

Abstract
The recently identified C-terminal splice variant of the human 5-HT4 receptor, the h5-HT4(d) receptor, was stably expressed in a CHO cell line at 493±25 fmol mg−1 protein. We analysed its pharmacological properties by measuring binding affinities and 5-HT4 ligand-induced cyclic AMP production.
The pharmacological binding profile determined in competition studies with the specific antagonist [3H]-GR113808 revealed a rank order of affinity of 5-HT4 ligands for the h5-HT4(d) receptor that was consistent with those previously reported for other 5-HT4 receptor isoforms.
In adenylyl cyclase functional assays, the h5-HT4(d) receptor displayed equipotent coupling for all 5-HT4 agonists tested (EC50 in the range of 1–6 nM). EC50 values were lower than those previously obtained with the 5-HT4(e) receptor stably expressed in CHO cells indicating that the 5-HT4(d) receptor was more efficiently coupled to its effector than the 5-HT4(e) receptor isoform. Moreover, in terms of agonist efficacy (Emax), the benzamide derivative, renzapride displayed full agonist properties at the h5-HT4(d) receptor (same Emax as 5-HT) whereas it was previously shown to be a partial agonist at the h5-HT4(e) receptor.
A constitutive activity of the h5-HT4(d) receptor was observed in CHO cells in the absence of any 5-HT4 ligand. Surprisingly, two 5-HT4 ligands, SB204070 and RS39604 which are described as highly potent antagonists in various biological models, revealed partial agonist properties at the h5-HT4(d) receptor.
We conclude that C-terminal tails of 5-HT4 receptor isoforms may directly influence their functional properties.
Keywords: Human, serotoninergic receptors, 5-HT4 ligands, inverse agonism, G-protein coupled receptors, adenylyl cyclase, benzamides
Introduction
Serotonin [5-hydroxytryptamine, 5-HT] mediates a large diversity of physiological effects both in the central nervous system and the periphery of vertebrates, through a complex mechanism involving at least seven classes of receptor (5-HT1-5-HT7) (Saxena, 1995). Each family of receptors is characterized by distinct molecular and pharmacological properties and subdivided in several receptor subtypes, conferring an additional level of complexity in the system (Gerhardt & Van Heerikhuizen, 1997).
5-HT4 receptors were first characterized by their ability to stimulate cyclic AMP production in mouse colliculi neurones and by the inability of classical 5-HT1, 5-HT2 and 5-HT3 antagonists to inhibit this function (Dumuis et al., 1988). Since that discovery, numerous pharmacological studies have been done in a wide variety of tissues and species indicating that 5-HT4 receptors were highly distributed in the central nervous system (Eglen et al., 1995) and peripheral tissues of vertebrates including alimentary tract, urinary bladder, heart and adrenal gland (Hedge & Eglen, 1996). Despite the existence of a typical 5-HT4 pharmacological profile, heterogeneity in the potency and intrinsic activities of 5-HT4 ligands has been reported in different biological models. For instance, two substituted benzamides, cisapride and renzapride behaved as full and potent agonists in mouse colliculi neurones (Dumuis et al., 1989) but were only partial in guinea-pig hippocampal membranes (Bockaert et al., 1990), rat oesophagus (Baxter et al., 1991), rat distal colon (Wardle & Sanger, 1993) and human heart atrium (Ouadid et al., 1991). The agonist ML10302 displayed a poor agonistic effect in receptor cloned from human atrium (Blondel et al., 1997) whereas it was full agonist in rat oesophagea (Langlois et al., 1994). Altogether, these observations make it difficult to appreciate the functional potency of a given 5-HT4 ligand and suggest the existence of several 5-HT4 receptor subtypes with distinct intrinsic pharmacological properties.
A confirmation of the presence of multiple 5-HT4 receptors has been made with the molecular identification of five C-terminal splice variants of the human 5-HT4 receptor (h5-HT4) named h5-HT4(a), h5-HT4(b), h5-HT4(c), h5-HT4(d) and h5-HT4(e) (Blondel et al., 1997; 1998; Claeysen et al., 1997; 1999; Van den Wyngaert et al., 1997; Mialet et al., 2000). In addition, a novel h5-HT4 splice variant (h5-HT4(hb)) with an extra insertion of 14 amino acids in the second extracellular loop has been very recently cloned (Bender et al., 2000). These receptors belong to the family of seven transmembrane domain G-protein coupled family of receptors which activate adenylyl cyclase. Tissue distribution studies revealed some degree of specificity in the pattern of expression of the h5-HT4 receptor isoforms with h5-HT4(a), h5-HT4(b), h5-HT4(c) and h5-HT4(e) receptors being expressed in cardiac atria and brain (Blondel et al., 1998; Mialet et al., 2000). Interestingly, expression of the h5-HT4(d) receptor was only restricted to the gut (Blondel et al., 1998). This distinct spatial distribution may explain the difference in efficacy of 5-HT4 ligands on 5-HT4 receptor-mediated responses in various tissues. In addition, the coupling of each 5-HT4 receptor isoform to its effector may be influenced by the cellular environment. In support of this hypothesis, we have recently shown that functional potency of h5-HT4(e) receptor ligands was dependent on the cellular context in which the receptor was expressed (Mialet et al., 2000). It is important therefore to analyse the effects of different compounds at individual 5-HT4 receptor subtypes measuring the same second messenger response in cells of the same genetic background. This need is further highlighted by the recent increased interest in the therapeutic utility of selective 5-HT4 ligands to treat a variety of disorders such as cardiac arrythmias (Kaumann, 1994; Rahme et al., 1999), neurodegenerative diseases (Reynolds et al., 1995; Wong et al., 1996), irritable bowel syndrome (Hedge & Eglen, 1996), and urinary incontinence (Boyd & Rohan, 1994).
To date there has been no detailed and comprehensive pharmacological study to characterize and compare the effects of different 5-HT4 ligands on different h5-HT4 receptor isoforms. The present study was therefore undertaken to determine the pharmacological profile of the h5-HT4(d) receptor and compare the potencies and intrinsic activities of 5-HT4 serotoninergic agents using the elevation of cyclic AMP as a functional measure of compound activity. The h5-HT4(d) receptor subtype is of particular interest for several reasons. Firstly, in contrast to h5-HT4(a), h5-HT4(b) and h5-HT4(e) receptors which are human counterparts of rat (Gerald et al., 1995; Claeysen et al., 1999) and mouse (Claeysen et al., 1996; 1999) 5-HT4 receptors, the h5-HT4(d) receptor isoform has not yet been described in any other species. Secondly, expression of the h5-HT4(d) receptor was only restricted to the gut (Blondel et al., 1998) and therefore it could serve as a drug target for the treatment of gastrointestinal associated disorders such as the irritable bowel syndrome. Finally, the h5-HT4(d) receptor isoform corresponds to an ultrashort form of the receptor with a truncation of the carboxyl terminus with only two amino acids after the splicing site (Blondel et al., 1998).
