal calf serum and 20% endothelial growth medium. After serum starvation for 24 h the quiescent HUVEC were treated with AngII (0.1–1 μM) in the presence or absence of the AT1 blocker CV11974 [CV, 1 μM, (candesartan)] or the AT2 blocker PD123319 (PD, 1 μM) for 6 h.
Detection of endothelial superoxide production
Endothelial superoxide formation was determined by the cytochrome c method. HUVEC were incubated in a HEPES (20 mM)-Tyrode buffer containing cytochrome c (40 μM) with or without SOD (200 u ml−1). After 30 min the supernatant was removed and the SOD-inhibitable reduction of cytochrome c measured at 550 nm. The superoxide production was calculated from the difference between samples with or without SOD (ε550 nM=21.1 mM−1 cm−1). NADH induced superoxide production in cell lysates was measured by adding NADH (100 μM) to aliquots of cell lysates.
Immunoprecipitation and Western blot analysis
HUVEC were lysed in modified ice cold RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 0.1% (w v−1) SDS, 0.5% DOC, 1% NP40, 10 μg ml−1 leupeptin, 10 μg ml−1 pepstatin, 10 μM PMSF, 2 mM orthovanadate, 100 mM NaF). 10 μg ml−1 anti-SHP antibody was added to the lysates for 1 h at 4°C. Immunocomplexes of SHP-1 were precipitated by addition of protein G-agarose beads and allowed to equilibrate for 12 h. The precipitates were divided in two halves and either used for tyrosine phosphatase assay or for Western blotting. For the latter, the immunoprecipitated SHP-1 were separated on a SDS–PAGE following standard procedures and transferred onto nitrocellulose membrane. After incubation with the SHP-1 antibody and an alkaline phosphatase conjugated secondary antibody the loading of SHP-1 was detected using the NBT/BCIP system.
Tyrosine phosphatase assay
For measurements of cellular tyrosine phosphatase activity a commercially available kit (Promega) was used. Endothelial cells were lysed in 100 μl lysis buffer (20 mM Tris HCl pH 7.2, 2 mM EDTA, 1% Triton-X100, 1 mM PMSF, 0.1% 2-mercaptoethanol, 1 μg ml−1 aprotinin, 1 μg ml−1 leupeptin). The phosphatase reaction mix, containing 5 μl of the tyrosine phosphopeptide DADE(pY)LIPQQG, 40 μl storage buffer and 5 μl of the sample were incubated for 45 min at 37°C. PO43− concentration was determined at 595 nm using a ELISA reader (TECAN) and normalized to protein content of the sample.
Reverse transcriptase PCR
For RT–PCR 1 μg total RNA was reverse transcribed in (mM): Tris/HCl (pH 8.3) 50, KCl 75, MgCl2 3, DTT 10, dNTP 0.5, specific reverse primer 3 and 100 uM-MLV RT (Promega). 35 cycles of PCR were performed in (mM): Tris/HCl (pH 8.8) 50, KCl 50, MgCl2 1.5, DTT 2, 0.1% TritonX-100, dNTP 0.2, 0.7 μM specific primers and 2,5 u Taq Pol (AmpliTaqGold, Promega). The used primers were (AT1) 5′- GCTATGGAATACCGCTGGCCCTTTGG-3′ (265–290) and 5′- GGCATGGCCGTGTCCACAATATCTGC-3′ (854-829); (AT2): 5′- CCAGCGGTCTTCACTTCGGG-3′ (44–63) and 5′- GCCAGAACAACAGCAGCTGC-3′ (791–772) from human AngII receptor mRNA.
Drugs
CV11974 (Candesartan) was kindly provided by Astra Chemicals (Mülndal, Sweden). PD123319 was obtained as a gift from Parke Davis (Ann Arbor, U.S.A.). SOD was purchased from Boehringer MA (Germany). Endothelial cell growth medium was from PromoCell (Heidelberg, Germany) and orthovanadate from Alexis (San Diego, U.S.A.). Anti-phosphotyrosine was purchased from Upstate (Lake Placid, U.S.A.), anti-SHP-1 from Santa Cruz (Santa Cruz, U.S.A.) and the tyrosine phosphatase activity assay kit from Promega (Madison, U.S.A.). All other drugs were obtained from Sigma (Deisenhofen, Germany).
