

Abstract
Apoptosis and mitochondrial dysfunction are thought to be involved in the aetiology of neurodegenerative diseases. We have tested an orally active anti-apoptotic molecule (CGP 3466B) that binds to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in an animal model with motoneuron degeneration, i.e. a mouse mutant with progressive motor neuronopathy (pmn).
In pmn/pmn mice, CGP 3466B was administered orally (10–100 nmol kg−1) at the onset of the clinical symptoms (2 weeks after birth). CGP 3466B slowed disease progression as determined by a 57% increase in life-span, preservation of body weight and motor performance.
This improvement was accompanied by a decreased loss of motoneurons and motoneuron fibres as well as an increase in retrograde transport. Electron microscopic analysis showed that CGP 3466B protects mitochondria which appear to be selectively disrupted in the motoneurons of pmn/pmn mice.
The data support evaluation of CGP 3466B as a potential treatment for motor neuron disease.
Keywords: Apoptosis, motoneuron disease, pmn mice, non-peptidic molecule, mitochondria, CGP 3466B, GAPDH
Introduction
Motor neuron disease is a general term that groups various diseases with a common manifestation, that is the death of motoneurons in the spinal cord, brain stem and cortex. Amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) are the most frequent forms of motor neuron disease and in general have a very poor prognosis. Neurotrophic factors have been considered as a possible treatment for these diseases because of their ability to act on neuronal survival and to prevent apoptosis in various injury paradigms. However their clinical potential has so far been disappointing probably because of their inability to cross the blood brain barrier (BBB) (for reviews see Rothstein, 1996; Morrison & Morrison, 1999). A new surge of studies has suggested a potential of small molecules with ‘trophic' activity which not only cross the BBB but are also orally bioavailable.
One of these is riluzole which was originally shown to inhibit glutamate release in the CNS and subsequently to rescue motoneurons in an animal model with familial amyotrophic lateral sclerosis (FALS) (Gurney et al., 1996). Riluzole is now used as a standard therapy in the treatment of ALS (Wokke, 1996). Other small organic molecules such as CEP-1347 promote motoneuron survival in vitro and in vivo (Maroney et al., 1998; Glicksman et al., 1998). SR57746A, another nonpeptide compound, improves motor function in the pmn/pmn (progressive motor neuronopathy) mouse following oral treatment (Duong et al., 1998).
Here we report on the ability of CGP 3466B, an orally delivered anti-apoptotic compound, to act as a neuroprotective agent in a mouse mutant (pmn) with a spontaneous recessive mutation resulting in a loss of motoneurons and motor fibres. CGP 3466B is structurally related to both classical tricyclic antidepressent agents and monoamine oxidase inhibitors but lacks the pharmacological properties relevant to their anti-depressant effects (Kragten et al., 1998). It has been shown to rescue human neuroblastoma and PC-12 cells from apoptotic death (Kragten et al., 1998; Carlile et al., 2000) and also cerebellar granule cells in vitro from death induced by cytosine arabinoside (Paterson et al., 1998a), and hippocampal pyramidal cells from ischaemic and kainate-induced cell death in vivo (Paterson et al., 1998b).
In pmn/pmn mice, CGP 3466B improves several parameters of the disease. It prevents mitochondrial disruption, stimulates axonal transport and concomitantly slows down motoneuron cell death and axonal degeneration. These effects lead to a 57% increase in the life-span of the animals as well as an improvement of motor function and preservation of body weight.
Methods
pmn/+ breeder mice were obtained from the laboratory of Dr J.L. Guénet (Institut Pasteur, Paris, France); the litters (usually 6–8 animals) contained statistically 25% pmn/pmn homozygotes. The pmn/pmn phenotype was determined by the inability to grasp with their hindlimbs (Sagot et al., 1995a). Animals were treated immediately after their identification (i.e. beginning at days 13–15). Pmn/pmn mice had access to food and water ad libitum during all the experiments. The overall study concerns 79 animals issued from 46 litters.
Administration
Dibenzo[b,f]oxepin-10-ylmethyl-methyl-prop-2-ynyl-amine (CGP 3466B; Novartis, Basel, Switzerland) was delivered orally by direct injection into the stomach (100 μl mouse−1) using a 1 ml glass syringe with a 15G rounded-end needle (Meditec Basel, Switzerland). CGP 3466B was initially dissolved in 20% ethanol at 1 μg μl−1 and diluted thereafter in water to achieve the final concentration. Each animal was weighed before treatment and the administered dose calculated appropriately. Four concentrations of CGP 3466B have been tested with a delivery schedule of three times per week (Monday, Wednesday and Friday) at 1 nmol kg−1 (six mice), 10 nmol kg−1 (seven mice), 100 and 1000 nmol kg−1 (nine mice each). The vehicle group (seven animals) received 0.5% ethanol (i.e. the highest concentration of ethanol obtained in the CGP 3466B cohorts). An additional group of 12 mice treated with 100 nmol kg−1 of CGP 3466B was done with a delivery schedule of five times a week (Monday, Tuesday, Wednesday, Thursday and Friday). For the riluzole treatment group (six mice), the compound (RBI, Natick MA, U.S.A.) was diluted in an acidic solution as described by Gurney et al. (1996) and used at a final concentration of 100 μg ml−1 in the drinking water; the solution was changed three times per week.
After the identification of the pmn/pmn mice, treatments were randomly attributed between CGP 3466B (10 and 100 nmol kg−1), 0.5% ethanol, riluzole and untreated pmn/pmn mice in a first cohort of experiments and between CGP 3466B at 1 nmol kg−1, 100 nmol kg−1 (five times per week) and 1000 nmol kg−1 in the second cohort.
Life-span
Life-span results were submitted to a Kaplan-Meier test (SPSS for Windows version 5.0; equality of survival distribution) as previously described (Sagot et al., 1995a). The survival of untreated pmn/pmn mice (n=22) was compared to those with mice treated with CGP 3466B 100 nmol kg−1 (five times per week, n=12; three times per week, n=9) and with vehicle (0.5% EtOH, n=8).
