

Abstract
Sarcolemmal Na+/H+ exchanger (NHE) activity is increased by stimulation of Gq protein-coupled receptors (GqPCRs), but the roles of other GPCRs are largely unknown. We determined the effects of N-[(1S,trans)-2-hydroxycyclopentyl]adenosine (GR79236), a selective agonist of the GiPCR adenosine A1 receptor, on sarcolemmal NHE activity in adult rat ventricular myocytes (n=8–10 per group). NHE activity was indexed by the H+ efflux rate after intracellular acidification, measured by microepifluorescence. GR79236 alone (0.01–10 μM) had no effect on NHE activity. However, co-administration of GR79236 inhibited, in a concentration-dependent manner, the stimulation of NHE activity by the α1-adrenoceptor agonist phenylephrine (10 μM). The inhibitory effect of GR79236 (10 μM) was abolished by (1) the selective A1 antagonist 1,3-dipropyl-8-cyclopentylxanthine (0.1 μM), confirming an A1 receptor-mediated action, and (2) pre-treatment with pertussis toxin (5 μg ml−1 for 60 min), indicating a Gi protein-mediated mechanism. Our data suggest the existence of inhibitory crosstalk between the GiPCR adenosine A1 receptor and the GqPCR α1-adrenoceptor in the regulation of sarcolemmal NHE activity.
Keywords: G protein-coupled receptors, adenosine A1 receptor, α1-adrenoceptor, cardiac myocyte, Na+/H+ exchanger
Introduction
The sarcolemmal Na+/H+ exchanger (NHE) is the ubiquitously expressed product of the NHE1 gene (Fliegel & Dyck, 1995) and is an important H+ extrusion mechanism that contributes to the control of intracellular pH (pHi) in cardiac myocytes (Leem et al., 1999). Nevertheless, increased activity of the sarcolemmal NHE has been implicated in the development of myocardial injury and dysfunction during ischaemia and reperfusion, and NHE1-selective pharmacological inhibitors have been shown to be cardioprotective in this setting in numerous animal studies (Avkiran, 1999a). Recent clinical data suggest that one such inhibitor, cariporide, may provide cardioprotective benefit in patients with anterior myocardial infarction who receive early reperfusion by direct coronary angioplasty (Rupprecht et al., 2000) and in high-risk patients who undergo global myocardial ischaemia and reperfusion during coronary artery bypass graft (CABG) surgery (Théroux et al., 2000).
Sarcolemmal NHE activity is regulated primarily by pHi and increases markedly in response to intracellular acidosis (Leem et al., 1999); it is also subject to stimulation by agents that act via Gq protein-coupled receptors (GqPCRs), such as α1-adrenoceptor (α1-AR) agonists, endothelin, thrombin and angiotensin II (reviewed by Avkiran, 1999b). However, there is little known about the regulation of sarcolemmal NHE activity by receptors outside the GqPCR family.
Adenosine is an adenine nucleoside that has been shown to possess cardioprotective efficacy in animal models of myocardial ischaemia and reperfusion (Lasley & Mentzer, 1996). The therapeutic potential of adenosine has also recently been tested in humans, as an adjunct to thrombolysis in patients with acute myocardial infarction (Mahaffey et al., 1999) and as an additive to cardioplegia in patients undergoing CABG surgery (Mentzer et al., 1999), with encouraging results. Although adenosine can potentially afford myocardial protection during ischaemia and reperfusion through the stimulation of multiple adenosine receptors in a variety of cell types, direct stimulation of myocardial A1 receptors appears to be an important component of such protection (Lasley & Mentzer, 1996). Indeed, hearts from transgenic mice with cardiac-specific overexpression of the A1 receptor have been shown to exhibit reduced susceptibility to ischaemia and reperfusion-induced injury (Matherne et al., 1997). The mechanisms through which the myocardial A1 receptor, which is a member of the GiPCR family, exerts protection are unclear.
Our recent work has shown that stimulation of another GiPCR, the angiotensin AT2 receptor, inhibits sarcolemmal NHE activation via the GqPCR angiotensin AT1 receptor (Gunasegaram et al., 1999). The possibility exists that adenosine A1 receptors may also initiate signalling events that negatively regulate sarcolemmal NHE activity. Therefore, the present study was undertaken to determine the effects of adenosine A1 receptor stimulation by the selective agonist N-[(1S,trans)-2-hydroxycyclopentyl]adenosine (GR79236) (Gurden et al., 1993) on sarcolemmal NHE activity, in the resting state and following GqPCR stimulation. Preliminary results of the study have been published in abstract form (Yokoyama & Avkiran, 2000).