In this paper, we stably expressed the h5-HT4(d) receptor in Chinese hamster ovary (CHO) cells and showed that it displayed a typical 5-HT4 binding profile with the radiolabelled specific antagonist [3H]-GR113808 and a series of 5-HT4 displacing ligands. However, by comparison with our previous functional study in CHO cells stably expressing the h5-HT4(e) receptor (Mialet et al., 2000), we report striking differences in its potency in stimulating cyclic AMP formation in response to various standard 5-HT4 ligands.
Methods
Cell culture
Cell culture materials and reagents were obtained from Life Technologies (Cergy Pontoise, France). CHO cell line was purchased from ATCC (Rockville, U.S.A.) and was grown at 37°C and 5% CO2 in HamsF12 medium supplemented with 10% foetal calf serum, 10 mM HEPES (pH 7.4) and antibiotics.
Stable expression of the h5-HT4(d) receptor in CHO cells
The full coding region of h5-HT4(d) receptor was subcloned into the expression vector pRC/CMV containing the neomycin selection gene (Invitrogen, Carlsbad, CA, U.S.A.). Briefly, confluent cells were transfected with 10 μg of the expression vector by electroporation using a gene pulser transfection apparatus (Biorad, Ivry sur Seine, France; setting 960 μF, 250 V). Forty-eight hours after the transfection period neomycin (1.25 mg ml−1) was added to the dishes for selection. The antibiotic-containing medium was replaced every 2–3 days over 2 weeks. Isolated colonies were selectively trypsinized for further selection, subcloning and propagation of cell clones. h5-HT4(d) receptor expressing clones were detected both by their ability to stimulate cyclic AMP production after treatment with 5-HT and to bind a specific 5-HT4 antagonist, [3H]-GR 113808.
Cyclic AMP radioimmunoassay
For measurement of intracellular cyclic AMP production, stably transfected CHO cells grown to confluence in 24-well plates were incubated overnight with Ham-F12 medium containing 5% dialysed-FCS. At the beginning of the assay, CHO cells were preincubated for 15 min in serum-free medium supplemented with 5 mM theophylline, 10 μM pargyline and 1 μM GR127935 to block the activity of endogenous 5-HT1B receptors. 5-HT4 antagonists were also incubated during this preincubation period. 5-HT or other serotoninergic agonists were then added for an additional 15 min. The reaction was stopped by aspiration of the medium and addition of 50 μl ice-cold perchloric acid (20%). After a 30 min period, neutralization buffer was added (HEPES 25 mM, KOH 2N) and supernatant was extracted after centrifugation at 10,000 r.p.m. for 5 min, cyclic AMP was quantified using a radioimmunoassay kit (cyclic AMP competitive radioimmunoassay. Immunotech, Marseille, France). Student's t-tests were performed using the QuickTTest software.
Membrane preparation and radioligand binding assays
Membrane preparation and radioligand binding assays were performed as previously described (Blondel et al., 1998; Mialet et al., 2000). Briefly, cells grown at confluence were washed twice with Phosphate-Buffered Saline (PBS) and centrifuged at 300×g for 5 min. The resulting pellet was resuspended in 1 ml of ice-cold HEPES buffer (50 mM, pH 7.4), centrifuged at 40,000×g for 15 min at 4°C. The final pellet was resuspended in 1 ml HEPES buffer and protein concentrations were determined by the method of Bradford (1976).
Radioligand binding assays were performed in 500 μl buffer (50 mM HEPES, pH 7.4) containing 20 μl of [3H]-GR113808, 50 μg of membrane preparation and 20 μl of displacing drug. Saturation experiments were performed using [3H]-GR113808 at nine concentrations ranging from 0.01–4 nM. Non specific binding was measured in the presence of 10 μM ML10375 and subtracted from total binding to determine the affinity of [3H]-GR113808 for its receptor (Kd, nM) and the total number of receptors (Bmax, fmol mg−1 protein). Competition assays were performed in the presence of nine concentrations of the displacing ligands (10−12–10−4 M) and a concentration of 0.2 nM of [3H]-GR113808. Incubations were performed at 25°C for 30 min and the reaction was terminated by rapid filtration through Whatman GF/B filter paper using the Brandel model 48R cell harvester. Radioactivity was measured using a Beckman model LS 6500C liquid scintillation counter. Binding data were analysed by computer-assisted nonlinear regression analysis (Prism; GraphPad Software, San Diego, CA, U.S.A.).
Drugs
GR113808 ([1 - [2 - (methylsulphonyl)amino]ethyl] - 4 -piperidinyl]methyl1-methyl-1H-indole-3 - carboxylate) and GR127935 (N-[4-methoxy-3-(4 -methyl-1-piperazinyl)phenyl] - 2′- methyl -4′ - (5-methyl - 1,2,4-oxadiazol - 3 - yl)[1,1 - biphenyl] - 4- carboxamide) were gifts from Glaxo Research Group (Ware, Hertfordshire, U.K.). [3H] - GR113808 was purchased from Amersham ( Orsay, France). ML10302 (2 - piperidinoethyl 4 - amino - 5 - chloro - 2-methoxybenzoate hydrochloride) and ML10375 (2 - [cis - 3,5 - dimethylpiperidino]ethyl 4 -amino-5-chloro-2-methoxybenzoate) were synthesized as previously described (Langlois et al., 1994; Yang et al., 1997). 5 - HT (5 - hydroxytryptamine) and 5 - MeOT (5 - methoxytryptamine) were from Aldrich (L'Isle d'Abeau Chesnes, France). BIMU1 (endo - N - 8 - methyl - 8-azabicyclo[3.2.1]oct-3-yl)-2,3- dihydro-3-ethyl-2-oxo-1H-benzimidazole-1-carboxamide) and cisapride (cis-4-amino-5-chloro-N-[1-[3 - (4-fluoro - phenoxy) propyl] -3-methoxy - 4 - piperidinyl] - 2 - methoxy benzamide) were synthesized in our laboratory. Renzapride (BRL 24924) ((±)-endo-4-amino -5- chloro-2- methoxy-N- (1- azabicyclo[3.3.1]non -4 -yl)benzamide monohydrochloride) and SB204070 (8-amino-7-chloro -(N-butyl -4- piperidyl)- methylbenzo -1, 4-dioxan-5-carboxylate hydrochloride) were generously given by SmithKline Beecham (Harlow, U.K.). RS23597 (3-(piperidine-1-yl)-propyl - 4 - amino - 5 - chloro - 2 - methoxybenzoate hydrochloride), RS39604 (1 -(5-chloro -2(3,5 -dimethoxy)benzyloxy -4- aminophenyl) -3 - (N - (methylsulphamido) ethyl - 4 - piperidyl) propanone) and RS67333 (1-(4-amino-5-chloro-2-methoxyphenyl)-3-(1-n-butyl-4-piperidinyll-propanone) were from Tocris Interchim (Montluçon, France).