Statistical analysis
Statistical comparisons with and without treatment within the same experimental group were performed using the Wilcoxon signed rank test for paired observations. Differences were considered significant at an error probability of P<0.05. For descriptive means all results are expressed as means±s.e.mean.
Results
Basal superoxide production in endothelial cells
Quiescent cells produced 0.27±0.03 nmol superoxide mg protein−1 min−1 (n=10). Since a pretreatment with the NO-synthase inhibitor NG-nitro-L-arginine (L-NA, 30 μM) increased the basal superoxide production by a further 48.5% (n=10, P<0.01), all further experiments were performed in the presence of L-NA to avoid modulating effects of NO. Proliferating cells kept in serum had an about 3 fold higher basal superoxide formation (0.27±0.03 vs 0.84±0.07 nmol superoxide mg protein−1 min−1, n=10, P<0.01).
Different effects of AT1 and AT2 stimulation on endothelial superoxide formation
AngII (100 nM) did not affect the superoxide formation in proliferating cells (0.75±0.05 vs 0.85±0.04 nmol superoxide mg protein−1 min−1, n=11, n.s.). In quiescent cells, however, exposure to AngII (10–100 nM) for 6 h significantly enhanced the superoxide formation by 30.0 and 37.1% (n=12, P<0.05), respectively, whereas at a higher concentration (1 μM) of AngII did not significantly affect the superoxide formation. The AngII (0.1 μM) mediated superoxide formation was mediated by AT1 receptor, since the AT1 blocker CV (1 μM) abolished the AngII induced effect (Figure 1, n=24, P<0.01). In contrast, blockade of the AT2 by PD (1 μM) augmented the superoxide formation by 73.7% of control (Figure 1, n=24, P<0.01). Although this effect was highly significant in some subset of cells, PD123319 showed no significant effects on AngII induced superoxide formation. In the latter cells, however, AngII induced a markedly stronger increase in superoxide production (29.7% vs 95.0%) supporting the inhibitory role of AT2 receptors on superoxide formation. The flavoprotein enzyme inhibitor diphenyleniodonium chloride (DPI, 10 μM) as well as the direct NAD(P)H oxidase blocker phenylarsine oxide (PAO, 1 μM) (Le Cabec & Maridonneau, 1995) significantly attenuated the AngII induced superoxide formation by 79.3 and 67.3%, respectively (n=6, P<0.05). As shown in Figure 2, pre-stimulation of the AT1 receptor in intact cells also augmented the NADH and NADPH-dependent superoxide formation after cell lysis by 88.7% (n=10, P<0.01) and 65.1% (n=17, P<0.05), respectively. This was, in turn, prevented by preincubation of the intact cells with the AT1 blocker CV whereas an AT2 blockade failed to have any effects.
Figure 1.
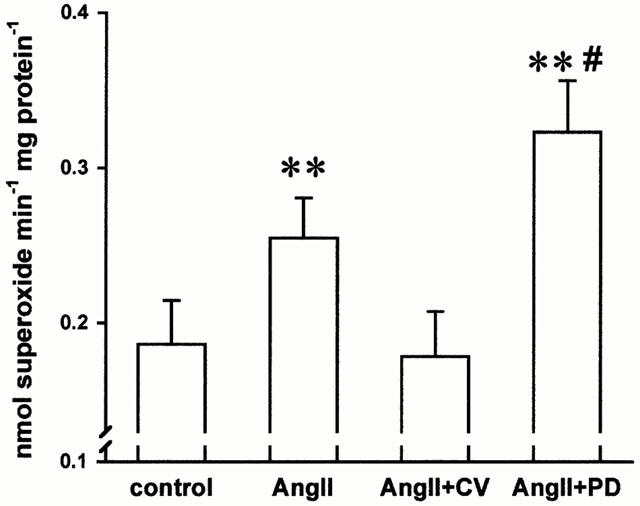
Angiotensin II (AngII, 100 nM incubation for 6 h increased the superoxide formation in intact endothelial cells. This was inhibited by the AT1 receptor blocker CV11974 (CV, 1 μM) whereas an AT2 receptor (PD123319, 1 μM) blockade further increased the superoxide formation. (n=24, **P<0.01 vs. control, #P<0.05 vs angiotensin II).