Body weight evolution
The body weight evolution from the 15th day until death was expressed as the cumulative sum of the variation in the percentage of the initial body weight:
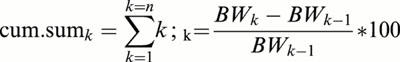 |
where BW=body weight; BWk1=body weight at the beginning of the treatment. The animals were always weighed at the end of the afternoon (16.00 h). Body weight evolution was compared between vehicle-treated (n=7), riluzole (n=6) and CGP 3466B-treated mice (100 nmol kg−1) (three times per week, n=9; five times per week, n=12).
Behavioural test
Mice were tested every 2 days for their ability to walk; this was determined by a direct observation of their capacity to walk upright instead of dragging their hind limbs or walking on the backs of their toes. Results are expressed as a percentage of the animals passing the test as a function of age. The values in 12-day-old animals were obtained before the beginning of the treatment. Animals were treated either with vehicle (0.5% EtOH) or with CGP 3466B (100 nmol kg−1) administered three (n=8) or five times (n=12) per week.
Fluorogold labelling of motoneurons
Mice were anaesthetized with 250 μg tribromoethanol (Aldrich) gm−1 body weight in PBS and the left sciatic nerve was sectioned. The proximal stump was capped with a tube containing a fluorogold solution (Fluorochrome, Englewood, CO, U.S.A., final concentration 2.5%) in PBS (75 mM, pH 7.4). Twenty-four hours after surgery, the animals were perfused transcardially with 4% paraformaldehyde in PBS and then processed for histological analysis; labelled motoneurons were counted using a fluorescent microscope. Pmn/pmn mice were treated either with vehicle (n=7) or with CGP 3466B (100 nmol kg−1 p.o.) (n=5). Control wild-type mice of the same age were treated in the same manner with vehicle (n=13) or with CGP 3466B (n=5).
Histological procedures
Moribund mice were deeply anaesthetized in pentobarbital (30 mg kg−1) and perfused. The brainstem was embedded in paraffin and serially sectioned (8 μm). Following cresyl violet staining, every fifth section was counted and only motoneurons with prominent nucleoli were included. No correction factor was used for the expression of motoneuron counts. The myelinated axons in the phrenic nerve were estimated as previously described (Sagot et al., 1995a) except that the sections were stained with Sudan Black instead of cresyl violet.
Electron microscopy
Mice were deeply anaesthetized with pentobarbital (30 mg kg−1) and perfused transcardially with 3% glutaraldehyde in 0.1 M phosphate buffer at pH 7.4. The lumbar part of the spinal cord was dissected and processed as described by Buchs et al. (1994). Ultra-thin sections of approximately 60 nm were stained and examined with a PHILIPS CM10 electron microscope at 80 kV. Swollen mitochondria with a large vacuole or a clear disruption of the cristae were counted as abnormal. Four mice were analysed per group in a ‘blind' manner, M represents the total number of mitochondria that were counted. In pmn/pmn mice, 37 motoneurons (M=2011) were analysed; in CGP 3466B-treated pmn/pmn mice, 38 motoneurons were analysed (M=2199) and in the control wild-type (non-pmn/pmn) mice, 22 motoneurons were analysed (M=1726).
Statistics
Multiple groups were compared using a one-way ANOVA with a Dunnett t-test. When two populations were compared, the unpaired Student's t-test was used. In all cases±refers to s.e.mean.
Results
CGP 3466B but not riluzole increases the survival of pmn/pmn mice
CGP 3466B and vehicle (0.5% EtOH) were administered orally three times per week (Monday, Wednesday and Friday). Pmn/pmn mice which received the vehicle alone did not survive significantly longer than untreated pmn/pmn. In contrast, at 10 nmol kg−1 as well as 100 nmol kg−1, CGP 3466B significantly slowed down the disease progression by 30% as compared to untreated or vehicle-treated pmn/pmn mice (P<0.05) (Figure 1).
Figure 1.
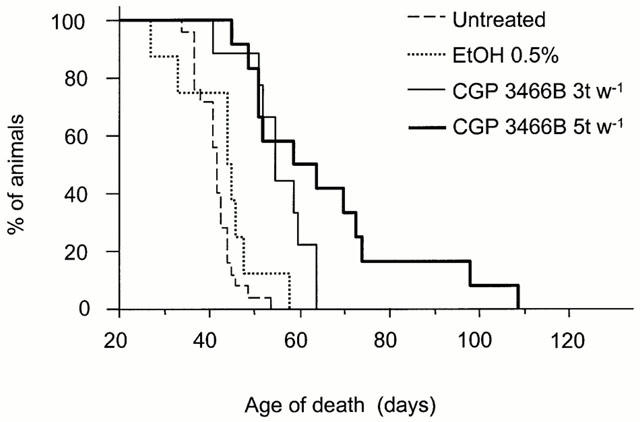
Survival of untreated pmn/pmn mice (n=22) and mice treated with CGP 3466B 100 nmol kg−1 (five times per week: 5 t w−1, n=12; three times per week: 3 t w−1, n=9) and with vehicle (0.5% EtOH, n=8). Note the highly significant effect of CGP 3466B (5 t w−1) on pmn/pmn survival (57% increase as compared to untreated mice, P<0.0001).
Since no difference was found between these two doses of CGP 3466B, a higher dose of CGP 3466B was used in order to determine whether it could increase the life-span even further (Table 1). Interestingly, at 1 μmol kg−1, the positive effects obtained at the lower doses were completely lost. Animals treated with a lower dose of CGP 3466B (1 nmol kg−1) did not survive longer than untreated or vehicle-treated pmn/pmn mice. As a result, the therapeutic window appeared to be in the range of 10 to 100 nmol kg−1. Other workers (Carlile et al., 2000) have also reported that CGP 3466B produced a bell-shaped concentration response curve in PC-12 cells undergoing apoptosis either due to withdrawal of NGF or of serum.
Table 1.