Results
Binding studies of the h5-HT4(d) receptor
The cDNA encoding the h5-HT4(d) receptor was stably expressed in CHO cells at 493±25 fmol mg−1 protein. Saturation analysis revealed a single saturable binding site with high affinity for [3H]-GR113808 (Figure 1). Non-specific binding increased linearly with increasing ligand concentration (Figure 1). The Kd value for the transfected h5-HT4(d) receptor was 0.30±0.05 nM, respectively (Figure 1, Table 1) and correlated with those obtained in COS-7 cells transiently transfected with the different h5-HT4 receptors (Blondel et al., 1998). It was also close to those found in CHO and C6-glial cells stably expressing the h5-HT4(e) receptor (Mialet et al., 2000).
Figure 1.
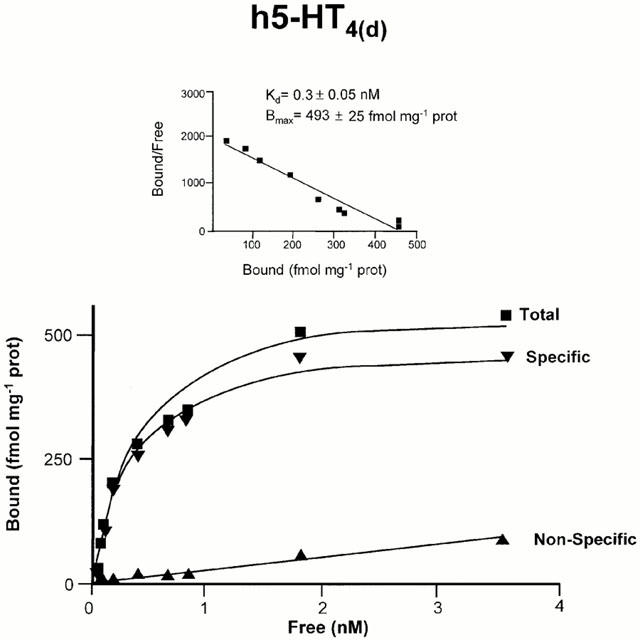
Saturation analysis of [3H]-GR113808 binding to the h5-HT4(d) receptor stably expressed in CHO cells. Membranes harvested from stably transfected CHO cells expressing the h5-HT4(d) receptor were incubated with nine concentrations of [3H]-GR113808 for 30 min at 25°C. Non-specific binding was determined with 10 μM ML10375. Results are from single experiments but are representative of three such experiments.
Table 1.
Affinities of various 5-HT4 ligands that compete with the binding of 0.2 nM [3H]-GR113808 in CHO cells stably expressing the h5-HT4(d) receptor

Since the amino sequences of 5-HT4 receptor splice variants are identical in the transmembrane regions (Blondel et al., 1998; Gerald et al., 1995), it was anticipated that there would be no major pharmacological differences at the level of binding between the h5-HT4(d) receptor isoform and the h5-HT4(e) receptor previously studied in the same cellular system (Mialet et al., 2000). This was verified with a number of standard 5-HT4 receptor ligands (Table 1). A range of 5-HT4 receptor agonists and antagonists completely inhibited the specific binding of [3H]-GR113808 in CHO membranes expressing the h5-HT4(d) receptor (Figure 2). All the displacement curves were monophasic, giving a Hill coefficient not different from 1. The data summarized in Table 1 demonstrate that the pharmacological profile of the h5-HT4(d) receptor in terms of rank order of affinity of the different ligands tested is similar to those found for native 5-HT4 receptors as studied in vivo in human atria (Kaumann et al., 1996), rat striatum (Langlois et al., 1994; Yang et al., 1997), mouse colliculi (Ansanay et al., 1996), or after expression of cloned human, mouse or rat 5-HT4 receptor isoforms in COS-7 (Gerald et al., 1995; Blondel et al., 1997; 1998; Claeysen et al., 1996; 1999) and CHO cell lines (Mialet et al., 2000). The rank order of apparent agonist and antagonist affinities were, respectively, RS67333>ML10302>BIMU1>5-HT=cisapride>renzapride>5-MeOT for the h5-HT4(d) receptor (Figure 2, Table 1). However, we found that BIMU1 had a lower affinity (about 2 fold) for the h5-HT4(d) receptor than the h5-HT4(e) receptor (Ki=64 nM) stably transfected in CHO cells (Table 1, Mialet et al., 2000).
Figure 2.
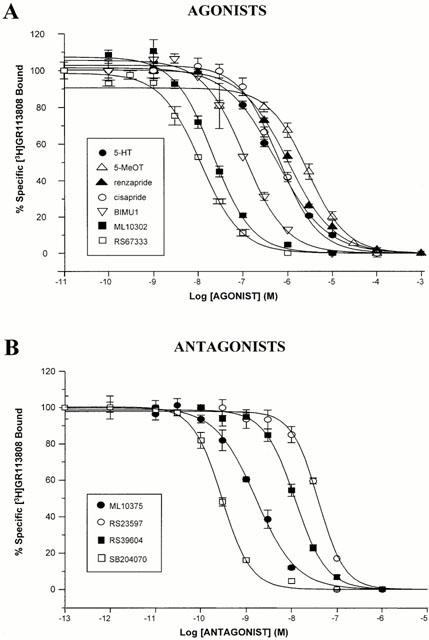
Inhibition of specific [3H]-GR113808 binding to the h5-HT4(d) receptor expressed in CHO cells. Membranes from CHO cells stably expressing the h5-HT4(d) (A,B) receptor were incubated with 0.2 nM [3H]-GR113808 in the presence of increasing concentrations of 5-HT4 agonists (A) or antagonists (B). Non specific binding was defined by 10 μM ML10375. Data are presented as a percentage of specific binding in the absence of displacing drug. Results are from single experiments but are representative of three such experiments using a range of nine concentrations of ligands. Data were analysed by computer-assisted non-linear regression analysis (GraphPad, Prism Software). The corresponding Ki values are presented in Table 1.