Figure 2.
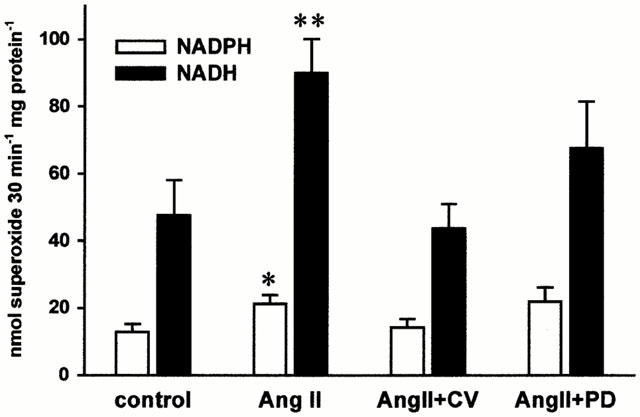
In separate experiments the pretreated endothelial cells were resuspended in a lysis buffer and either the NADPH or NADH (100 μM each) dependent superoxide formation measured (n=17 for NADPH and n=10 for NADH; *P<0.05 and **P<0.01 vs control). In contrast to intact cells an AT2 receptor inhibition did not affect the AT1 receptor mediated stimulation of NADH/NADPH dependent superoxide production.
Tyrosine kinases mediate AT1 induced endothelial superoxide production
As shown in Figure 3 the AngII induced superoxide formation could be prevented by the tyrosine kinase inhibitor genistein (30 μM, n=8, P<0.05) while the protein kinase C inhibitor staurosporine (100 nM) was not effective. The latter concentration of staurosporine inhibited phorbol 12-myristate 13-acetate induced superoxide formation in cells cultured in same conditions (Sohn et al., 1999). In separate experiments, the more specific PKC-blocker chelerythrine chloride (10 μM) was tested. It also did not exert a significant effect on AngII induced superoxide formation (control: 0.23±0.04 vs AngII: 0.36±0.04 and AngII+chelerythrine chloride: 0.35±0.05 nmol superoxide mg protein−1 min−1, n=12, n.s). Neither genistein nor staurosporine alone affected the basal superoxide formation (0.28±0.03 vs 0.26±0.02 and 0.31±0.02 nmol superoxide mg protein−1 min−1, n=13, n.s.).
Figure 3.
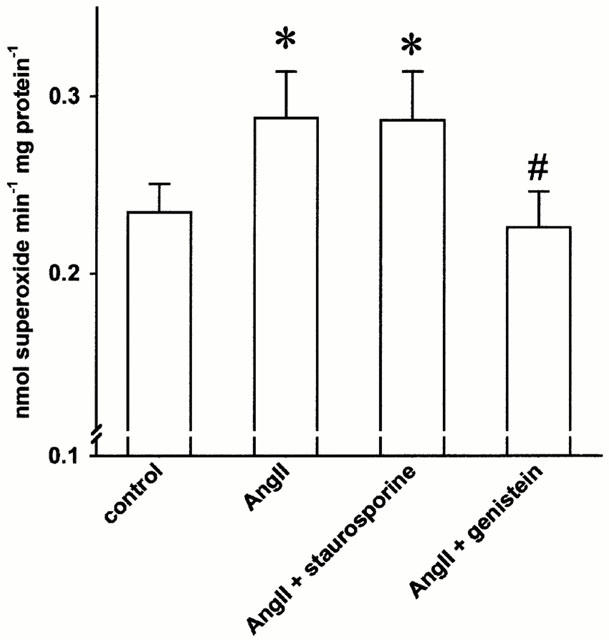
The angiotensin II (AngII, 100 nM) induced endothelial superoxide formation was abolished in the presence of the tyrosine kinase inhibitor genistein (30 μM, n=11, *P<0.05 vs control, #P<0.05 vs angiotensin II) while the protein kinase C inhibitors staurosporine and chelerythrine chloride (10 μM, see text) were not effective.