Methods
This investigation was performed in accordance with the Home Office ‘Guidance on the Operation of the Animals (Scientific Procedures) Act 1986', published by HMSO, London.
Isolation of ventricular myocytes
Ventricular myocytes were isolated from hearts of adult (200–250 g body weight) male Wistar rats (B&K Universal, Hull, U.K.) by enzymatic digestion, as previously described (Yasutake et al., 1996; Yokoyama et al., 1998; Gunasegaram et al., 1999; Snabaitis et al., 2000).
Determination of sarcolemmal NHE activity
Sarcolemmal NHE activity was determined in single myocytes loaded with the pH-sensitive fluoroprobe cSNARF-1, using an established microepifluorescence technique (Yasutake et al., 1996; Yokoyama et al., 1998; Gunasegaram et al., 1999; Snabaitis et al., 2000). Cells were superfused with bicarbonate-free Tyrode's solution (34°C) throughout each experiment and the rate of acid efflux (JH) was used as the index of sarcolemmal NHE activity. JH was determined at an intracellular pH (pHi) of 6.90 (JH6.9), during recovery from intracellular acidosis that was induced by transient exposure to NH4Cl (see below).
Experimental protocols
Myocytes were subjected to intracellular acidosis by a 3-min exposure to 20 mmol/l NH4Cl (first acid pulse), which was repeated 15 min later (second acid pulse) (Yasutake et al., 1996; Yokoyama et al., 1998; Gunasegaram et al., 1999; Snabaitis et al., 2000). In control cells, both acid pulses occurred in the absence of any drug. When studying the effects of GR79236 (gift from GlaxoWellcome, Stevenage, U.K.) alone, this was present during the second acid pulse. When studying the effects of GR79236 on the response to phenylephrine or thrombin (both from Sigma, Poole, U.K.), the GqPCR agonist was given during the second acid pulse and GR79236 was present from 6 min before this pulse. 1,3-Dipropyl-8-cyclopentylxanthine (DPCPX; Sigma), when used, was given concomitantly with GR79236. Stock solutions of drugs, except DPCPX, were dissolved in deionized water and were diluted (<1000 fold) in Tyrode's solution to obtain appropriate final concentrations; the DPCPX stock solution was dissolved in dimethylsulphoxide (final concentration 0.005%). Phenylephrine solutions contained 1 μM atenolol (Sigma), to preclude β1-AR-mediated actions (Yokoyama et al., 1998). When required, cells were pre-treated with 5 μg ml−1 pertussis toxin (Sigma) for 60 min, in order to inactivate Gi proteins (Obayashi et al., 1997).
Statistical analysis
Data are expressed as mean±s.e.mean. Each protocol comprised 4–8 study groups and experiments within it were carried out in a randomized manner (n=8–10 myocytes per group, obtained from 5–15 hearts). For inter-group comparison of the changes in JH6.9 (ΔJH6.9) in response to vehicle or drug(s), data were subjected to ANOVA; if a significant difference was found, further analysis was by Dunnett's test, to compare each treatment group with the control group. P<0.05 was considered significant.
Results
Effects of GR79236 alone
JH6.9 values during recovery from the first acid pulse in control cells and in those that received 0.01, 0.1, 1 or 10 μM GR79236 (n=10 cells per group, from 10 hearts) were 3.56±0.31, 3.28±0.45, 3.71±0.70, 4.36±0.77 and 3.06±0.41 mM min−1, respectively (NS). There was no significant change in JH6.9 during the second acid pulse in any group, with ΔJH6.9 values of 13±19, 18±25, 13±20, 4±12 and 10±31%, respectively (NS). These findings indicate that acute exposure to GR79236 does not significantly affect sarcolemmal NHE activity in the resting state.