Life-span, motoneuron cell loss and axonal degeneration in pmn/pmn mice
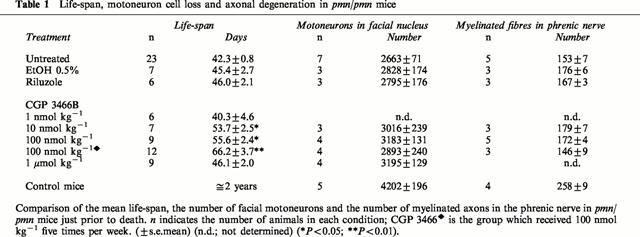
To test whether more frequent administration of CGP 3466B would further improve survival, mutant mice were treated with 100 nmol kg−1 five times per week (Monday, Tuesday, Wednesday, Thursday and Friday) instead of three times per week. Indeed, survival was prolonged by about 10 days by the former as compared to the latter treatment; two mice survived over 90 days (Table 1 and Figure 1). Overall there was a 57% increase in the life-span of the pmn/pmn mice treated with CGP 3466B five times per week as compared to untreated mice.
To compare the efficacy of CGP 3466B in pmn/pmn mice with a substance presently being used as a treatment for ALS, we tested the effect of riluzole on the life-span of pmn/pmn mice. Riluzole was added directly to the drinking water as described by Gurney et al. (1996). No effect on survival was observed (Table 1). Oral administration of riluzole (40 mg kg−1 every 2nd day) also was ineffective (data not shown).
CGP 3466B prevents weight loss of pmn/pmn mice
Following the appearance of the disease symptoms, the body weight of untreated or vehicle-treated mutant mice ceased to increase (Figure 2, figure shows vehicle treatment). As the disease progressed, the mice lost weight; during the last 3–5 days, a severe loss occurred which was pre-symptomatic of death. Pmn/pmn mice treated with CGP 3466B at 100 nmol kg−1 three times per week not only stabilized but also increased their body weight by an average of 20%; this effect was even more pronounced with a treatment of five times per week and persisted throughout the life-span of the animals (Figure 2). Some individual animals increased their weight by 50%. Interestingly, such an effect was not observed with riluzole (Figure 2) or other doses of CGP 3466B (1 nmol kg−1, 1 μmol kg−1) and even at 10 nmol kg−1 which had been shown to be effective on the life-span (data not shown).
Figure 2.
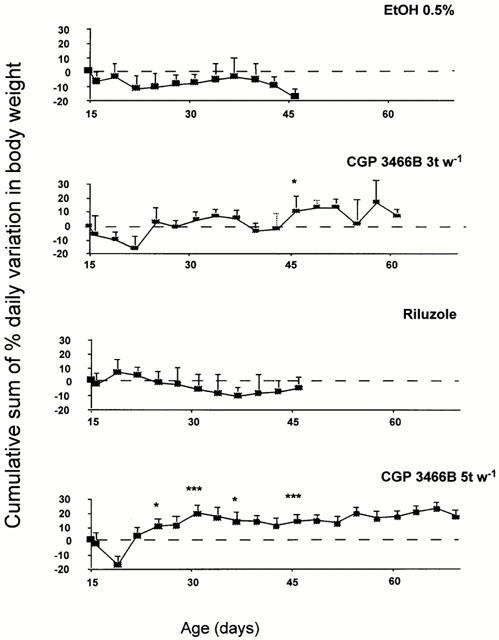
CGP 3466B prevented weight loss in pmn/pmn mice. In contrast to vehicle or riluzole-treated mice which lose body weight during the disease progression, CGP 3466B-treated mice (100 nmol kg−1; five times per week: 5 t w−1 (n=12), three times per week: 3 t w−1, n=9) regained and stabilized their body weight. From the 26th day until death, the cumulative sum of the daily variation in the body weight was significantly different between CGP 3466B (100 nmol kg−1; 5 t w−1; n=12) and EtOH (n=7) or riluzole-treated mice (n=6) (*P<0.05; ***P<0.005).
CGP 3466B preserves motor function in pmn/pmn mice
The effects of CGP 3466B were not restricted to life-span and body weight but also concerned motor function. With disease progression, untreated or vehicle-treated pmn/pmn mice gradually lost their ability to walk and dragged their hind limbs (Figure 3). At the end of their life-span, less than 20% of the mice were able to walk. When CGP 3466B was administered five times per week at 100 nmol kg−1, the surviving 62 day-old treated mice walked normally; at this age all mice in the other treatment groups had already died.
Figure 3.
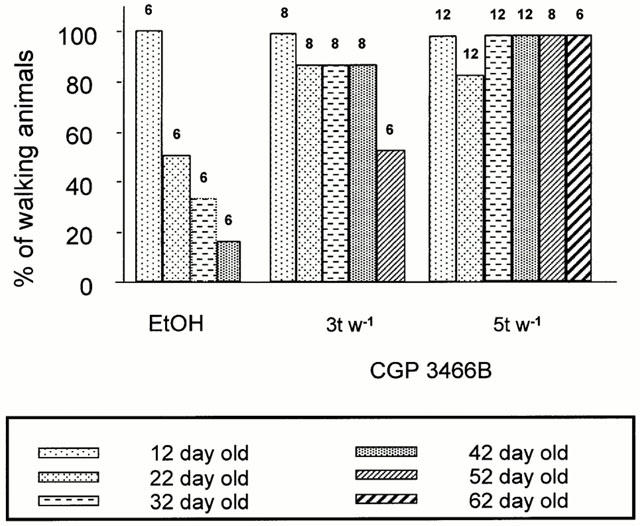
Preservation of motor function in CGP 3466B-treated mice (100 nmol kg−1) three times per week (3 t w−1) or five times per week (5 t w−1). The percentage of pmn/pmn mice that can walk was plotted as a function of age and according to the treatment they received. Animals were treated either with vehicle (0.5% EtOH) or with CGP 3466B (100 nmol kg−1) three (n=8) or five times (n=12) per week. The numbers above the columns indicate the number of animals in each group at a given time.