AT2 mediates activation of PTPs
In quiescent cells the PTP activity was enhanced by 49.8% compared to proliferating cells (n=18, P<0.05, data not shown). Selective stimulation of AT2 receptors for 15 minutes (AngII: 0.1 μM) in the presence of the AT1 receptor blocker CV (1 μM) significantly increased the PTP-activity in quiescent cells by a further 29.8% (Figure 4A, P<0.05, n=10). To evaluate the potential role of a particular PTP in AT2 receptor mediated inhibition of superoxide formation, we assessed the activity of SHP-1, which is involved in AT2 receptor mediated JAK2 dephosphorylation (Marrero et al., 1998). Stimulation of the AT2 receptor markedly increased the SHP-1 activity by 293.4% (Figure 4B, n=6, P<0.05). Equal loading of SHP-1 of the PTP-assay was confirmed by Western blotting of immunoprecipitated SHP-1 (see Figure 4B inlet).
Figure 4.
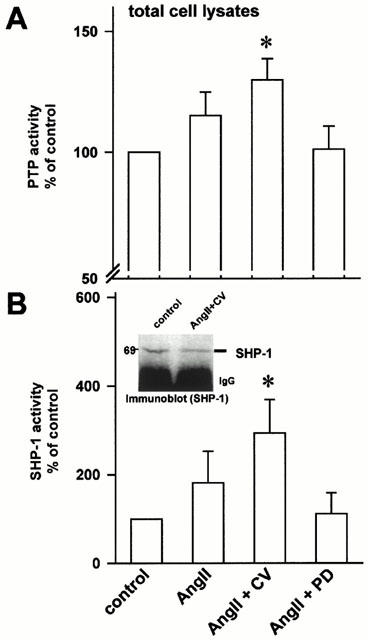
(A) Activity of cellular phosphotyrosine phosphatases (PTP). Selective stimulation of endothelial AT2 receptors (in the presence of the AT1 receptor blocker CV, 1 μM) increased the activity of phosphotyrosine phosphatases measured in total endothelial cell lysates (n=10, *P<0.05 vs control). (B) In separate experiments, SHP-1 was immunoprecipitated from cell lysates and its activity determined. AT2 receptor stimulation markedly enhanced the SHP-1 activity (n=6, *P<0.05 vs control). Inlet, Immunoblot of SHP-1. Aliquots from immunoprecipitated SHP-1 were subjected to SDS–PAGE and blotted onto nitrocellulose membrane. Equal loading of SHP-1 was confirmed by specific antibody against SHP-1.
PTP inhibition prevents AT2 mediated effects on endothelial superoxide production
Using the PTP inhibitor vanadate we studied whether changes in PTP activity modulate endothelial superoxide production. We showed previously (Sohn et al., 1999) that vanadate at a concentration of 100 μM significantly increases tyrosine phosphorylation of various proteins in endothelial cells. The same concentration of vanadate also enhanced the basal endothelial superoxide generation (by 35.9%, P<0.05, Figure 5). Vanadate further augmented the AT1 induced superoxide formation (P<0.05), while an AT2 receptor blockade did not further affect the superoxide production in the presence of vanadate (n=15, Figure 5).
Figure 5.
