

Abstract
GPR10 is a novel G-protein coupled receptor that is the human orthologue of rat Unknown Hypothalamic Receptor-1 (UHR-1). Human prolactin-releasing peptide (PrRP) has been identified as an endogenous ligand for GPR10, and occurs as 31 and 20 amino acid forms. The present study characterizes the binding of [125I]-PrRP-20 to HEK293 cells stably expressing GPR10 receptors.
Specific binding of [125I]-PrRP-20 was saturable, and analysis suggested evidence of both high and low affinity sites, with KD values of 0.026±0.006 and 0.57±0.14 nM respectively, and Bmax values of 3010±400 and 8570±2240 fmol mg protein−1 respectively. Kinetic studies were unable to distinguish two sites, but single site analysis of association and dissociation data produced a KD of 0.012 nM.
Competition studies revealed that human and rat PrRP-20 and PrRP-31 all display high affinity for GPR10. A range of other drugs which are known ligands at receptors which share limited homology with GPR10 were also tested. None of the drugs tested, including the RF-amide neuropeptide FF, demonstrated any affinity for GPR10.
Human PrRP-20 failed to alter basal or forskolin-stimulated levels of intracellular cyclic AMP in HEK293-GPR10 cells, suggesting that GPR10 does not couple via either Gs or Gi.
Functional studies using measurements of intracellular calcium confirmed that human and rat PrRP-20 and PrRP-31 are all potent, full agonists at the GPR10 receptor. The response was blocked both by thapsigargin, indicating mobilization of intracellular Ca2+ stores.
These studies indicate that [125I]-PrRP-20 is a specific, high affinity radioligand for GPR10. The availability of this radioligand binding assay will be a valuable tool for the investigation of the key features involved in PrRP binding and studies on the localization and function of GPR10.
Keywords: Prolactin-releasing peptide; GPR10 receptor; G-protein coupled receptor; UHR-1 receptor, [125I]-PrRP-20; radioligand binding
Introduction
The G-protein coupled receptor superfamily is characterized by seven distinct hydrophobic regions, which are each 20–30 amino acids in length and form transmembrane domains (Schoneberg et al., 1999). This superfamily is thought to contain over 800 members, many of which have been discovered recently with the development of genome and cDNA research. The rapid advancement in the discovery of putative G-protein coupled receptors had led to changes in the drug discovery process. An orphan receptor of unknown function can now be used as bait to go ‘fishing' for its ligand. This is in contrast to the traditional approach of identification of a ligand, characterization of the tissue pharmacology and subsequent cloning of a receptor, and has been described as a ‘reverse' pharmacology approach (Libert et al., 1991). G-protein coupled receptors have a good history as drug targets – several hundred drugs launched in the past three decades are directed at known G-protein coupled receptors (Wilson et al., 1998).
GPR10 is a novel G-protein coupled receptor identified by polymerase chain reaction and genomic DNA library screening (Marchese et al., 1995). It is a human counterpart of rat Unknown Hypothalamic Receptor-1 (UHR-1) (Welch et al., 1995) and shares low, but significant homology with the neuropeptide Y receptor family (30% at the amino acid level). A receptor initially termed hGR3 (Hinuma et al., 1998) is essentially identical to GPR10. Identification of a ligand for the receptor was carried out utilizing an arachidonic acid release assay using Chinese Hamster Ovary (CHO) cells transfected with hGR3/GPR10, in which bovine hypothalamic extracts were positive. Chromatographic purification of the extracts led to the identification of two peptide sequences with identical N-terminal regions, one of which appears to be a truncated form of the other (Hinuma et al., 1998). The peptides are both members of the structurally related RF-amide family which includes the morphine modulating peptide neuropeptide-FF (Yang et al., 1985). The generic peptide identified was shown to increase prolactin secretion from anterior pituitary cells derived from lactating rats and was hence named prolactin releasing peptide or PrRP. Studies showed that the PrRP preproprotein can be cleaved at two different positions to give rise to two forms of 31 or 20 amino acids; PrRP-31 and PrRP-20 respectively. Rat PrRP has also been identified and occurs as 31 or 20 amino acid forms; these peptides are highly conserved between species (Figure 1) (Hinuma et al., 1998).