Functional coupling of the h5-HT4(d) receptor isoform
Functional coupling of the recombinant h5-HT4(d) receptor isoforms to adenylyl cyclase was investigated by measuring cyclic AMP production in response to various 5-HT4 receptor ligands (Figure 3). All 5-HT4 agonists used in these experiments stimulated cyclic AMP production in a dose dependent manner in expressing the 5-HT4(d) receptor isoform (Figure 3A). However, we were not able to rank EC50 values of 5-HT4 agonists obtained from adenylyl cyclase functional assays in CHO cells. Indeed, EC50 values of ML10302, BIMU1, cisapride, 5-HT and renzapride were similar (Table 2). Furthermore, they were lower than those previously obtained with the 5-HT4(e) receptor indicating that the 5-HT4(d) receptor was more efficiently coupled to its effector than the 5-HT4(e) receptor isoform (Table 2, Mialet et al., 2000). At the 5-HT4(d) receptor, 5-MeOT was the most potent ligand to induce cyclic AMP production since its EC50 value was 1.1±0.2 nM. With regard to agonist efficiencies, renzapride was the only agonist found to display full agonist properties at the 5-HT4(d) receptor (Figure 3A, Table 2).
Figure 3.
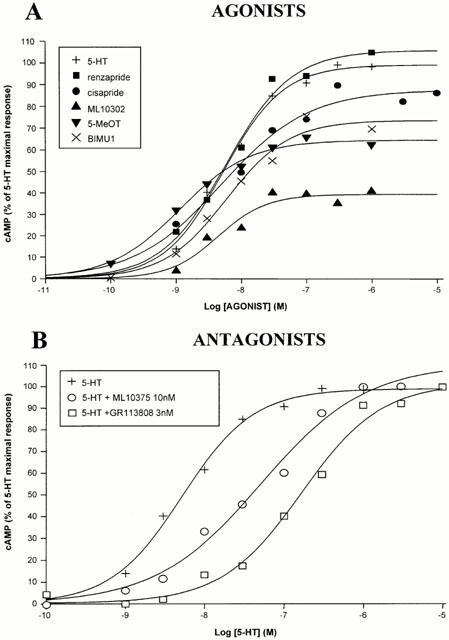
Stimulation of cyclic AMP accumulation in CHO cells stably expressing the h5-HT4(d) receptor by various 5-HT4 ligands. Cyclic AMP measurements were performed on the h5-HT4(d) receptor (A,B) as described in Methods. In (A), the cells were incubated for 15 min with increasing concentrations of agonists and cyclic AMP production was then quantified. In (B), the cells were preincubated 15 min with a concentration of antagonists corresponding to ∼10 fold Ki value as measured in binding experiments (see Table 1); increasing concentrations of 5-HT were then added for an additional 15 min before cyclic AMP was measured. Values are expressed as the percentage of 5-HT maximal response. Each point is the mean of at least three independent experiments, each performed in triplicate. EC50 and Emax values are presented in Table 2.
Table 2.
Effects of 5-HT4 ligands on cyclic AMP production in CHO cells expressing the h5-HT4(d) receptor

5-HT4 antagonists were tested for their ability to antagonize 5-HT-induced cyclic AMP production (Figure 3B; Table 2). GR113808 (3 nM) and ML10375 (10 nM) produced a rightward shift in 5-HT dose response curve (Figure 3B) and were approximately equipotent (Table 2). Surprisingly, we were unable to generate a dose-response curve for 5-HT in the presence of the two highly specific 5-HT4 antagonists, SB204070 and RS39604 (see below).
Constitutive activity of the h5-HT4(d) receptor
A constitutive activity of the h5-HT4(d) receptor was observed in the absence of any 5-HT4 ligand (Figure 4). Indeed, the absolute value for cyclic AMP production in untransfected CHO cells and under basal conditions was 4.3±0.8 pmoles cyclic AMP/well (Figure 4). In CHO cells stably expressing the h5-HT4(d) receptor isoform, cyclic AMP content reached 17.5±2.8 pmoles/well, (Figure 4). This effect was not due to the presence of 5-HT in culture medium since cells were cultured overnight before the assay with dialysed FCS-containing medium. These data clearly indicate that expression of the h5-HT4(d) receptor induced a spontaneously active receptor state.
Figure 4.
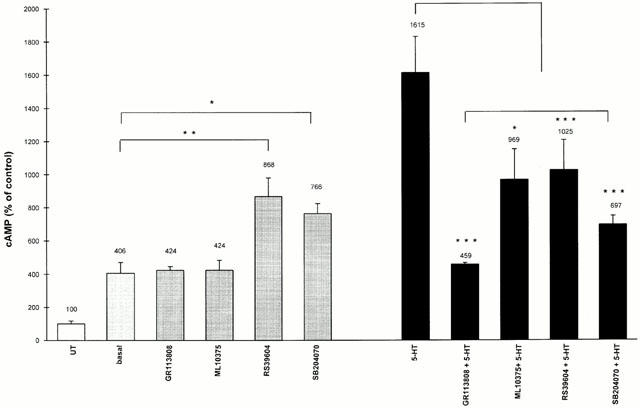
Functional effects of potent 5-HT4 receptor antagonists on basal and 5-HT-stimulated 5-HT4(d) receptors. The effects of 5-HT, GR113808, ML10375, SB204070 and RS39604 in the absence or in the presence of 5-HT on cyclic AMP production were determined in CHO cells expressing the h5-HT4(d) receptor. Results are expressed as percentage of control (untransfected CHO cells, UT). GR113808, ML10375, SB204070, RS39604 and 5-HT were used at a concentration of 1 μM. Values are mean±s.e.mean of three independent experiments performed in triplicate. *P<0.05, **P<0.01, ***P<0.001 versus indicated values by t-test.
Since constitutive activation of G-protein-coupled receptors is often related to inverse agonist properties, we tested the effects of potent selective 5-HT4 receptors antagonists on the intrinsic activity of the h5-HT4(d) receptor (Figure 4). ML10375 (1 μM) and GR113808 (1 μM) which behaved as inverse agonists at the h5-HT4(e) receptor (Mialet et al., 2000) were neutral antagonists and had no effect on the constitutive activity of the h5-HT4(d) receptor (Figure 4). These experiments show that the observed inverse agonist effect of GR113808 and ML10375 is dependent on the considered isoform.
In addition, RS39604 and SB204070 usually described as potent 5-HT4 antagonists in various biological models such as mouse colliculi neurones (Claeysen et al., 1998) acted as partial agonists for the recombinant h5-HT4(d) receptor (Figure 4). Indeed, concentration response curves clearly showed that SB204070 (Figure 5A) and RS39604 (Figure 5B) were able to stimulate cyclic AMP production at the h5-HT4(d) receptor. These effects resulted from 5-HT4 receptor activation because they were antagonized by the selective neutral 5-HT4 antagonist ML10375 (1 μM) (Figure 5). However, all the 5-HT4 antagonists tested in the study, significantly inhibited 5-HT-induced cyclic AMP production at the h5-HT4(d) receptor (Figure 4).
Figure 5.