CGP 3466B stimulates retrograde labelling of motoneurons in pmn/pmn mice
We then examined the ability of a single oral administration of the compound to compensate for the impairment in axonal transport which has previously been reported in pmn/pmn mice (Sagot et al., 1998). Seven minutes after administration of CGP 3466B (100 nmol kg−1 p.o.) the sciatic nerve was cut and a tube containing the retrograde tracer, fluorogold, was applied onto the nerve. Twenty-four hours later, a significant increase in the number of labelled motoneurons was observed in CGP 3466B-treated as compared to vehicle-treated pmn/pmn mice (733±41, n=5 versus 548±46, n=7, P<0.05). No effects were observed in control wild-type animals of the same age (1288±70, n=5 for CGP 3466B-treated controls, versus 1205±43, n=13, for vehicle-treated controls).
CGP 3466B slows but does not abolish motoneuron cell loss in pmn/pmn mice
To evaluate whether increased survival and improved motor function were accompanied by an increase in motoneuron survival in CGP 3466B-treated mice, motoneurons in the facial nucleus and myelinated fibres in the phrenic nerve were counted. In moribund mice, the numbers of motoneurons or myelinated axons in the phrenic nerve were not significantly different between the various groups (see Table 1). However, when the results for the motoneurons were plotted as a function of age, the distribution domain of the untreated, vehicle and riluzole-treated animals was clearly different from the domain encompassed by CGP 3466B-treated animals (Figure 4). Indeed, a linear regression analysis showed that the loss of motoneurons per day was −16.3 for CGP 3466B-treated mice (χ2=0.56; P<0.001) versus −35.7 for untreated/vehicle/EtOH treated mice (χ2=0.76; P<0.001); furthermore, there was a loss of myelinated axons in the phrenic nerve per day with a linear regression of −1.2 for CGP 3466B-treated mice (χ2=0.91; P<0.001) versus −2.0 for untreated/vehicle/EtOH treated mice (χ2=0.88; P<0.001). This strongly suggests that the rate of loss of motoneurons and myelinated fibres in CGP 3466B-treated animals was decreased but not arrested.
Figure 4.
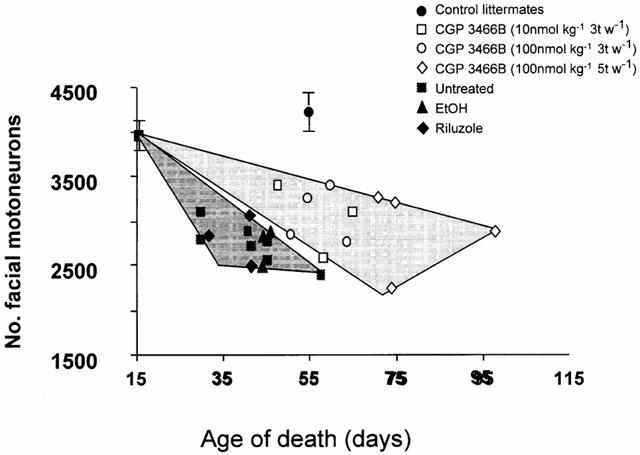
CGP 3466B slows down but does not stop facial motoneuron cell death. The number of facial motoneurons was determined just prior to death. A distribution domain was drawn which started at the value obtained in 16-day-old animals and which contained all values for untreated (n=7), EtOH (n=3) and riluzole (n=3) treated pmn/pmn mice; another domain was drawn for the values obtained from pmn/pmn mice treated with CGP 3466B (10 nmol kg−1, 3 t w−1, n=3); 100 nmol kg−1, 3 t w−1, n=4 and 100 nmol kg−1, 5 t w−1, n=4). Despite the absence of difference in total number of facial motoneurons, the two domains did not overlap suggesting that CGP 3466B slowed down the motoneuron cell loss in pmn/pmn mice. This was confirmed (see Results) by counting the number of facial motoneurons in age-mated pmn/pmn mice untreated or treated with CGP 3466B. Control littermates (n=5).
To confirm this observation, another group of CGP 3466B-treated mice (100 nmol kg−1, five times per week) was perfused at an age when the mice normally die (i.e. 42 days of age) and the number of motoneurons in the facial nucleus counted. In this case, the number of facial motoneurons in CGP 3466B-treated mice was significantly higher than in age-mated untreated, vehicle or riluzole-treated pmn/pmn mice (respectively, 3346±127, n=6 versus 2663±71, n=6; 2828±174, n=3 and 2795±176, n=3. P<0.05 for all). These results confirm that CGP 3466B enhanced motoneuron survival in pmn/pmn mice.
CGP 3466B improves mitochondrial integrity in pmn/pmn mice
Electron microscopic analysis of spinal cord sections revealed that in 42 day-old untreated pmn/pmn mice, a large number of the mitochondria displayed an interrupted mitochondrial membrane. By analysing 2011 mitochondria in 37 motoneurons, we could show that 47.2±4.5% displayed an abnormal appearance (Figures 5A, B and 6A). Meanwhile, in cells other than motoneurons, only 5.5±1% of the mitochondria were abnormal (32 cells and 1006 mitochondria analysed). In healthy mice, the percentage of abnormal mitochondria differed only slightly between motoneurons (17.7±3.2% in 22 motoneurons and 1726 mitochondria analysed) and other cells (3.4±0.5% in 22 cells and 683 mitochondria analysed; P<0.01), probably reflecting the relative fragility of motoneurons in general (Figures 5C and 6A).
Figure 5.