Figure 1.

Amino acid sequences of short and long forms of prolactin releasing peptide (PrRP) in rat and human.
PrRP protein has been detected by immunocytochemistry in the rat hypothalamus (Maruyama et al., 1999), medulla oblongata (Iijima et al., 1999) and the pituitary (Matsumoto et al., 1999), whilst expression of PrRP mRNA has been shown in the hypothlamus and brainstem (Roland et al., 1999). The distribution of the peptide suggests that it may have other roles in addition to its prolactin-releasing ability. The mRNA for the corresponding receptor for PrRP is expressed in rat pituitary, cerebellum, brainstem, hypothalamus, thalamus and spinal cord (Welch et al., 1995; Hinuma et al., 1998; Roland et al., 1999), although autoradiographic studies have shown receptor binding only in thalamus and hypothalamus (Roland et al., 1999).
The signal transduction pathways for GPR10 are not clear, yet Kimura et al. (2000) have demonstrated that in both GH3 rat pituitary cells and primary cultures of rat anterior pituitary, PrRP activates extracellular signal-regulated protein kinase (ERK) in an almost wholly pertussis toxin (PTX) sensitive manner. This suggests that at least part of the coupling of GPR10 is through Gi/Go. They also demonstrated that PrRP causes activation of c-Jun N-terminal protein kinase (JNK). This response is protein kinase C (PKC) dependent, as it is inhibited by pre-incubation with phorbol esters and insensitive to PTX treatment.
We have used a radio-iodinated form of human PrRP-20 to characterize the binding of the GPR10 receptor stably expressed in HEK293 cells (Wilson et al., 1998). [125I]-PrRP-20 binds to the receptor with high affinity and displays high levels of specific binding. Saturation studies suggest [125I]-PrRP-20 binds to two sites with different affinities. Competition studies reveal that both rat and human PrRPs display high affinity for GPR10 receptors and are equipotent at displacing [125I]-PrRP-20. Measurements of intracellular cyclic AMP levels in HEK293-GPR10 cells reveal that PrRP-20 has no effect on either basal levels or forskolin-stimulated levels of cyclic AMP, suggesting that in this system GPR10 couples via neither Gs or Gi. Functional studies on the four peptides via measurements of intracellular calcium levels in HEK293-GPR10 cells reveal that all four peptides are equipotent, full agonists at the GPR10 receptor, with potencies which agree well with their binding affinities. This response is blocked by pre-incubation with thapsigargin, suggesting that it is a result of mobilization of intracellular calcium.
The availability of this radioligand binding assay will be a useful tool for the identification of potential non-peptide ligands which bind to the receptor. [125I]-PrRP-20 may be used for further investigations of the tissue distribution of GPR10 and UHR-1 via autoradiography studies.
Methods
Membrane preparation
Human embryonic kidney cells (HEK293) stably transfected with GPR10 were harvested with PBS, pelleted and stored at −80°C until further use. Membranes were prepared using a modification of the method of Miyamoto et al. (1994); all procedures were carried out at 4°C. In brief, cells were washed in 30 vols (w v−1) of PBS with 0.2 mM EDTA. The suspension was homogenized using an Ultra-Turrax homogenizer and the subsequent homogenates centrifuged at 39,000×g for 15 min. The resultant pellets were resuspended in 30 volumes of buffer containing 10 mM Na2CO3, 1 mM EDTA, 0.5 mM phenylmethylsulphonyl fluoride (PMSF), 1 μg ml−1 pepstatin and 1×Complete™ serine and cysteine protease inhibitor tablet 250 ml−1 (pH 7.4). The suspension was then homogenized and centrifuged at 1000×g for 10 min, the supernatant decanted and centrifuged at 48,000×g for 20 min. The resultant pellets were resuspended in buffer containing 20 mM Tris-HCl, 0.25 M sucrose, 2 mM EDTA, 0.5 mM PMSF, 1 μg ml−1 pepstatin and 1×Complete™ serine and cysteine protease inhibitor tablet 250 ml−1 (pH 7.4) to a volume of approximately 48×106 cells ml−1 and stored at −80°C until used.