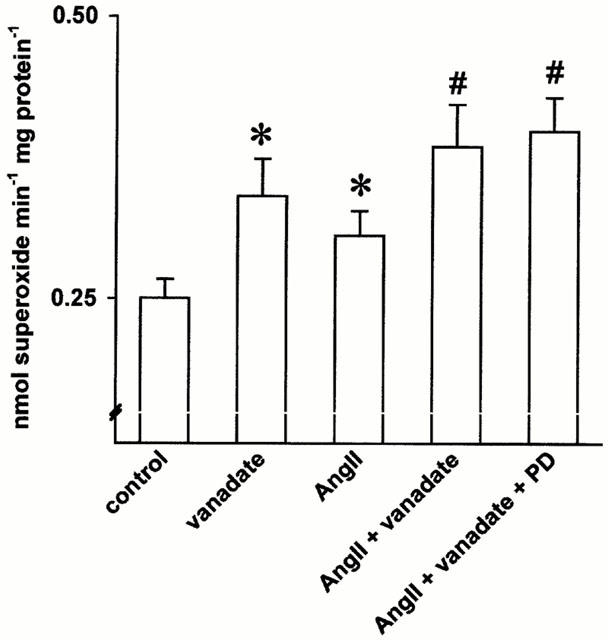
Effect of the phosphotyrosine phosphatase inhibitor vanadate on angiotensin II induced endothelial superoxide formation. Vanadate (100 μM) further augmented the angiotensin II mediated superoxide formation. AT2 receptor blockade (PD123319, 1 μM), however, did not affect the superoxide production in the presence of vanadate (n=15, *P<0.05 vs control, #<0.05 vs angiotensin II).
AngII receptor subtypes in HUVEC
The expression of both AngII receptor subtypes was confirmed by RT–PCR analysis (Figure 6). In contrast to AT1, the AT2 receptor was found to be not expressed in some populations of HUVEC.
Figure 6.
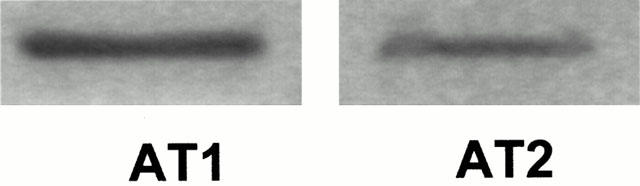
Expression of AT1 and AT2 receptors in HUVEC. RT–PCR analysis showed the expression of both angiotensin II receptor subtypes in HUVEC.
Discussion
This study shows that AngII via AT1 receptor induces superoxide formation in endothelial cells (Zhang et al., 1999). This is in agreement with previous studies in vascular smooth muscle cells (Griendling et al., 1994) and fibroblasts (Pagano et al., 1998). The inhibitory effects of DPI and PAO (Le Cabec & Maridonneau, 1995), as well as the NADH-sensitivity of superoxide formation suggest that a NAD(P)H dependent oxidase was the main source of endothelial superoxide formation in our experiments (Jones et al., 1996; Al-Mehdi et al., 1998).
The regulation of this superoxide generating system in endothelial cells is not fully understood. Increases in intracellular calcium and/or activation of protein kinase C seem to play a role (Matsubara & Ziff, 1986). Our results indicate, however, that the AT1 mediated superoxide formation in HUVEC includes a tyrosine phosphorylation dependent step (Sohn et al., 2000), since genistein, but not staurosporine, completely abolished the effect of AngII. This is in agreement with studies in neutrophils (Dusi et al., 1996) and fibroblasts (Thannickal et al., 1998) showing tyrosine phosphorylation being an important pathway in the activation of free radical production.
On the other hand, a blockade of the AT2 receptor resulted in a further increase of the AT1 mediated superoxide formation, suggesting an antagonistic function of both receptor subtypes. Such an antagonism have been reported previously on bFGF induced proliferation of cultured rat coronary endothelial cells (Stoll et al., 1995). Likewise, long-term treatment with AngII resulted in an increase of the capillary density in the rat cremaster muscle, which was prevented by blockade of AT1, but was further augmented by an AT2 inhibition (Munzenmaier & Greene, 1996). Since AT2 activation increases PTP activity in various cells (Tsuzuki et al., 1996; Bedecs et al., 1997; Horiuchi et al., 1999) we studied whether PTPs play a role in regulation of endothelial superoxide production. In this study, the PTP-inhibitor vanadate significantly increased the superoxide generation suggesting that, in turn, an increased activity of PTP results in an attenuation of superoxide formation. Direct measurements of PTP-activity confirmed that AT2 stimulation indeed activates PTP. Accordingly, in quiescent cells with a low rate of superoxide formation, the PTP activity was per se elevated. Tsuzuki et al. (1996) observed in R3T3 cells an AT2 mediated inhibition of cell proliferation which was associated with an activation of PTP. Possible candidates might represent, in particular, the vanadate sensitive SHP-1 which is involved in termination of proliferation related signalling by growth factors or by AngII (Bedecs et al., 1997; Marrero et al., 1998; Klingmuller et al., 1995). We demonstrated that the AT2 induced inhibition of superoxide production is associated with a marked increase of SHP-1 activity. Although these studies do not establish a causal relationship, the findings are highly suggestive for a role of SHP-1 in regulation of endothelial superoxide formation. This is further supported by recent findings in neutrophils showing that an initiation of the respiratory burst was followed by a declined activity of SHP-1 (Brumell et al., 1997).