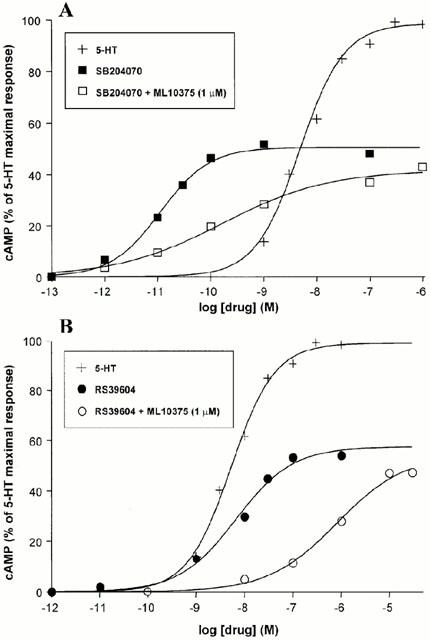
Concentration response curves of SB204070 and RS39604 on cyclic AMP production in CHO cells expressing the h5-HT4(d) receptor. Cyclic AMP measurements were performed on the h5-HT4(d) receptor as described in Methods. Values are expressed as the percentage of 5-HT maximal response. Neutral antagonist ML10375 (1 μM) was added 15 min before agonists, (A) cells were incubated for 15 min with increasing concentrations of 5-HT (+) or SB204070 (▪). EC50 and Emax values of SB204070 were 0.011±0.002 nM and 50.6±1.5%, respectively. In the presence of ML10375 (1 μM) □, EC50 and Emax values were 0.16±0.06 nM and 42±2%, respectively, (B) cells were incubated for 15 min with increasing concentrations of 5-HT (+) or RS39604 •. EC50 and Emax values of RS39604 were 6.6±1.8 nM and 57.4±2.0%, respectively. In the presence of ML10375 (1 μM) (○), EC50 and Emax values were 807±472 nM and 54±6%, respectively. Each point is the mean of at least three independent experiments, each performed in triplicate.
Discussion
h5-HT4 receptor isoforms are generated by splicing events occurring both in the second extracellular loop and the C- terminus of the h5-HT4 receptor just after the amino acid Leu358 (Blondel et al., 1997; 1998; Bender et al., 2000; Claeysen et al., 1997; 1999; Mialet et al., 2000; Van den Wyngaert et al., 1997). Unfortunately it is not possible to study individual h5-HT4 receptor in native tissues due to the lack of complete segregation of the receptors in the central nervous system and the periphery (Blondel et al., 1998; Mialet et al., 2000). The use of recombinant expression systems is therefore a useful approach and also allows the study of human receptors. To analyse the influence of the C-terminal tail on pharmacological properties of the h5-HT4(d) receptor, binding studies and the functional profile of this recombinant h5-HT4 receptor isoform stably expressed in the CHO cell line was determined using a number of standard 5-HT4 agonists and antagonists.
Saturation binding assays with the specific radiolabelled antagonist [3H]-GR113808 showed a Kd value for the h5-HT4(d) receptor (Figure 1) which is in good agreement with those reported in human atria (Kaumann et al., 1996), in human brain membranes (Waeber et al., 1993), and in COS-7 and CHO cell lines transfected with other h5-HT4 receptor splice variants (Bender et al., 2000; Blondel et al., 1998; Claeysen et al., 1997, Mialet et al., 2000, Van den Wyngaert et al., 1997). The pharmacological binding profile determined in competition studies revealed a rank order of affinity of 5-HT4 compounds for the h5-HT4(d) receptor (Figure 2, Table 1) that was consistent with those previously reported in various biological models such as rat striatum and mouse colliculi (Ansanay et al., 1996; Yang et al., 1997). In addition, it matched that obtained in CHO cells expressing the h5-HT4(e) receptor (Mialet et al., 2000). The ligand found to have the highest binding affinity for the h5-HT4(d) receptor was the benzodioxane derivative, SB204070 (Table 1). On the other hand, 5-HT and 5-MeOT showed the lowest affinity (Table 1). These results correlate well with those obtained in binding experiments with the h5-HT4(e) receptor expressed in CHO and C6-glial cells (Mialet et al., 2000) and suggest that C-terminal tails of h5-HT4 receptor splice variants do not influence binding properties of 5-HT4 ligands.
Interestingly, we found that the rank order of potencies of 5-HT4 agonists did not parallel their rank order of binding affinities. Recently, we also noted significantly lower agonist potencies relative to their binding affinities for the h5-HT4(e) receptor stably expressed in CHO and C6-glial cells (Table 2 from Mialet et al., 2000). This is not specific for the h5-HT4 receptor since similar observations have already been reported for rat 5-HT4(a) and 5-HT4(b) receptors, and human 5-HT7 receptors when expressed in different cell lines (Adham et al., 1998; Gerald et al., 1995). A possible explanation for the discrepancy between agonist functional potency and binding affinity is that [3H]-GR113808 labels a different agonist affinity state of the receptor compared to that mediating the functional response. Alternatively, it has been speculated that some ligands might differentially activate a receptor reserve (Gerald et al., 1995).
In contrast to binding studies, very striking differences in functional studies were observed between the pharmacological profile of the h5-HT4(d) receptor and that previously determined for the h5-HT4(e) receptor in the same cellular system (Table 2, Mialet et al., 2000). Overall, the h5-HT4(d) receptor displayed equipotent coupling for all the agonists tested in this study (EC50 in the range of 1–6 nM, Figure 3, Table 2). On the other hand, the h5-HT4(e) receptor isoform was less potent than the h5-HT4(d) receptor to increase cyclic AMP accumulation in response to 5-HT4 agonists (EC50 in the range of 7–180 nM, Table 2, Mialet et al., 2000). For instance, renzapride was almost 20 times more potent at the h5-HT4(d) receptor than we previously observed at the h5-HT4(e) receptor (Table 2). These results appear not to be dependent on receptor expression since according to a previous study from our laboratory the h5-HT4(e) receptor was expressed in CHO cells at a density which was close to that of the h5-HT4(d) receptor found in this study (Mialet et al., 2000).
Our data show that structural differences in the C-terminal tails of h5-HT4 receptors influence and contribute to the specificity of their functional pharmacological profile. The second and third intracellular loops of G protein coupled receptors (GPCRs) are defined as being the regions that physically interact with G proteins and are required for effective coupling. This has been shown for various GPCRs such as the α2-adrenergic receptor (Eason & Ligget, 1996) and the 5-HT7 receptor (Obosi et al., 1997). But recent data indicate that C-terminal sequences of GPCRs also contribute to the potency of coupling between the receptor and its effector, as reported for the dopamine D1 (Jensen et al., 1996; Sugamori et al., 1998) and the angiotensin II receptors (Sano et al., 1997). The fact that the h5-HT4(e) receptor is less coupled to Gs proteins than the h5-HT4(d) receptor also suggest that these splice variants could be coupled either to distinct isoforms of Gs proteins with different coupling efficiencies to adenylyl cyclase or to different signal transduction cascades. In support of this hypothesis, Namba et al. (1993) have shown that the isoforms of the prostaglandin EP3 receptor, which differ only at their C-terminal tails and are produced by alternative splicing, couple to different G proteins to activate different messenger systems without changing ligand binding specificity. The possibility that h5-HT4 receptor splice variants might regulate distinct signalling pathways is currently under investigation.