[125I]-PrRP-20 binding assays
HEK293-GPR10 receptor expressing cell membranes were incubated with [125I]-PrRP-20 in buffer containing 20 mM Tris-HCl, 5 mM Mg-Acetate, 2 mM EGTA, 0.5 mM PMSF, 1 μg ml−1 pepstatin and 1×Complete™ serine and cysteine protease inhibitor tablet 250 ml−1 and 0.1% (w v−1) BSA (pH 7.4) at 25°C for 90 min. The total assay volume was 0.5 ml. The reaction was terminated by rapid filtration through Whatman GF/B glass fibre filters, followed by rapid washing of the filters with 5×1 ml aliquots of ice cold buffer containing 50 mM Tris-HCl, 10 mM MgCl2 (pH 7.4). Bound radioactivity was determined by gamma counting. Non-specific binding was defined as that remaining in the presence of 0.1 μM rat PrRP-31. Saturation studies were carried out by incubating membranes (4 μg protein well−1) with a range of concentrations of [125I]-PrRP-20 (0.01–5 nM). Specific binding data was analysed using the program Radlig (Biosoft) to provide estimates of KD and Bmax values. Protein content was assayed using the Bradford method (Bradford, 1976) using bovine serum albumin as a standard. Association kinetic studies were performed by measuring specific binding of [125I]-PrRP-20 (0.2 nM) at 0.5–90 min after addition of membranes (2 μg protein well−1). For dissociation studies, membranes were pre-incubated with [125I]-PrRP-20 (0.2 nM) for 90 min. Specific binding was then measured at 5–200 min after the addition of 0.1 μM PrRP-31. Kinetic data was analysed by GraFit (Erithacus Software) to provide estimates of Kon and Koff values. Competition studies were performed by incubating cell membranes (2 μg protein well−1) with [125I]-PrRP-20 (0.2 nM) and a range of concentrations of the test compound. Competition curves were analysed by non-linear least-squares fitting to a four parameter logistic equation by Microsoft Excel in order to determine IC50 values (Bowen & Jerman, 1995). Ki values were then derived from the IC50 values using a nominal KD value of 0.1 nM (which takes into account binding to both high and low affinity sites obtained from saturation studies) (Cheng & Prussoff, 1973). Results are given as means (±s.e.mean) of at least three independent experiments.
Calcium mobilization assays
Intracellular calcium was monitored using the fluorescent dye Fluo 4AM in a Fluorometric Imaging Plate Reader (FLIPR, Molecular Devices, U.K.). HEK293-GPR10 cells were cultured in poly-D-lysine coated 96-well microtitre plates 24 h before use at as a seeding density of 52,000 cells well−1. Prior to assay on FLIPR, cells were incubated with Fluo 4AM (1 μM) for 60 min at 37°C in Hank's buffered saline solution containing 0.1% BSA and 2.5 mM probenicid. Extracellular dye was then removed by washing three times with 150 μl Hank's buffered saline solution containing 2.5 mM probenicid without BSA. Compounds were tested for agonist activity in FLIPR by adding 40 μl of test solution to a plate volume of 120 μl at 37°C. Peak stimulation (minus basal) was plotted versus concentration of test compound and iteratively curve fitted using a four parameter logistic equation (Grafit, Erithacus Software) to assess agonist potency and maximal response.
Cyclic AMP assays
Intracellular cyclic AMP levels were determined with flashplates (SMP004, New England Nuclear). Briefly, HEK293-GPR10 cells were dispensed into 96-well flashplates (50,000 cells well−1) at 37°C and incubated with 0.5 mM isobutylmethylxanthene (IBMX) for 15 min before PrRP-20 and / or forskolin (30 μM) were added simultaneously to a final volume of 100 μl. After 15 min the incubation was terminated and plates processed according to the manufacturers instructions. Plates were counted in a TopCount scintillation counter (Packard).
Materials
HEK-293 cells stably transfected with GPR10 were produced by SmithKline Beecham Pharmaceuticals.