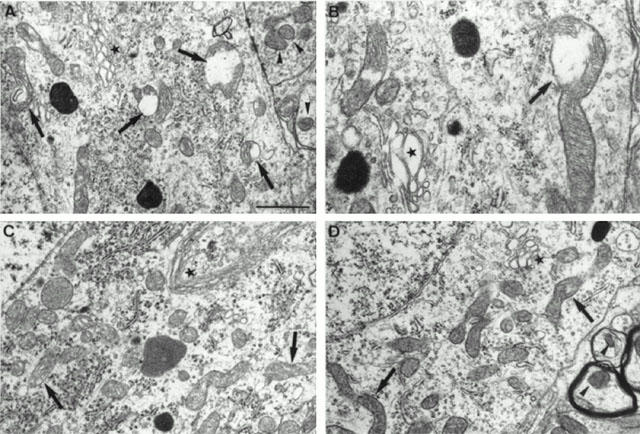
Electron microscopic photographs of lumbar spinal cord motoneurons from untreated pmn/pmn mice (A,B), control wild-type (C) and CGP 3466-treated pmn/pmn mice (100 nmol kg−1, 5 t w−1) (D). (A,B) In 42 day-old untreated pmn/pmn mice, a large number of mitochondria in the motoneurons displayed interrupted mitochondrial membranes (arrows) meanwhile outside the motoneurons they were well-preserved (arrowhead). Note the very large and swollen Golgi apparatus (star). (C) In age-mated healthy control wild-type mice, the mitochondria were well-preserved inside the motoneurons (arrow) as well in the other cells. Note the thin shape of the Golgi apparatus (star). (D) In 42-day-old pmn/pmn mice treated with CGP 3466B, most of the mitochondria were in excellent condition in the motoneuron cell body as well as in the dendritic structures. Despite the treatment with CGP 3466B the Golgi apparatus remained very swollen (star). Scale bar: 1 μm for A, C, D and 0.5 μm for B.
Figure 6.
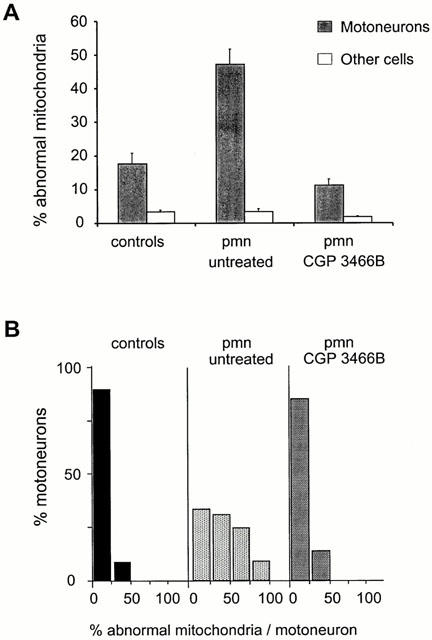
CGP 3466B protects mitochondria in pmn/pmn mice. The number of damaged mitochondria in lumbar spinal cord motoneurons was counted in electron microscopic photographs of ultra-thin sections. (A) The percentage of abnormal mitochondria in motoneurons and other cell types in control wild-type, pmn/pmn and pmn/pmn CGP 3466B-treated mice. (B) The results were expressed as a percentage of spinal motoneurons belonging to a defined class (i.e. 0–25, 25–50, 50–75, 75–100% of broken mitochondria).
In age-matched pmn/pmn mice treated with CGP 3466B, the percentage of abnormal mitochondria was not significantly different from the value obtained in control wild-type mice (11.2±1.9% in 38 motoneurons and 2199 mitochondria analysed) (P<0.05). The values for cells other than motoneurons were lower (1.8±0.3% in 38 cells and 1024 mitochondria analysed) (Figure 6A).
In untreated pmn/pmn mice, 35% of the motoneurons contained more than 50% abnormal mitochondria (2011 mitochondria analysed in 37 cells) (Figure 6B). In contrast, in age-mated pmn/pmn mice treated with CGP 3466B (100 nmol kg−1, five times per week), more than 85% of the motoneurons showed less than 25% broken mitochondria (2199 mitochondria analysed in 38 cells), a profile close to that obtained in control wild-type mice (1726 mitochondria analysed in 22 cells) (Figures 5D and 6B).
To eliminate the possibility that the mitochondrial pattern observed in pmn/pmn mice resulted from an artifact related to the perfusion fixation, spinal cord slices (1 mm in thickness) in control wild-type and pmn/pmn mice were fixed only by immersion in the fixative. These two fixation procedures produced similar results with respect to the number of abnormal mitochondria in both wild-type controls and in pmn/pmn (data not shown).
Discussion
In the present study we have examined the effects of CGP 3466B, an orally active, nonpeptidic molecule, in an animal model with motoneuron degeneration (pmn/pmn mice). In these mice, oral delivery of CGP 3466B was not only able to increase the life-span by 57% but also to maintain the body weight and certain motor functions such as the ability to walk. Although more animals must be examined, there was a tendancy towards a decrease in both the loss of motoneurons and myelinated axons, a preservation of motoneuronal mitochondria and an improved rate of axonal transport.
Pmn/pmn mice as a model for motoneuron diseases
Although the pmn mutation does not have any human equivalent in the family of motor neuron diseases to date (Brunialti et al., 1995), it shares several analogies to human motor neuron diseases as well as to other mouse models of ALS. These include impairment of axonal transport (Sagot et al., 1998) and specific motoneuron degeneration (Zhang et al., 1997; for review Sagot et al., 1997). The relevance of motoneuron apoptosis in pmn/pmn mice remains an open issue since there is no clear consensus about the type of cell death in ALS nor in other mouse models of motor neuron disease. In fact in SOD transgenic mice, the phenotypic expression of the disease depends on the particular mutation that is expressed (Gurney et al., 1994; Ripps et al., 1995; Brujin et al., 1997).
In this study, we additionally show specific disruption of mitochondria in the motoneurons in pmn/pmn mice. Since the non-neuronal cells in the spinal cord do not demonstrate such morphological alterations, these results would suggest that mitochondrial impairments in the motoneurons may be associated with the aetiology of the disease process. Massive mitochondrial motoneuron degeneration has also been found in SOD transgenic mice (Kong & Xu, 1998) despite the fact that the cell death in these animals is not driven by an apoptotic mechanism (Gurney et al., 1994). Nevertheless, it is known that mitochondrial abnormalities can lead to various types of cell death depending on the cellular context (for reviews see Lemasters et al., 1998; Montal, 1998; Kroemer et al., 1998). Due to the highly reproducible, short life span, the selective mitochondrial degeneration and the apparent apoptotic loss of motoneurons (Sendtner et al., 1997), the pmn/pmn mouse qualifies as a useful model for examining substances with anti-apoptotic properties.