Incubation of intact HUVEC with AngII also increased the NADH dependent superoxide formation in cell homogenates, which was mediated by AT1 receptors. AT2 receptors do not seem to affect the NAD(P)H oxidase assembly but rather control its activity via modulating PTP which seem not to function anymore in cell lysates. Accordingly, inhibition of PTP by vanadate abolished the effect of an AT2 blockade on superoxide generation in intact cells but this effect did not persist after lysis of the cells.
Some investigators were unable to detect an effect on AngII induced superoxide formation by AT2 receptor blockade. The expression of the AT2 receptor is quite variable in different tissues and appears not only to be dependent on the cell types but also on in vitro culture conditions (Ichiki et al., 1995; Dimmeler et al., 1997; Pueyo & Michel, 1997). Although the HUVEC were kept under similar conditions in our study the expression of AT2 was not found in some populations. However, it supports our hypothesis of an inhibitory role of AT2 receptors on superoxide formation, that in HUVEC lacking the AT2 receptor, the AngII induced superoxide formation was markedly stronger.
In conclusion, the present experiments demonstrate that AngII receptor subtypes differentially modulate endothelial superoxide formation. While AT1 activates superoxide formation, AT2 receptors appear to attenuate this AT1-induced effect, most likely by its functional coupling to activation of PTP, probably SHP-1. The relative distribution of both receptor subtypes on the same cell type, which may vary following ischemia or in myocardial hypertrophy (Bartunek et al., 1999; Horiuchi et al., 1999) may therefore influence endothelial superoxide formation and hence NO dependent dilation.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (SFB 553 Teilprojekt B2 and DFG Ga 431/4–1). H.Y. Sohn was a fellow of the German Cardiac Society. We thank Paula H. Sohn for helpful discussion.
Abbreviations
- AngII
Angiotensin II
- CV
CV11974
- DPI
diphenyleniodonium chloride
- HUVEC
human umbilical vein endothelial cells
- L-NA
NG-nitro-L-arginine
- PAO
phenylarsine oxide
- PD
- PTP
phosphotyrosine phosphatases
- SHP-1
src homology 2 domain containing phosphatases
- SOD
superoxide dismutase
References
- AL-MEHDI A.B., ZHAO G., DODIA C., TOZAWA K., COSTA K., MUZYKANTOV V., ROSS C., BLECHA F., DINAUER M, FISHER A.B. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+ Circ. Res. 1998;83:730–737. doi: 10.1161/01.res.83.7.730. [DOI] [PubMed] [Google Scholar]
- BARTUNEK J., WEINBERG E.O., TAJIMA M., ROHRBACH S, LORELL B.H. Angiotensin II type 2 receptor blockade amplifies the early signals of cardiac growth response to angiotensin II in hypertrophied hearts. Circulation. 1999;99:22–25. doi: 10.1161/01.cir.99.1.22. [DOI] [PubMed] [Google Scholar]