Human PrRP-20, human PrRP-31, rat PrRP-20 and rat PrRP-31 were synthesized by SmithKline Beecham Pharmaceuticals. Other drugs and chemicals were purchased from Sigma-Aldrich (Poole, U.K.), Tocris Cookson (Bristol, U.K.), Bachem (Saffron Walden, U.K.), Phoenix Pharmaceuticals (CA, U.S.A.), Boehringer Mannheim (Mannheim, Germany), Fisher Scientific (Loughborough, U.K.), BDH (Poole, U.K.) and Molecular Probes (Leiden, Holland). [125I]-PrRP-20 (specific activity 2000 Ci mmol−1) was synthesized by Amersham Pharmacia (Cardiff, U.K.).
Results
Saturation studies
Specific binding of [125I]-PrRP-20 represented more than 90% of total binding and was saturable, whereas non-specific binding increased linearly with radioligand concentration (Figure 2a). [125I]-human PrRP-31 was also tested and showed similar levels of specific binding (data not shown); only [125I]-PrRP-20 was used in subsequent studies. Analysis of binding data revealed that [125I]-PrRP-20 bound to two sites on the HEK293-GPR10 membranes with KD values of 0.026±0.006 and 0.57±0.14 nM, with respective Bmax values of 3010±400 and 8570±2240 fmol mg protein−1 (Figure 2b). Each individual experiment showed a two site fit to be statistically better than a one site fit (P<0.05, F-test).
Figure 2.
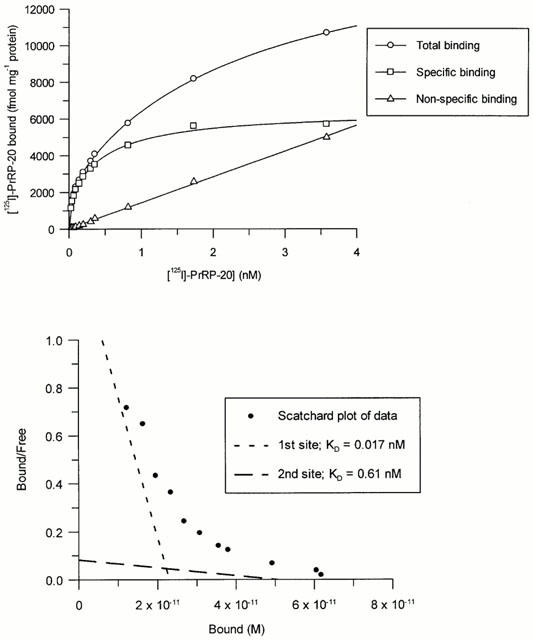
(a) Total, non-specific and specific binding of [125I]-PrRP-20 to membranes from HEK293 cells expressing GPR10 receptors with increasing radioligand concentration. Data represents a single experiment (each point determined in quadruplicate), which was replicated six times with similar results. (b) Scatchard transformation of the data from (a).
Kinetic studies
Association studies indicated that equilibrium was reached within 90 min. Although association curves appeared to be biphasic (Figure 3a and inset linear transformation), analysis of the data did not reveal any significant difference between single and two site fits. Dissociation of [125I]-PrRP-20 upon addition of excess cold PrRP-31 was very slow and appeared monophasic, with 50% of specific binding being dissociated after 120 min (Figure 3b and inset linear transformation). Single site analysis of these results gave a Kobs of 0.077±0.008 min−1 and a Koff of 0.0048±0.0002 min−1, leading to a calculated Kon value of 396±54 min−1 μM−1. The KD derived from this data was 0.012 nM, which is similar to the value for the high affinity site derived from the saturation studies.
Figure 3.
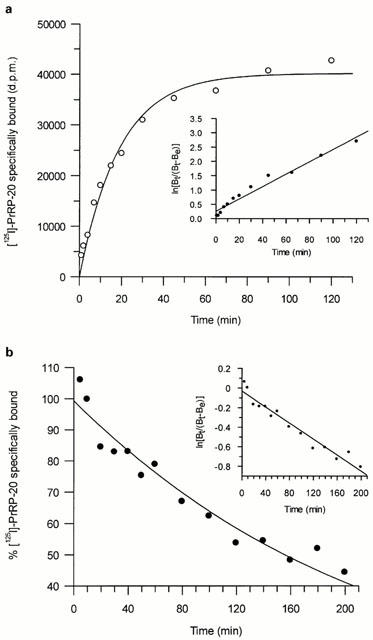
(a) Time course for association of [125I]-PrRP-20 binding to HEK293-GPR10 receptor expressing membranes. Data represents a single experiment, which was replicated five times with similar results. The inset shows the data transformed as a semi-log plot (correlation coefficient r=0.98), where Bt is the specific binding at time t and Be is the specific binding measured at equilibrium. (b) Time course for the dissociation of [125I]-PrRP-20 binding from HEK293-GPR10 receptor expressing membranes. Data represents a single experiment, which was replicated three times with similar results. The inset shows the data transformed as semi-log plot, where Bt is the specific binding at time t and Be is the specific binding measured at equilibrium.