Effects of GR79236 on the response to phenylephrine
We next determined whether exposure to GR79236 affects the stimulation of sarcolemmal NHE activity by phenylephrine. JH6.9 values during the first acid pulse did not differ significantly between the eight study groups (n=10 cells per group, from 15 hearts) and were as follows: 3.18±0.48 mM min−1 in control cells; 3.21±0.44 mM min−1 in cells that received phenylephrine (10 μM) alone; 2.5±0.29, 2.78±0.50, 2.73±0.69 and 3.33±0.27 mM min−1, respectively, in cells that received phenylephrine (10 μM) in combination with 0.01, 0.1, or 10 μM GR79236; 3.80±0.69 and 3.13±0.49 mM min−1, respectively, in cells that received 1 or 10 μM GR79236 alone. Figure 1 illustrates the ΔJH6.9 values in these groups. Consistent with our earlier findings (Yokoyama et al., 1998; Snabaitis et al., 2000), phenylephrine alone produced a large and significant increase in sarcolemmal NHE activity. GR79236 inhibited the NHE-stimulatory effect of phenylephrine in a concentration-dependent manner, such that the α1-AR agonist no longer produced a significant increase in NHE activity when given in combination with 1 or 10 μM GR79236. Once again, 1 or 10 μM GR79236 alone failed to produce a significant change in sarcolemmal NHE activity. These data indicate that exposure to GR79236 inhibits α1-AR-mediated stimulation of sarcolemmal NHE activity.
Figure 1.
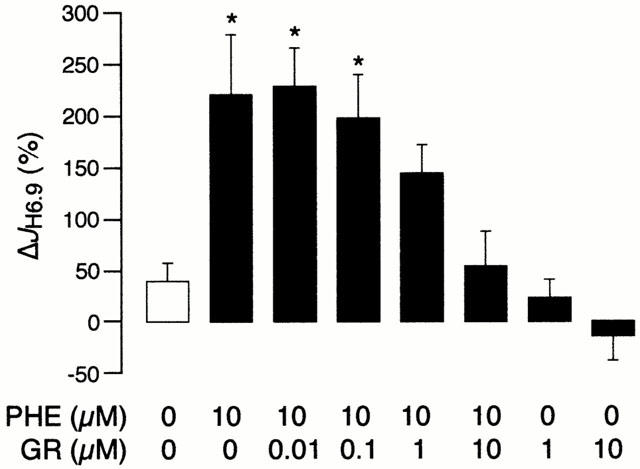
The change in H+ efflux rate at pHi 6.90 (ΔJH6.9) in the control group and in response to phenylephrine (PHE) and GR79236 (GR), alone or in combination. The protocol comprised 80 myocytes (n=10 per group) obtained from 15 hearts. *P<0.05 vs control.
Reversal of the inhibitory effect of GR79236 by DPCPX
In order to confirm that the inhibitory effect of GR79236 on the α1-adrenergic response was mediated via the A1 receptor, rather than through a non-specific action, we tested the reversibility of this effect by the selective A1 receptor antagonist DPCPX. JH6.9 values during the first acid pulse did not differ significantly between the five study groups (n=9 cells per group, from seven hearts) and ranged between 2.48±0.44 and 3.08±0.30 mM min−1. Figure 2 illustrates the ΔJH6.9 values. As expected, 10 μM phenylephrine produced a significant increase in NHE activity and this response was again abolished when phenylephrine was given in combination with 10 μM GR79236. However, when GR79236 was given concomitantly with 0.1 μM DPCPX, the A1 agonist was no longer able to inhibit α1-AR-mediated stimulation of sarcolemmal NHE activity. DPCPX alone was without effect. These findings confirm that the inhibitory effect of GR79236 on α1-adrenergic stimulation of sarcolemmal NHE activity was mediated via the A1 receptor.
Figure 2.
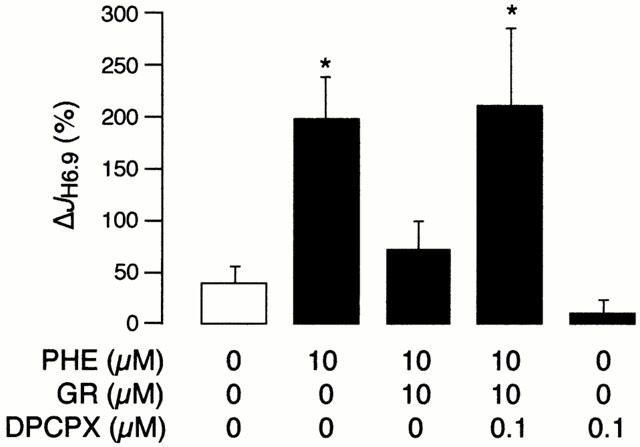
The change in H+ efflux rate at pHi 6.90 (ΔJH6.9) in the control group and in response to phenylephrine (PHE), GR79236 (GR) and 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), alone or in combination. The protocol comprised 45 myocytes (n=9 per group) obtained from seven hearts. *P<0.05 vs control.