- BEDECS K., ELBAZ N., SUTREN M., MASSON M., SUSINI C., STROSBERG A.D, NAHMIAS C. Angiotensin II type 2 receptors mediate inhibition of mitogen-activated protein kinase cascade and functional activation of SHP-1 tyrosine phosphatase. Biochem. J. 1997;325:449–454. doi: 10.1042/bj3250449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRUMELL J.H., CHAN C.K., BUTLER J., BORREGAARD N., SIMINOVITCH K.A., GRINSTEIN S, DOWNEY G.P. Regulation of Src homology 2-containing tyrosine phosphatase 1 during activation of human neutrophils. Role of protein kinase C. J. Biol. Chem. 1997;272:875–882. doi: 10.1074/jbc.272.2.875. [DOI] [PubMed] [Google Scholar]
- DIMMELER S., RIPPMANN V., WEILAND U., HAENDELER J, ZEIHER A.M. Angiotensin II induces apoptosis of human endothelial cells. Protective effect of nitric oxide. Circ. Res. 1997;81:970–976. doi: 10.1161/01.res.81.6.970. [DOI] [PubMed] [Google Scholar]
- DUSI S., DELLA B.V., DONINI M., NADALINI K.A, ROSSI F. Mechanisms of stimulation of the respiratory burst by TNF in nonadherent neutrophils: its independence of lipidic transmembrane signaling and dependence on protein tyrosine phosphorylation and cytoskeleton. J. Immunol. 1996;157:4615–4623. [PubMed] [Google Scholar]
- GRIENDLING K.K., MINIERI C.A., OLLERENSHAW J.D, ALEXANDER R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- HEIN L., BARSH G.S., PRATT R.E., DZAU V.J, KOBILKA B.K. Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor gene in mice. Nature. 1995;377:744–747. doi: 10.1038/377744a0. [DOI] [PubMed] [Google Scholar]
- HELIN K., STOLL M., MEFFERT S., STROTH U., UNGER T., GRIENDLING K.K., MINIERI C.A., OLLERENSHAW J.D, ALEXANDER R.W. The role of angiotensin receptors in cardiovascular diseases. Ann. Med. 1997;29:23–29. doi: 10.3109/07853899708998740. [DOI] [PubMed] [Google Scholar]
- HORIUCHI M., AKISHITA M, DZAU V.J. Recent progress in angiotensin II type 2 receptor research in the cardiovascular system. Hypertension. 1999;33:613–621. doi: 10.1161/01.hyp.33.2.613. [DOI] [PubMed] [Google Scholar]
- ICHIKI T., KAMBAYASHI Y, INAGAMI T. Multiple growth factors modulate mRNA expression of angiotensin II type-2 receptor in R3T3 cells. Circ. Res. 1995;77:1070–1076. doi: 10.1161/01.res.77.6.1070. [DOI] [PubMed] [Google Scholar]
- JONES S.A., O'DONNELL V.B., WOOD J.D., BROUGHTON J.P., HUGHES E.J, JONES O.T. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am. J. Physiol. 1996;271:H1626–H1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- KLINGMULLER U., LORENZ U., CANTLEY L.C., NEEL B.G, LODISH H.F. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80:729–738. doi: 10.1016/0092-8674(95)90351-8. [DOI] [PubMed] [Google Scholar]
- LE CABEC V, MARIDONNEAU P.I. Complete and reversible inhibition of NADPH oxidase in human neutrophils by phenylarsine oxide at a step distal to membrane translocation of the enzyme subunits. J. Biol. Chem. 1995;270:2067–2073. doi: 10.1074/jbc.270.5.2067. [DOI] [PubMed] [Google Scholar]
- MARRERO M.B., VENEMA V.J., JU H., EATON D.C, VENEMA R.C. Regulation of angiotensin II-induced JAK2 tyrosine phosphorylation: roles of SHP-1 and SHP-2. Am. J. Physiol. 1998;275:C1216–C1223. doi: 10.1152/ajpcell.1998.275.5.C1216. [DOI] [PubMed] [Google Scholar]
- MATSUBARA T, ZIFF M. Superoxide anion release by human endothelial cells: synergism between a phorbol ester and a calcium ionophore. J. Cell. Physiol. 1986;127:207–210. doi: 10.1002/jcp.1041270203. [DOI] [PubMed] [Google Scholar]