Competition studies
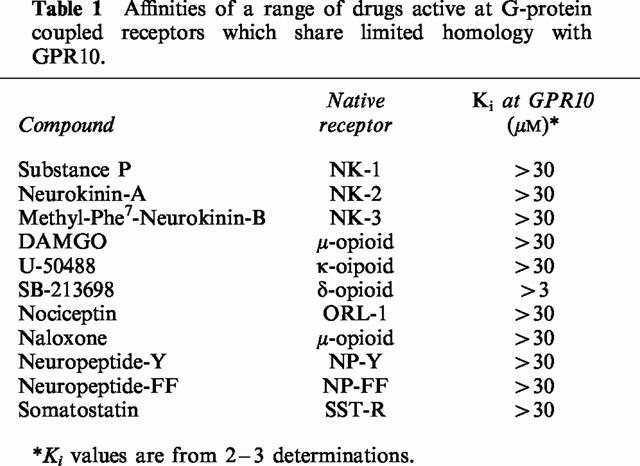
Human PrRP-20, human PrRP-31, rat PrRP-20 and rat PrRP-31 displayed high affinity for GPR10 receptors, with Ki values of 0.26±0.07, 1.03±0.41, 0.22±0.06 and 0.33±0.11 nM respectively (Figure 4). In contrast, a range of other drugs which are known ligands at receptors which share limited homology with GPR10 displayed no affinity for GPR10 receptors up to 30 μM (Table 1).
Figure 4.
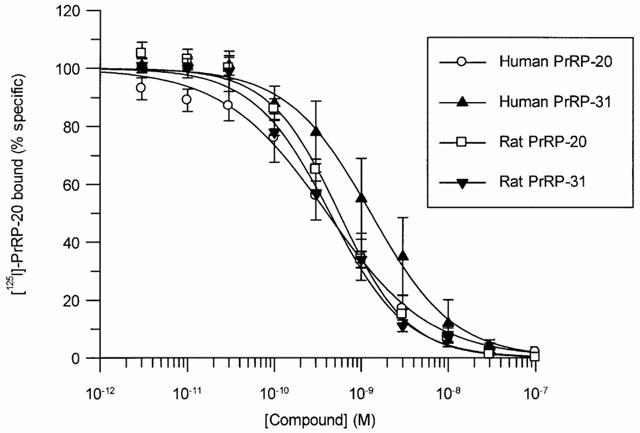
Competition for [125I]-PrRP-20 binding to HEK293-GPR10 expressing membranes by rat and human PrRP-20 and PrRP-31. [125I]-PrRP-20 (0.2 nM) was incubated in the presence of increasing concentrations of the compounds. Data are the mean of at least three independent experiments; vertical lines show s.e.mean.
Table 1.
Affinities of a range of drugs active at G-protein coupled receptors which share limited homology with GPR10.
Cyclic AMP assays
Ten μM PrRP-20 failed to elevate intracellular cyclic AMP levels in HEK293-GPR10 cells (2.96±0.42 pmoles 106 cells−1 compared to basal level of 4.09±0.57 pmoles 106 cells−1). Ten μM PrRP-20 also failed to significantly decrease cyclic AMP levels following stimulation with 30 μM forskolin (508±12.6 pmoles 106 cells−1 compared to control level of 502±17.8 pmoles 106 cells−1).
Calcium mobilization studies
Human PrRP-20, human PrRP-31, rat PrRP-20 and rat PrRP-31 were equipotent in stimulating Ca2+ mobilization in HEK293 cells stably transfected with the GPR10 receptor (Figure 5). EC50 values for the four peptides were 1.06±0.22, 1.54±0.26, 0.75±0.06 and 1.56±0.42 nM respectively. The response to human PrRP-20 was abolished by pre-incubation for 30 min with 1 μM thapsigargin with an IC50 value of 4.9±0.6 nM (data not shown).