Another interesting observation from this study is that RS39604 and SB204070 behaved as partial agonists in CHO cells expressing the h5-HT4(d) receptor although they were described as highly potent antagonists in various biological models like guinea-pig distal colon (Gaster & Sanger, 1994; Wardle et al., 1994), rat oesophagus (Hedge et al., 1995), and mouse colliculi neurones (Claeysen et al., 1998). Our results are in accordance with those of Claeysen et al. (1998) who found partial agonistic activities of these two 5-HT4 compounds in COS-7 cells expressing the h5-HT4(a) receptor.
In fact to explain the discrepancies between in vitro and in vivo data, one could speculate that several 5-HT4 receptor isoforms expressed in a given tissue could form heterodimers with distinct functional pharmacological properties. This molecular mechanism has been recently described for other GPCR such as the opioid receptors (Jordan & Devi, 1999). Yet, tissue specific proteins could interact with the intracellular C-terminal tails of h5-HT4 receptors and influence their intracellular signalling. It is interesting to note that the new cloned h5-HT4(e) receptor splice variant displays in its C-terminal end a consensus motif for PDZ-domain containing proteins (Claeysen et al., 1999; Mialet et al., 2000). These proteins with PDZ-domains act as molecular organizers that cluster receptors in the plasma membrane or mediate direct interaction with GPCR to influence their intracellular events (Saras & Heldin, 1996). Whether such mechanisms occur for h5-HT4 receptor splice variants and contribute to the specificity of their pharmacological profile in a given tissue has yet to be elucidated.
Finally, we found that the h5-HT4(d) receptors expressed in CHO cells were constitutively activated when expressed at physiological densities (Figure 4). Such constitutive coupling has already been reported for the h5-HT4(e) receptor stably expressed in CHO cell line (Mialet et al., 2000) and for recombinant mouse, rat and h5-HT4(e) receptors transfected in COS-7 cells (Claeysen et al., 1999). It has been shown that the constitutive activity of mouse 5-HT4 short splice variants is higher than that of the long C-terminal sequence variants (Claeysen et al., 1999). However, we found that the h5-HT4(d) receptor which has the shortest C-terminal tail displayed a similar constitutive activity (Figure 4) than the h5-HT4(e) receptor (Mialet et al., 2000). Therefore in our hands, we cannot conclude about the influence of the length of the C-terminal tail on constitutive activation.
Some interesting features were found with inverse agonist properties of several 5-HT4 ligands (Figure 4). The highly potent 5-HT4 antagonists, GR113808 and ML10375, were neutral antagonist at the 5-HT4(d) receptor but on the contrary acted as inverse agonists at the 5-HT4(e) receptor (Figure 4, Mialet et al., 2000). These results clearly indicate that the C-terminal tails of the 5-HT4 receptor isoforms may strongly influence the inverse agonist properties of a given 5-HT4 ligand.
In conclusion, we have determined the pharmacological profile of the h5-HT4(d) receptor stably expressed in CHO cells. We did not observe any major difference in the binding properties of 5-HT4 ligands between the h5-HT4(d) receptor and other h5-HT4 receptor isoforms. However, striking differences were observed in functional studies and we have presented evidence that C-terminal tails of 5-HT4 receptor isoforms may directly influence their functional properties. Further detailed pharmacological characterizations of other cloned h5-HT4 receptor splice variants in heterologous expression systems will facilitate the interpretation of in vivo responses to 5-HT4 ligands in native tissues.
Acknowledgments
We wish to thank Monique Gastineau and Yamina Dahmoune for their excellent technical assistance. We also thank Cécile Beaupère. Sheerazed Boulkroun and Carine Jacquard for their help in binding experiments. Frank Lezoualc'h was a recipient of a grant from the Fondation pour la Recherche Médicale. This work was supported by the Fondation de France.
Abbreviations
- 5-HT
5-hydroxytryptamine
- 5-MeOT
5-methoxytryptamine
- BIMU1
endo-N-8-methyl-8-azabicyclo[3.2.1]oct-3-yl)-2,3-dihydro-3-ethyl-2-oxo-1H-benzimidazole-1-carboxamide
- CHO cells
Chinese hamster ovary cells
- cisapride
cis-4-amino-5-chloro-N-[1-[3-(4-fluoro-phenoxy)propyl]-3-methoxy-4-piperidinyl]-2-methoxy benzamide
- GPCR
G-protein-coupled receptors
- GR113808
[1-[2-(methylsulphonyl)amino]ethyl]-4-piperidinyl]methyl1-methyl-1H-indole-3-carboxylate)
- GR127935
N-[4-methoxy-3-(4-methyl-1-piperazinyl)phenyl]-2′-methyl-4′-(5-methyl-1,2,4-oxadiazol-3-yl)[1,1-biphenyl]-4-carboxamide
- h5-HT4
human 5-HT4 receptor
- ML10302
2-piperidinoethyl 4-amino-5-chloro-2-methoxybenzoate hydrochloride
- ML10375
2-[cis-3,5-dimethylpiperidino]ethyl 4-amino-5-chloro-2-methoxybenzoate
- renzapride (BRL 24924)
(±)-endo-4-amino-5-chloro-2-methoxy-N-(1-azabicyclo[3.3.1]non-4-yl)benzamide monohydrochloride
- RS23597
3-(piperidine-1-yl)-propyl-4-amino-5-chloro-2-methoxybenzoate hydrochloride
- RS39604
1-(5-chloro-2(3,5-dimethoxy)benzyloxy-4-aminophenyl)-3-(N-(methylsulfamido)ethyl-4-piperidyl)propanone
- RS67333
1-(4-amino-5-chloro-2-methoxyphenyl)-3-(1-n-butyl-4-piperidinyl)-1-propanone
- SB204070