Comparison of the effects of CGP 3466B to those of neurotrophic factors in pmn/pmn mice
In previous studies, we have examined the effects of two neurotrophic factors, ciliary neurotrophic factor (CNTF) (Sagot et al., 1995a) and glial cell-derived neurotrophic factor (GDNF) (Sagot et al., 1996) and also the effects of the anti-apoptosis molecule Bcl-2 (Sagot et al., 1995b) on the life-span, the number of motoneuron cell bodies and myelinated axons and the retrograde transport properties of pmn/pmn mice. We observed that CNTF was able to improve all of these different parameters in these mutant mice as opposed to GDNF and Bcl-2 which could only act on rescuing the cell bodies of the motoneurons and not the axons, thus perhaps accounting for the incapacity of these two proteins to act on the life-span of the animals. These experiments demonstrate that there may be separate and distinct intracellular mechanisms responsible for preventing degeneration of the cell body and that of the axon.
In this study, we show that CGP 3466B acts in a manner similar to that of CNTF. Whereas CNTF had to be delivered by polymer-encapsulated cells (Sagot et al., 1995a), CGP 3466B was active following a simple oral administration technique. Both molecules could act on rescuing the motoneuron cell bodies and the myelinated axons which thus may reflect their ability to also increase the life-span of the animals. Furthermore, these studies show that CGP 3466B is capable of acting on the intracellular processes which are responsible for preventing the cell death of the motoneuron soma and also those which can impede the degeneration associated with the axons. As such CGP 3466B has more therapetuic potential as compared to GDNF and Bcl-2.
Other small molecular weight neuroprotective agents have recently been described. For example, the molecule BN 80933 has combined properties of inhibiting neuronal nitric oxide synthase and lipid peroxidation (Chabrier et al., 1999); it has been shown to reduce brain damage induced by head trauma in mice, global ischaemia in gerbils and transient focal ischaemia in rats (Chabrier et al., 1999). We have recently shown that oral administration of this molecule in pmn/pmn mice can improve their life-span by 40% (Kato et al. unpublished data).
Another non-peptide neurotrophic compound SR 57746A that is orally active can improve the life-span of pmn/pmn mice by 50% more than vehicle-treated controls. This compound was also capable of improving the motor function and to increase the surface area of the sciatic nerve in these same animals (Duong et al., 1998).
The neuroprotective properties of these three substances (CGP 3466B, BN 80933 and SR 57746A) underscores the importance of small molecular weight molecules that can pass the blood brain barrier and which can be administered orally. It is possible that the future of neuroprotective agents lies in the use of small molecules rather than in large molecular weight proteins such as neurotrophic factors which have limited clinical potential due to their inability to cross the blood brain barrier and also to their short half-life in the circulation.
Possible mechanisms of CGP 3466B action in pmn/pmn mice?
We do not know by which mechanism CGP 3466B protects mitochondria in motoneurons. However, deprenyl, a compound related to CGP 3466B, stabilizes the mitochondrial membrane potential and thus prevents apoptosis associated with serum or NGF withdrawal in PC-12 cells (Wadia et al., 1998). Deprenyl also increases the expression of Bcl-2 (Tatton & Chalmers-Redman, 1996) which is known to facilitate the closure of permeability transition pores in mitochondria (Zamzami et al., 1998). In addition, deprenyl has been shown to decrease hydroxyl radicals (Wu et al., 1993) and also to increase SOD activity in rodent striatum exposed to MPTP (Kitani et al., 1994). However, deprenyl was less effective in pmn/pmn mice than CGP 3466B (Sagot et al. unpublished observations). This may relate to its metabolism to amphetamine and methamphetamine, both of which antagonize deprenyl's effects on neuronal survival (Tatton et al., 1996).
Whether in pmn/pmn mice the action of CGP 3466B on mitochondria is mediated by its interaction with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a multi-functional enzyme (Kragten et al., 1998), is not known. However, the recent reports suggesting a role of this enzyme in neuronal apoptosis and the protective action of CGP 3466B in various in vitro or in vivo paradigms where apoptosis and GAPDH are implied (Wadia et al., 1998; Paterson et al., 1998a,1998b; Carlile et al., 2000), argue in favour of this hypothesis. Indeed, GAPDH mRNA and protein are up-regulated during apoptosis and antisense oligonucleotides directed against GAPDH can delay cell death (Ishitani et al., 1996). GAPDH appears to act as a mediator of cell death by translocating to the nucleus (Sawa et al., 1997). Recently it has been shown that CGP 3466B can decrease the nuclear accumulation of GAPDH in PC-12 cells subjected to an apoptotic stimuli and simultaneously convert GAPDH from its usual tetrameric form to a dimeric form (Carlile et al., 2000).
At higher doses of CGP 3466B the beneficial effect observed in pmn/pmn mice is lost. Similar findings have been observed by Carlile et al. (2000) who showed that higher doses of this same molecule (10−5 M) were less efficient at preventing apoptosis in PC-12 cells as compared to lower doses (10−9 M). This indicates that other targets than GAPDH or other functions of GAPDH were affected at these high concentrations. Finally, since GAPDH is ubiquitously distributed, we do not exclude the possibility that CGP 3466B affects cells other than motoneurons.
Taken together, these results suggest that CGP 3466B plays an important role in preventing neuronal degeneration via mechanisms related to the energy status of the motoneurons perhaps via the ubiquitous enzyme GADPH. Because mitochondrial dysfunction and apoptotic cell death are thought to be involved in other neurodegenerative disorders such as Hungtinton's disease and Parkinson's disease (for review Beal, 1996; Stefanis et al., 1997; Leist & Nicotera, 1998), the present results with CGP 3466B would merit an investigation on its role in other in vivo situations where neuronal degeneration has the same characteristics.