Reversal of the inhibitory effect of GR79236 by pertussis toxin
In order to probe the signalling mechanisms distal to the A1 receptor that mediate the inhibitory effect of GR79236, we examined the consequences of inactivating the Gi protein, by a 60-min pertussis toxin pre-treatment. JH6.9 values during the first acid pulse were similar between the eight study groups (n=8 cells per group, from 14 hearts) within this protocol and ranged between 2.81±0.47 and 3.30±0.40 mM min−1. Figure 3 illustrates the ΔJH6.9 values. Our observations in cells pre-treated with vehicle for 60 min were essentially identical to those illustrated in Figure 1; 10 μM phenylephrine significantly increased sarcolemmal NHE activity and this response was abolished by 10 μM GR79236 (Figure 3, top panel). In contrast, in cells pre-treated with pertussis toxin for 60 min, 10 μM GR79236 was no longer able to inhibit the stimulation of sarcolemmal NHE activity by 10 μM phenylephrine (Figure 3, bottom panel). It appears therefore that Gi protein activation is a critical step in the signalling mechanisms downstream of the A1 receptor that mediate the inhibitory effect of GR79236 on α1-adrenergic stimulation of sarcolemmal NHE activity.
Figure 3.
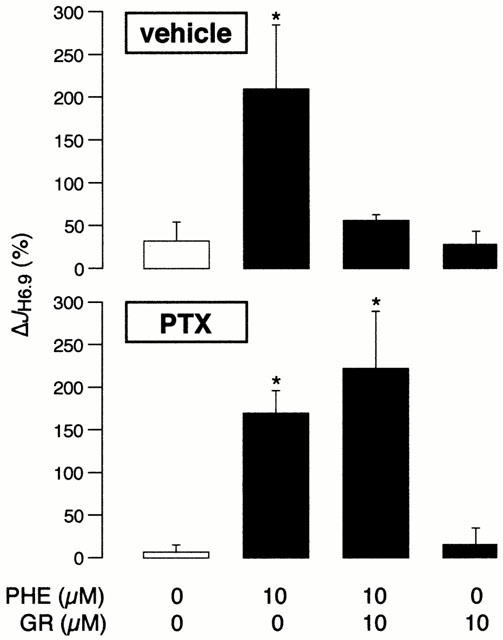
The change in H+ efflux rate at pHi 6.90 (ΔJH6.9) in the control group and in response to phenylephrine (PHE) and GR79236 (GR), alone or in combination. The protocol comprised 64 myocytes (n=8 per group) obtained from 14 hearts. The cells were pre-treated for 60 min with either vehicle (top panel) or 5 μg ml−1 pertussis toxin (PTX; bottom panel). *P<0.05 vs control.
Effects of GR79236 on the response to thrombin
Finally, we determined whether GR79236 could inhibit the stimulation of sarcolemmal NHE activity by another GqPCR agonist, namely thrombin (Yasutake et al., 1996). In this protocol also, JH6.9 values during the first acid pulse were similar between the four study groups (range from 3.75±0.75 to 4.13±0.71 mM min−1; n=8 cells per group, from five hearts). As expected from our earlier work (Yasutake et al., 1996), relative to control (ΔJH6.9 14±19%), 5 u ml−1 thrombin significantly increased sarcolemmal NHE activity (ΔJH6.9 126±34%); however, this response was attenuated by 1 μM GR79236 (ΔJH6.9 50±22%) and abolished by 10 μM GR79236 (ΔJH6.9 26±16%). These data suggest that GR79236 can inhibit the stimulation of sarcolemmal NHE activity by multiple GqPCR agonists.