8-amino-7-chloro-(N-butyl-4-piperidyl)-methylbenzo-1,4-dioxan-5-carboxylate hydrochloride
References
- ADHAM N., ZGOMBICK J.M., BARD J., BRANCHEK T.A. Functional characterisation of the recombinant human 5-hydroxytryptamine7(a) receptor isoform coupled to adenylate cyclase stimulation. J. Pharmacol. Exp. Ther. 1998;287:508–514. [PubMed] [Google Scholar]
- ANSANAY H., SEBBEN M., BOCKAERT J., DUMUIS A. Pharmacological comparison between [3H]- GR113808 binding sites and functional 5-HT4 receptors in neurons. Eur. J. Pharmacol. 1996;298:165–174. doi: 10.1016/0014-2999(95)00786-5. [DOI] [PubMed] [Google Scholar]
- BAXTER G.S., CRAIG D.A., CLARKE D.E. 5-HT4 receptors mediate relaxation of the rat oesophageal tunica muscularis mucosae. Naunyn-Schmied. Arch. Pharmacol. 1991;343:439–446. doi: 10.1007/BF00169544. [DOI] [PubMed] [Google Scholar]
- BENDER E., PINDON A., VAN OERS I., ZHANG Y.B., GOMMEREN W., VERHASSELT P., JURZAK M., LEYSEN J., LUYTEN W. Structure of the human serotonin 5-HT4 receptor gene and cloning of a novel 5-HT4 splice variant. J. Neurochem. 2000;74:478–489. doi: 10.1046/j.1471-4159.2000.740478.x. [DOI] [PubMed] [Google Scholar]
- BLONDEL O., GASTINEAU M., DAHMOUNE Y., LANGLOIS M., FISCHMEISTER R. Cloning, expression and pharmacology of four human 5-HT4 receptor isoforms produced by alternative splicing in the carboxyl terminus. J. Neurochem. 1998;70:2252–2261. doi: 10.1046/j.1471-4159.1998.70062252.x. [DOI] [PubMed] [Google Scholar]
- BLONDEL O., VANDECASTEELE G., GASTINEAU M., LECLERC S., DAHMOUNE Y., LANGLOIS M., FISCHMEISTER R. Molecular and functional characterisation of a 5-HT4 receptor cloned from human atrium. FEBS Lett. 1997;412:465–474. doi: 10.1016/s0014-5793(97)00820-x. [DOI] [PubMed] [Google Scholar]
- BOCKAERT J., SEBBEN M., DUMUIS A. Pharmacological characterisation of 5-hydroxytryptamine4(5-HT4) receptors positively coupled to adenylate cyclase in adult guinea pig hippocampal membranes: effect of substituted benzamide derivatives. Mol. Pharmacol. 1990;37:408–411. [PubMed] [Google Scholar]
- BOYD I.W., ROHAN A.P. Urinary disorders associated with cisapride. Med. J. Aust. 1994;160:579–580. [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CLAEYSEN S., FAYE P., SEBBEN M., LEMAIRE S., BOCKAERT J., DUMUIS A. Cloning and expression of human 5-HT4S receptors. Effect of receptor density on their coupling to adenylyl cyclase. Neuroreport. 1997;8:3189–3196. doi: 10.1097/00001756-199710200-00002. [DOI] [PubMed] [Google Scholar]
- CLAEYSEN S., FAYE P., SEBBEN M., TAVIAUX S., BOCKAERT J., DUMUIS A. 5-HT4 receptors: cloning and expression of new splice variants. Ann. N. Y. Acad Sci. 1998;861:49–56. doi: 10.1111/j.1749-6632.1998.tb10172.x. [DOI] [PubMed] [Google Scholar]
- CLAEYSEN S., SEBBEN M., BECAMEL C., BOCKAERT J., DUMUIS A. Novel brain-specific 5-HT4 receptor splice variants show marked constitutive activity: role of the C-terminal intracellular domain. Mol Pharmacol. 1999;55:910–920. [PubMed] [Google Scholar]
- CLAEYSEN S., SEBBEN M., JOURNOT L., BOCKAERT J., DUMUIS A. Cloning, expressing and pharmacology of the mouse 5-HT41. receptor. FEBS Lett. 1996;398:19–25. doi: 10.1016/s0014-5793(96)01132-5. [DOI] [PubMed] [Google Scholar]
- DUMUIS A., BOUHELAL R., SEBBEN M., CORY R., BOCKAERT J. A non classical 5-hydroxytryptamine receptor positively coupled with adenylate cyclase in the central nervous systeme. Mol. Pharmacol. 1988;34:880–887. [PubMed] [Google Scholar]
- DUMUIS A., SEBBEN M., BOCKAERT J. The gastrointestinal prokinetic benzamide derivatives are agonists at the non-classical 5-HT receptor (5-HT4) positively coupled to adenylate cyclase in neurons. Naunyn-Schmied. Arch. Pharmacol. 1989;340:403–410. doi: 10.1007/BF00167041. [DOI] [PubMed] [Google Scholar]
- EASON M.G., LIGGET S.B. Chimeric mutagenesis of putative G-protein coupling domains of the α2A-adrenergic receptor. J. Biol. Chem. 1996;271:12826–12832. doi: 10.1074/jbc.271.22.12826. [DOI] [PubMed] [Google Scholar]
- EGLEN R.M., WONG E.H.F., DUMUIS A., BOCKAERT J. Central 5-HT4 receptors. Trends Pharmacol. Sci. 1995;16:391–397. doi: 10.1016/s0165-6147(00)89081-1. [DOI] [PubMed] [Google Scholar]
- GASTER L.M., SANGER G.J. SB204070: 5-HT receptor antagonists and their potential therapeutic utility. Drugs Future. 1994;19:1109–1121. [Google Scholar]
- GERALD C., ADHAM N., KAO H.T., OLSEN M.A., LAZ T.M., SCHECHTER L.E., BARD J.E., VAYSSE P.J.J., HARTIG P.R., BRANCHEK T.A., WEINSHANK R.L. The 5-HT4 receptor: molecular cloning and pharmacological characterisation of two splice variants. EMBO J. 1995;14:2806–2815. doi: 10.1002/j.1460-2075.1995.tb07280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERHARDT C.C., VAN HEERIKHUIZEN H. Functional characteristics of heterologously expressed 5-HT receptors. Eur. J. Pharmacol. 1997;334:1–23. doi: 10.1016/s0014-2999(97)01115-1. [DOI] [PubMed] [Google Scholar]
- HEGDE S., BONHAUS D.W., JOHNSON L.G., LEUNG E., CLARK R.D., EGLEN R.M. RS39604: a potent, selective and orally active 5-HT4 receptor antagonist. Br. J. Pharmacol. 1995;115:1087–1095. doi: 10.1111/j.1476-5381.1995.tb15922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEGDE S., EGLEN R. Peripheral 5-HT4 receptors. FASEB J. 1996;10:1398–1407. doi: 10.1096/fasebj.10.12.8903510. [DOI] [PubMed] [Google Scholar]