- MÜGGE A., ELWELL J.H., PETERSON T.E, HARRISON D.G. Release of intact endothelium-derived relaxing factor depends on endothelial superoxide dismutase activity. Am. J. Physiol. 1991;260:C219–C225. doi: 10.1152/ajpcell.1991.260.2.C219. [DOI] [PubMed] [Google Scholar]
- MUNZENMAIER D.H, GREENE A.S. Opposing actions of angiotensin II on microvascular growth and arterial blood pressure. Hypertension. 1996;27:760–765. doi: 10.1161/01.hyp.27.3.760. [DOI] [PubMed] [Google Scholar]
- OHARA Y., PETERSON T.E, HARRISON D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAGANO P.J., CHANOCK S.J., SIWIK D.A., COLUCCI W.S, CLARK J.K. Angiotensin II induces p67phox mRNA expression and NADPH oxidase superoxide generation in rabbit aortic adventitial fibroblasts. Hypertension. 1998;32:331–337. doi: 10.1161/01.hyp.32.2.331. [DOI] [PubMed] [Google Scholar]
- PUEYO M.E, MICHEL J.B. Angiotensin II receptors in endothelial cells. Gen. Pharmacol. 1997;29:691–696. doi: 10.1016/s0306-3623(97)00021-9. [DOI] [PubMed] [Google Scholar]
- SCHIEFFER B., PAXTON W.G., MARRERO M.B, BERNSTEIN K.E. Importance of tyrosine phosphorylation in angiotensin II type 1 receptor signaling. Hypertension. 1996;27:476–480. doi: 10.1161/01.hyp.27.3.476. [DOI] [PubMed] [Google Scholar]
- SOHN H.Y., GLOE T., KELLER M., SCHOENAFINGER K, POHL U. Sensitive superoxide detection in vascular cells by the new chemiluminescence dye L-012. J. Vasc. Res. 1999;36:456–464. doi: 10.1159/000025688. [DOI] [PubMed] [Google Scholar]
- SOHN H.Y., KELLER M., GLOE T., MORAWIETZ H., RUECKSCHLOSS U, POHL U. The small G-protein Rac mediates depolarization induced superoxide formation in human endothelial cells. J. Biol. Chem. 2000;275:18745–18750. doi: 10.1074/jbc.M000026200. [DOI] [PubMed] [Google Scholar]
- STOLL M., STECKELINGS U.M., PAUL M., BOTTARI S.P., METZGER R, UNGER T. The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J. Clin. Invest. 1995;95:651–657. doi: 10.1172/JCI117710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THANNICKAL V.J., ALDWEIB K.D, FANBURG B.L. Tyrosine phosphorylation regulates H2O2 production in lung fibroblasts stimulated by transforming growth factor beta1. J. Biol. Chem. 1998;273:23611–23615. doi: 10.1074/jbc.273.36.23611. [DOI] [PubMed] [Google Scholar]
- TSUZUKI S., MATOBA T., EGUCHI S, INAGAMI T. Angiotensin II type 2 receptor inhibits cell proliferation and activates tyrosine phosphatase. Hypertension. 1996;28:916–918. doi: 10.1161/01.hyp.28.5.916. [DOI] [PubMed] [Google Scholar]
- WARNHOLTZ A., NICKENIG G., SCHULZ E., MACHARZINA R., BRASEN J.H., SKATCHKOV M., HEITZER T., STASCH J.P., GRIENDLING K.K., HARRISON D.G., BOHM M., MEINERTZ T, MÜNZEL T. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin- angiotensin system. Circulation. 1999;99:2027–2033. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- ZHANG H., SCHMEISSER A., GARLICHS C.D., PLOTZE K., DAMME U., MÜGGE A, DANIEL W.G. Angiotensin II-induced superoxide anion generation in human vascular endothelial cells: role of membrane-bound NADH-/NADPH-oxidases. Cardiovasc. Res. 1999;44:215–222. doi: 10.1016/s0008-6363(99)00183-2. [DOI] [PubMed] [Google Scholar]