Acknowledgments
This work was supported by the Association Française contre les Myopathies (France) and the Swiss National Science Foundation. We thank Dr J.C. Martinou for reading this manuscript and F. Pillonel for the assistance with the photographs. Nicolas Toni was sponsored by a grant from the Swiss National Foundation (N8 31-40815-94) to Dr D. Muller.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- BBB
blood brain barrier
- CNTF
ciliary neurotrophic factor
- FALS
familial amyotrophic lateral sclerosis
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GDNF
glial-cell derived neurotrophic factor
- pmn
progressive motor neuronopathy
- SMA
spinal muscular atrophy
References
- BEAL M.F. Mitochondria, free radicals, and neurodegeneration. Curr. Opin. Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- BRUIJN L.I., BECHER M.W., LEE M.K., ANDERSON K.L., JENKINS N.A., COPELAND N.G., SISODIA S.S., ROTHSTEIN J.D., BORCHELT D.R., PRICE D.L., CLEVELAND D.W. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- BRUNIALTI A.L., POIRIER C., SCHMALBRUCH H., GUENET J.L. The mouse mutation progressive motor neuronopathy (pmn) maps to chromosome 13. Genomics. 1995;29:131–135. doi: 10.1006/geno.1995.1223. [DOI] [PubMed] [Google Scholar]
- BUCHS P.A., STOPPINI L., PARDUCZ A., SIKLOS L., MULLER D. A new cytochemical method for the ultrastructural localization of calcium in the central nervous system. J. Neurosci. Methods. 1994;54:83–93. doi: 10.1016/0165-0270(94)90162-7. [DOI] [PubMed] [Google Scholar]
- CARLILE G.W., CHALMERS-REDMAN M.E., TATTON N.A., PONG A., BORDEN K.E., TATTON W.G. Reduced apoptosis after nerve growth factor and serum withdrawal: conversion of tetrameric glyceraldehyde-3-phosphate dehydrogenase to a dimer. Mol. Pharmacol. 2000;57:2–12. [PubMed] [Google Scholar]
- CHABRIER P.-E., AUGUST M., SPINNEWYN B., AUVIN S., CORNET S., DEMERLE-PALLARDY C., GUILMARD-FAVRE C., MARIN J.-G., PIGNOL B., GILLARD-ROUBERT V., ROUSSILLOT-CHARNET C., SCHULZ J., VIOSSAT I., BIGG D., MONCADA S. BN 80933, a dual inhibitor of neuronal nitric oxide synthase and lipid peroxidation: A promising neuroprotective strategy. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10824–10829. doi: 10.1073/pnas.96.19.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUONG F., FOURNIER J., KEANE P.E., GUENET J.L., SOUBRIE P., WARTER J.M., POINDRON P. The effect of the nonpeptide neurotrophic compound SR 57746A on the progression of the disease state of the pmn mouse. Br. J. Pharmacol. 1998;123:811–817. doi: 10.1038/sj.bjp.0701885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLICKSMAN M.A., CHIU A.Y., DIONNE C.A., HARTY M., KANEKO M., MURAKATA C., OPPENHEIM R.W., PREVETTE D., SENGELAUB D.R., VAUGHT J.L., NEFF N.T. CEP-1347/KT7515 prevents motor neuronal programmed cell death and injury-induced dedifferentiation in vivo. J. Neurobiol. 1998;35:361–370. [PubMed] [Google Scholar]
- GURNEY M.E., CUTTING F.B., ZHAI P., DOBLE A., TAYLOR C.P., ANDRUS P.K., HALL E.D. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann. Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- GURNEY M.E., PU H., CHIU A.Y., DAL CANTO M.C., POLCHOW C.Y., ALEXANDER D.D., CALIENDO J., HENTATI A., KWON Y.W., DENG H.-X., CHEN W., ZHAI P., SUFIT R.L., SIDDIQUE T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- ISHITANI R., SUNAGA K., HIRANO A., SAUNDERS P., KATSUBE N., CHUANG D.-M. Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. J. Neurochem. 1996;66:928–935. doi: 10.1046/j.1471-4159.1996.66030928.x. [DOI] [PubMed] [Google Scholar]
- KITANI K., KANAI S., CARRILLO M.C., IVY G.O. (−)Deprenyl increases the life span as well as activities of superoxide dismutase and catalase but not of glutathione peroxidase in selective brain regions in Fischer rats. Ann. N. Y. Acad. Sci. 1994;717:60–71. doi: 10.1111/j.1749-6632.1994.tb12073.x. [DOI] [PubMed] [Google Scholar]
- KONG J., XU Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRAGTEN E., LALANDE I., ZIMMERMANN K., ROGGO S., SCHINDLER P., MÜLLER D., VAN OOSTRUM J., WALDMEIER P., FÜRST P. Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(−)-deprenyl. J. Biol. Chem. 1998;273:5821–5828. doi: 10.1074/jbc.273.10.5821. [DOI] [PubMed] [Google Scholar]
- KROEMER G., DALLAPORT A.B., RESCHE-RIGON M. The mitochondrial death/life regulator in apoptosis and necrosis. Ann. Rev. Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- LEIST M., NICOTERA P. Apoptosis, excitotoxicity, and neuropathology. Exp. Cell Res. 1998;239:183–201. doi: 10.1006/excr.1997.4026. [DOI] [PubMed] [Google Scholar]
- LEMASTERS J.J., NIEMINEN A.L., QIAN T., TROST L.C., ELMORE S.P., NISHIMURA Y., CROWE R.A., CASCIO W.E., BRADHAM C.A., BRENNER D.A., HERMAN B. The mitochondrial permeability transition in cell death : a common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- MARONEY A.C., GLICKSMAN M.A., BASMA A.N., WALTON K.M., KNIGHT J.R.E., MURPHY C.A., BARTLETT B.A., FINN J.P., ANGELES T., MATSUDA Y., NEFF N.T., DIONNE C.A. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J. Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONTAL M. Mitochondria, glutamate neurotoxicity and the death cascade. Biochim. Biophys. Acta. 1998;1366:113–126. doi: 10.1016/s0005-2728(98)00124-8. [DOI] [PubMed] [Google Scholar]