Figure 5.
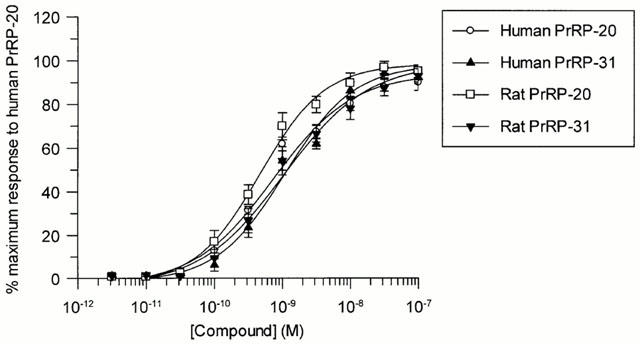
Concentration dependent stimulation of Ca2+ mobilization by human PrRP-20, human PrRP-31, rat PrRP-20 and rat PrRP-31 in HEK293 cells expressing GPR10. Data shown are the mean of six experiments; vertical lines show s.e.mean.
Discussion
These studies indicate that [125I]-PrRP-20 is a specific, high affinity radioligand for the GPR10 receptor. Saturation studies suggest the existence of two distinct binding sites for [125I]-PrRP-20 which bind with sub-nanomolar affinity (KD values of 0.026 and 0.57 nM respectively). Kinetic studies were unable to discern two distinct binding sites although a trend towards biphasic association was observed. Single site analysis of the data produced a KD of 0.012 nM, which is similar to the value for the higher affinity site obtained from saturation studies. The KD of the higher affinity site is similar to that reported by Hinuma et al. (1998) for bovine PrRP-31 binding to hGR3 / GPR10 and UHR-1. The KD of the lower affinity site is similar to the affinities of human and rat PrRP-20 and PrRP-31 determined by competition for of [125I]-PrRP-20 binding.
Studies on HEK293-GPR10 cells using Flashplate technology showed that human PrRP-20 had no effect on basal levels of intracellular cyclic AMP. This suggests that GPR10 does not couple through Gs, which would activate adenylyl cyclase to increase the cyclic AMP concentration. Additionally, PrRP-20 failed to decrease cyclic AMP levels after stimulation with forskolin, indicating that GPR10 does not couple via Gi (which would inhibit adenylyl cyclase to lower cyclic AMP levels). In calcium mobilization studies, all four PrRP peptides were shown to be potent, full agonists at the GPR10 receptor. EC50 values for these peptides agreed with their affinities as determined by competition for [125I]-PrRP-20 binding. Further calcium mobilization studies showed that the response to PrRP-20 was completely abolished by pre-incubation for 30 min with 1 μM thapsigargin, an inhibitor of the endoplasmic reticulum Ca2+-ATPase. This suggests that response to PrRP peptides is a result of mobilization of intracellular calcium stores. These data are in contrast to that of Kimura et al. (2000), which show that PrRP stimulates extracellular signal-regulated protein kinase (ERK) in both GH3 rat pituitary tumour cells and primary cultures of rat anterior pituitary. They demonstrate that the effect is largely pertussis toxin sensitive, indicating that the greater part of the coupling is via Gi/Go. Additionally, they show that ERK activation is independent of intracellular calcium. However, they also demonstrate that activation of c-Jun N-terminal protein kinase (JNK) by PrRP is fully dependent on protein kinase C (PKC). This suggests the possibility of GPR10 also signalling through a Gq pathway to stimulate PLC to produce diacylglycerol (DAG) and activate PKC. Comparison of the findings of Kimura et al. (2000) with ours suggest that the coupling of GPR10 is dependent on the cell system in which it expressed and the pathway being monitored.