- JENSEN A.A., PEDERSEN U.B., DIN N., ANDERSEN P.H. The dopamine D1 receptor family: structural and functional aspects. Biochem. Soc. Trans. 1996;24:163–169. doi: 10.1042/bst0240163. [DOI] [PubMed] [Google Scholar]
- JORDAN B.A., DEVI L.A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAUMANN A.J. Do human atrial 5-HT4 receptors mediate arrhythmias. Trends Pharmacol. Sci. 1994;15:451–455. doi: 10.1016/0165-6147(94)90058-2. [DOI] [PubMed] [Google Scholar]
- KAUMANN A.J., LYNHAM J.A., BROWN A.M. Comparison of the densities of 5-HT4 receptors, β1- and β2-adrenoceptors in human atrium: functional implications. Naunyn-Schmied. Arch. Pharmacol. 1996;353:592–595. doi: 10.1007/BF00169181. [DOI] [PubMed] [Google Scholar]
- LANGLOIS M., ZHANG L., YANG D., BRÉMONT B., SHEN S., MANARA L., CROCI T. Design of a potent 5-HT4 receptor agonist with nanomolar affinity. Biomed. Chem. Lett. 1994;4:1433–1436. [Google Scholar]
- MIALET J., BERQUE-BESTEL I., EFTEKHARI P., GASTINEAU M., GINER M., DAHMOUNE Y., DONZEAU-GOUGE P., HOEBEKE J., LANGLOIS M., SICSIC S., FISHMEISTER R., LEZOUALC'H F. Isolation of the serotoninergic 5-HT4(e) receptor from human heart and comparative analysis of its pharmacological profile in C6-glial and CHO cells. Br. J. Pharmacol. 2000;129:771–781. doi: 10.1038/sj.bjp.0703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAMBA T., SUGIMOTO Y., NEGISHI M., IRIE A., USHIKUBI F., KAKIZUKA A., ITO S., ICHIKAWA A., NARUMIYA S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993;365:166–170. doi: 10.1038/365166a0. [DOI] [PubMed] [Google Scholar]
- OBOSI L.A., HEN R., BEADLE D.J., BERMUDEZ I., KING L.A. Mutational analysis of the mouse 5-HT7 receptor: importance of the third intracellular loop for receptor-G-protein interaction. FEBS Lett. 1997;412:321–324. doi: 10.1016/s0014-5793(97)00813-2. [DOI] [PubMed] [Google Scholar]
- OUADID H., SEGUIN J., DUMUIS A., BOCKAERT J., NARGEOT J. Serotonin increases calcium current in human atrial myocytes via the newly described 5-hydroxytryptamine 4 receptors. Mol. Pharmacol. 1991;41:346–351. [PubMed] [Google Scholar]
- RAHME M.M., COTTER B., LEISTAD E., WADHWA M.K., MOHABIR R., FORD A.P.D.W., EGLEN R.M., FELD G.K. Electrophysiological and antiarrhythmic effects of the atrial selective antagonist RS-100302 in experimental atrial flutter and fibrillation. Circulation. 1999;100:2010–2017. doi: 10.1161/01.cir.100.19.2010. [DOI] [PubMed] [Google Scholar]
- REYNOLDS G.P., MASON L., MELDRUM A., DE KECZER S., PARNES H., WONG E.H.F. 5- hydroxytryptamine (5-HT)4 receptors in post mortem human brain tissue: distribution, pharmacology and effects of neurodegenerative diseases. Br. J. Pharmacol. 1995;114:993–998. doi: 10.1111/j.1476-5381.1995.tb13303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANO T., OHYAMA K., YAMANO Y., NAKAGOMI Y., NAKAZAWA S., KIKYO M., SHIRAI H., BLANK J.S., EXTON J.H., INAGAMI T. A domain for G protein coupling in carboxyl-terminal tail of rat angiotensin II receptor type 1A. J. Biol. Chem. 1997;272:23631–23636. doi: 10.1074/jbc.272.38.23631. [DOI] [PubMed] [Google Scholar]
- SARAS J., HELDIN C.H. PDZ domains bind carboxy-terminal sequences of target proteins. Trends Biochem. Sci. 1996;21:455–458. doi: 10.1016/s0968-0004(96)30044-3. [DOI] [PubMed] [Google Scholar]
- SAXENA P.R. Serotonin receptors: subtypes, functional responses and therapeutic relevance. Pharmacol. Ther. 1995;66:339–368. doi: 10.1016/0163-7258(94)00005-n. [DOI] [PubMed] [Google Scholar]
- SUGAMORI K.S., SCHEIDELER M.A., VERNIER P., NIZNIK H.B. Dopamine D1B receptor chimeras reveal modulation of partial agonist activity by carboxy-terminal tail sequences. J. Neurochem. 1998;71:2593–2599. doi: 10.1046/j.1471-4159.1998.71062593.x. [DOI] [PubMed] [Google Scholar]
- VAN DEN WYNGAERT I., GOMMEREN W., VERHASSELT P., JURZAK M., LEYSEN J., LUYTEN W.C., BENDER E. Cloning and expression of a human serotonin 5-HT4 receptor cDNA. J. Neurochem. 1997;69:1810–1819. doi: 10.1046/j.1471-4159.1997.69051810.x. [DOI] [PubMed] [Google Scholar]
- WAEBER C., SEBBEN M., GROSSMAN C., JAVOY-AGID F., BOCKAERT J., DUMUIS A. [3H]- GR113808 labels 5-HT4 receptors in the human and guinea-pig brain. Neuroreport. 1993;4:1239–1242. doi: 10.1097/00001756-199309000-00007. [DOI] [PubMed] [Google Scholar]
- WARDLE K.A., ELLIS E.S., BAXTER G.S., KENNETT G.A., GASTER L.M., SANGER G.J. The effects of SB204070, a highly potent and selective 5-HT4 receptor antagonist, on guinea-pig distal colon. Br. J. Pharmacol. 1994;112:789–794. doi: 10.1111/j.1476-5381.1994.tb13148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARDLE K.A., SANGER G.J. The guinea-pig distal colon: a sensitive preparation for the investigation of 5-HT4 receptor-mediated contractions. Br. J. Pharmacol. 1993;110:1593–1599. doi: 10.1111/j.1476-5381.1993.tb14006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONG E.H., REYNOLDS G.P., BONHAUS D.W., HSU S., EGLEN R.M. Characterisation of [3H]- GR113808 binding to 5-HT4 receptors in brain tissues from patients with neurodegenerative disorders. Behav. Brain. Res. 1996;73:249–252. doi: 10.1016/0166-4328(96)00106-4. [DOI] [PubMed] [Google Scholar]
- YANG D., SOULIER J.L., SICSIC S., MATHÉ-ALLAINMAT M., BRÉMONT B., CROCI T., CARDAMONE R., AUREGGI G., LANGLOIS M. New esters of 4-amino-5-chloro-2-methoxybenzoic acid as potent agonists and antagonists for 5-HT4 receptors. J. Med. Chem. 1997;40:608–621. doi: 10.1021/jm960320m. [DOI] [PubMed] [Google Scholar]