- MORRISON B.M., MORRISON J.H. Amyotrophic lateral sclerosis associated with mutations in superoxide dismutase : a putative mechanism of degeneration. Brain Res. Rev. 1999;29:121–135. doi: 10.1016/s0165-0173(98)00049-6. [DOI] [PubMed] [Google Scholar]
- PATERSON I.A., FENNIG C.J., GELOWITZ D.L., WALDMEIER P., BOULTON A.A.CGP3466 prevents neuronal death in models of ischemia and seizure in vivo J. Neurochem. 1998b70S6C(Abstract) [Google Scholar]
- PATERSON I.A., ZHANG D., WARRINGTON R.C., BOULTON A.A. R-Deprenyl and R-2-heptyl-N-methylpropargylamine prevent apoptosis in cerebellar granule neurons induced by cytosine arabinoside but not low extracellular potassium. J. Neurochem. 1998a;70:515–523. doi: 10.1046/j.1471-4159.1998.70020515.x. [DOI] [PubMed] [Google Scholar]
- RIPPS M.E., HUNTLEY G.W., HOF P.R., MORRISON J.H., GORDON J.W. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTHSTEIN J.D. Therapeutic horizons for amyotrophic lateral sclerosis. Curr. Opin. Neurobiol. 1996;6:679–687. doi: 10.1016/s0959-4388(96)80103-6. [DOI] [PubMed] [Google Scholar]
- SAGOT Y., DUBOIS-DAUPHIN M., TAN S.A., DE BILBAO F., AEBISCHER P., MARTINOU J.-C., KATO A.C. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J. Neurosci. 1995b;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAGOT Y., ROSSE T., VEJSADA R., PERRELET D., KATO A.C. Differential effects of neurotrophic factors on motoneuron retrograde labeling in a murine model of motoneuron disease. J. Neurosci. 1998;18:1132–1141. doi: 10.1523/JNEUROSCI.18-03-01132.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAGOT Y., TAN S., HAMMING J., AEBISCHER P., KATO A.C. GDNF cannot prevent nerve degeneration in an animal model of motoneuron disease. J. Neurosci. 1996;16:2335–2341. doi: 10.1523/JNEUROSCI.16-07-02335.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAGOT Y., TAN S.A., BAETGE E., SCHMALBRUCH H., KATO A.C., AEBISCHER P. Polymer encapsulated cell lines genetically engineered to release ciliary neurotrophic factor can slow down progressive motor neuronopathy in the mouse. Eur. J. Neurosci. 1995a;7:1313–1322. doi: 10.1111/j.1460-9568.1995.tb01122.x. [DOI] [PubMed] [Google Scholar]
- SAGOT Y., VEJSADA R., KATO A.C. Clinical and molecular aspects of motoneurone diseases: animal models, neurotrophic factors and Bcl-2 oncoprotein. TiPS. 1997;18:330–337. doi: 10.1016/s0165-6147(97)01094-8. [DOI] [PubMed] [Google Scholar]
- SAWA A., KHAN A.A., HESTER L.D., SNYDER S.H. Glyceraldehyde-3-phosphate dehydrogenase : nuclear translocation participates in neuronal and nonneuronal cell death. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENDTNER M., GOTZ R., HOLTMANN B., THOENEN H. Endogenous ciliary neurotrophic factor is a lesion factor for axotomized motoneurons in adult mice. J. Neurosci. 1997;17:6999–7006. doi: 10.1523/JNEUROSCI.17-18-06999.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEFANIS L., BURKE R.E., GREENE L.A. Apoptosis in neurodegenerative disorders. Curr. Opin. Neurol. 1997;10:299–305. doi: 10.1097/00019052-199708000-00004. [DOI] [PubMed] [Google Scholar]
- TATTON W.G., CHALMERS-REDMAN R.M.E. Modulation of gene expression rather than monoamine oxidase inhibition : (−)-Deprenyl-related compounds in controlling neurodegeneration. Neurology. 1996;47:S171–S183. doi: 10.1212/wnl.47.6_suppl_3.171s. [DOI] [PubMed] [Google Scholar]
- TATTON W.G., JU W.Y.H., WADIA J., TATTON N.A.Reduction of neuronal apoptosis by small molecules : promise for new approaches to neurological therapy Neurodegeneration and Neuroprotection in Parkinson's Disease 1996London: Academic Press; 209–220.ed. Olanow, C.W., Jenner, P., Youdim, M.B.H. pp [Google Scholar]
- WADIA J.S., CHALMERS-REDMAN R.M.E., JU W.J.H., CARLILE G.W., PHILLIPS J.L., FRASER A.D., TATTON W.G. Mitochondrial membrane potential and nuclear changes in apoptosis caused by serum and nerve growth factor withdrawal: time course and modification by (−)-deprenyl. J. Neurosci. 1998;18:932–947. doi: 10.1523/JNEUROSCI.18-03-00932.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOKKE J. Riluzole [see comments] Lancet. 1996;348:795–799. doi: 10.1016/S0140-6736(96)03181-9. [DOI] [PubMed] [Google Scholar]
- WU R.M., CHIUEH C.C., PERT A., MURPHY D.L. Apparent antioxidant effect of I-deprenyl on hydroxyl radical formation and nigral injury elicited by MPP+ in vivo. Eur. J. Pharmacol. 1993;243:241–247. doi: 10.1016/0014-2999(93)90181-g. [DOI] [PubMed] [Google Scholar]
- ZAMZAMI N., BRENNER C., MARZO I., SUSIN S.A., KROEMER G. Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins. Oncogene. 1998;16:2265–2282. doi: 10.1038/sj.onc.1201989. [DOI] [PubMed] [Google Scholar]
- ZHANG B., TU P., ABTAHIAN F., TROJANOWSKI J.Q., LEE V.M. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J. Cell. Biol. 1997;139:1307–1315. doi: 10.1083/jcb.139.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]