All four forms of PrRP (human 20 and 31 and rat 20 and 31) bound to GPR10 receptors with high and similar affinity (Ki values ⩽1 nM). In contrast, no other ligand studied exhibited any affinity for the receptor up to 3 μM. This is in agreement with the low levels of homology between GPR10 and known G-protein coupled receptors. Indeed, the greatest identity (30% homology) is seen with the neuropeptide Y receptor family and here we have shown that neuropeptide Y has very low affinity for GPR10 (Ki>30 μM). In addition, the mammalian RF-amide neuropeptide-FF displays similarly low affinity for the receptor (Ki>30 μM). In further agreement with this, neuropeptide-FF was also found to be inactive at GPR10 in calcium mobilization studies (Wilson et al., 1998). These findings demonstrate the high selectivity of GPR10 for the prolactin releasing peptides.
PrRP mRNA and PrRP-like immunoreactivity have been detected in the brainstem, medulla oblongata and hypothalamus of rat brain (Iijima et al., 1999; Minumai et al., 1999; Matsumoto et al., 1999; Roland et al., 1999). Immunocytochemistry has also localized PrRP to the neural lobe of the pituitary (Maruyama et al., 1999). PrRP receptor mRNA expression has also been reported in rat pituitary, as well as in hypothalamus, thalamus, brainstem, cerebellum and spinal cord (Welch et al., 1995; Hinuma et al., 1998; Roland et al., 1999). These findings suggest that the PrRP-UHR-1/GPR10 system may influence CNS functions. The characterization of [125I]-PrRP-20 as a specific GPR10 ligand should allow further investigation of GPR10/UHR-1 binding in native tissues. Characterization of [125I]-PrRP-20 binding to UHR-1 expressed in HEK293 cells is currently in progress.
In conclusion, our results show that [125I]-PrRP-20 is a suitable ligand for studies on GPR10 receptors. The availability of the radioligand binding assay should assist in the identification of the key features involved in the binding of prolactin releasing peptides to GPR10 and allow further studies on the localization and function of these receptors.
Acknowledgments
We thank the Department of Biotechnology and Genetics for the cloning and expression of GPR10, the Department of Microbial and Cell Culture Sciences for production of transfected HEK293-GPR10 cells, the Department of Synthetic Isotope Chemistry for the initial iodination of [125I]-PrRP-20 and Jeannette Watson for critical review of the manuscript.
Abbreviations
- CHO
Chinese hamster ovary
- DAG
diacyglycerol
- EDTA
ethylene diamine tetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- ERK
extracellular signal-regulated kinase
- FLIPR
fluorometric light imaging plate reader
- HEK293
human embryonic kidney 293 cells
- IP3
inositol 1,4,5 trisphosphate
- JNK
c-Jun N-terminal protein kinase
- PKC
protein kinase C
- PMSF
phenylmethylsulphonyl fluoride
- PrRP
prolactin releasing peptide
- PTX
pertussis toxin
- UHR-1
unknown hypothalamic receptor-1
References
- BOWEN W.P., JERMAN J. Nonlinear regression using spreadsheets. Trends Pharmacol. Sci. 1995;16:413–417. doi: 10.1016/s0165-6147(00)89091-4. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- HINUMA S., HABATA Y., FUJII R., KAWAMATA Y., HOSOYA M., FUKUSUMI S., KITADA C., MASUO Y., ASANO T., MATSUMOTO H., SEKIGUCHI M., KUROKAWA T., NISHIMURA O., ONDA H., FUJINO M. A prolactin-releasing peptide in the brain. Nature. 1998;393:272–276. doi: 10.1038/30515. [DOI] [PubMed] [Google Scholar]
- IIJIMA N., KATAOKA Y., KAKIHARA K., BAMBA H., TAMADA Y., HAYASHI S., MATSUDA T., TANAKA M., HONJYO H., HOSOYA M., HINUMA S., IBATA Y. Cytochemical study of prolactin-releasing peptide (PrRP) in the rat brain. NeuroReport. 1999;10:1713–1716. doi: 10.1097/00001756-199906030-00016. [DOI] [PubMed] [Google Scholar]
- KIMURA A., OHMICHI M., TASAKA K., KANDA Y., IKEGAMI H., HAYAKAWA J., HISAMOTO K., MORISHIGE K., HINUMA S., KURACHI H., MURATA Y. Prolactin-releasing peptide activation of the prolactin promoter is differentially mediated by the extracellular signal-regulated protein kinase and c-Jun N-terminal protein kinase. J. Biol. Chem. 2000;275:3667–3674. doi: 10.1074/jbc.275.5.3667. [DOI] [PubMed] [Google Scholar]
- LIBERT F., VASSART G., PARMENTIER M. Current developments in G-protein coupled receptors. Curr. Opin. Cell. Biol. 1991;8:218–223. doi: 10.1016/0955-0674(91)90142-l. [DOI] [PubMed] [Google Scholar]
- MARCHESE A., HEIBER M., NGUYEN T., HENG H.H.Q., SALDIVIA V.R., CHENG R., MURPHY P.M., TSUI L.-C., SHI X., GREGOR P., GEORGE S.R., O'DOWD B.F., DOCHERTY J.M. Cloning and chromosomal mapping of three novel genes, GPR9, GPR10 and GPR14 encoding receptors related to interleukin 8, neuropeptide Y and somatostatin receptors. Genomics. 1995;29:335–344. doi: 10.1006/geno.1995.9996. [DOI] [PubMed] [Google Scholar]
- MARUYAMA M., MATSUMOTO H., FUJIWARA K., KITADA C., HINUMA S., ONDA H., FUJINO M., INOUE K. Immunocytochemical localisation of prolactin-releasing peptide in the rat brain. Endocrinology. 1999;140:2326–2333. doi: 10.1210/endo.140.5.6685. [DOI] [PubMed] [Google Scholar]
- MATSUMOTO H., MURAKAMI Y., HORIKOSHI Y., NOGUCHI J., HABATA Y., KITADA C., HINUMA S., ONDA H., FUJINO M. Distribution and characterisation of immunoreactive prolactin-releasing peptide (PrRP) in rat tissue and plasma. Biochem. Biophys. Res. Commun. 1999;257:264–268. doi: 10.1006/bbrc.1999.0463. [DOI] [PubMed] [Google Scholar]
- MINUMAI S., NAKATA T., TOKITA R., ONODERA H., IMAKI J. Cellular localization of prolactin releasing peptide messenger RNA in the rat brain. Neurosci. Lett. 1999;266:73–75. doi: 10.1016/s0304-3940(99)00263-3. [DOI] [PubMed] [Google Scholar]
- MIYAMOTO Y., HABATA Y., OHTAKI T., MASUDA Y., OGI K., ONDA H., FUJINO M. Cloning and expression of a complementary DNA encoding the bovine receptor for pituitary adenylate cyclase-activating polypeptide (PACAP) Biochim. Biophys. Acta. 1994;1218:297–307. doi: 10.1016/0167-4781(94)90181-3. [DOI] [PubMed] [Google Scholar]
- ROLAND B., SUTTON S., WILSON S., LOU L., PYATI J., HUVAR R., ERLANDER M., LOVENBERG W. Anatomical distribution of prolactin-releasing peptide and its receptor suggests additional functions in the central nervous system and periphery. Endocrinology. 1999;140:5736–5745. doi: 10.1210/endo.140.12.7211. [DOI] [PubMed] [Google Scholar]
- SCHONEBERG T., SCHULTZ G., GUDERMANN T. Structural basis of G-protein coupled receptor function. Mol. Cell. Endocrinol. 1999;151:181–193. doi: 10.1016/s0303-7207(99)00017-9. [DOI] [PubMed] [Google Scholar]
- WELCH S.K., O'HARA B.F., KILDUFF T.S., HELLER H.C. Sequence and tissue distribution of a candidate G-coupled receptor cloned from rat hypothalamus. Biochem. Biophys. Res. Commun. 1995;209:606–613. doi: 10.1006/bbrc.1995.1543. [DOI] [PubMed] [Google Scholar]
- WILSON S., BERGSMA D.J., CHAMBERS J.K., MUIR A.I., FANTOM K.G.M., ELLIS C., MURDOCK P., HERRITY N.C., STADEL J.M. Orphan G-protein coupled receptors: the next generation of drug targets. Br. J. Pharmacol. 1998;125:1387–1392. doi: 10.1038/sj.bjp.0702238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG H.Y., FRATTA W., MAJANE E.A., COSTA E. Isolation, sequencing, synthesis and pharmacological characterisation of two brain neuropeptides that modulate the action of morphine. P.N.A.S. 1985;82:7757–7761. doi: 10.1073/pnas.82.22.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]