Discussion
Although the anti-β1-adrenergic effect of adenosine A1 receptor stimulation (thought to be mediated primarily through a Gi protein-mediated reduction in adenylyl cyclase activity) is well established, the present data represent the first demonstration of an anti-α1-adrenergic effect of A1 receptor stimulation in cardiac myocytes. Specifically, our data have shown that the A1 agonist GR79236 inhibits α1-adrenergic stimulation of sarcolemmal NHE activity, a response that is mediated via the α1A-AR (Yokoyama et al., 1998). The inhibitory effect of GR79236 occurs via stimulation of the A1 receptor (based on its reversibility by the A1 antagonist DPCPX) and requires a functional Gi protein (based on its abolition following pertussis toxin pre-treatment). Importantly, the effect was not limited to inhibition of the α1-adrenergic response, since GR79236 also inhibited the stimulation of sarcolemmal NHE activity by thrombin, a response that is mediated by the thrombin receptor (Yasutake et al., 1996), now termed protease-activated receptor 1 (PAR1). Since both the α1A-AR and PAR1 are members of the GqPCR family, our data suggest the existence of a novel inhibitory crosstalk mechanism between the GiPCR adenosine A1 receptor and multiple GqPCRs, at least in rat ventricular myocytes. If confirmed, such a mechanism may have implications beyond the regulation of sarcolemmal NHE activity.
In view of the important role that sarcolemmal NHE activity is believed to play in the development of myocardial injury and dysfunction during ischaemia and reperfusion (Avkiran, 1999a), inhibition of GqPCR-mediated NHE activation is likely to contribute to the mechanisms underlying the cardioprotective effects of adenosine A1 receptor stimulation, by GR79236 (Louttit et al., 1999) and other interventions (Lasley & Mentzer, 1996). However, during ischaemia and reperfusion, sarcolemmal NHE activity may be stimulated not only by a variety of endogenous mediators (e.g. catecholamines, thrombin, endothelin) that act via GqPCRs, but also by factors such as oxidant stress and exposure to lipid metabolites (Avkiran, 1999b). Therefore, it would be of interest to determine whether A1 receptor stimulation attenuates the stimulation of sarcolemmal NHE activity by these additional factors.
Of relevance to the present work, increased sarcolemmal NHE activity and consequent increases in pHi and/or intracellular [Na+] have been suggested also to be causally involved in the positive inotropic and hypertrophic consequences of myocardial α1-AR stimulation. In the light of our data, it is reasonable to expect that adenosine A1 receptor stimulation may attenuate these effects. Indeed, there is preliminary evidence that, in rat ventricular myocytes stimulated at 0.5 Hz, GR79236 inhibits the positive effects of phenylephrine on (1) cell shortening under normal conditions, and (2) the recovery of cell shortening following intracellular acidosis (P. Krishnan and J.C. Kentish, King's College London, personal communication).
Our data suggest that the inhibitory effect of adenosine A1 receptor stimulation on the α1-adrenergic response is mediated via the Gi protein. However, the relevant mechanisms that are downstream of Gi protein activation are unclear. In view of the inability of GR79236 to significantly reduce NHE activity when given alone (e.g. Figures 1 and 3), the inhibitory effect is unlikely to reflect functional antagonism through an independent signalling pathway. Rather, it appears that A1 receptor stimulation may initiate events that interfere with the NHE-regulatory signalling mechanisms that lie distal to the α1A-AR. Our recent work has shown that α1A-AR-mediated stimulation of sarcolemmal NHE activity in rat ventricular myocytes requires the activation of both protein kinase C (PKC) and extracellular signal-regulated kinases 1 and 2 (Snabaitis et al., 2000). PKC activation has been shown to be necessary also for thrombin-induced stimulation of sarcolemmal NHE activity (Yasutake et al., 1996). In this context, it is interesting to note that stimulation of adenosine A1 receptors in rat isolated hearts and ventricular myocytes attenuates the effects of dioctanoylglycerol, a direct activator of PKC, on contractility and Ca2+ handling (Narayan et al., 1998). Furthermore, there is recent evidence that, in rat ventricular myocytes, activation of protein phosphatases contributes to the anti-β1-adrenergic mechanisms of A1 receptor stimulation (Narayan et al., 2000). It is likely that the novel inhibitory effects of adenosine A1 receptor stimulation reported here are mediated through changes in the activity of NHE-regulatory kinases and/or the phosphorylation status of their pertinent substrates, although this remains to be confirmed by further investigation.
Acknowledgments
This work was supported by the British Heart Foundation (BS/93002) and GlaxoWellcome.
Abbreviations
- AR
adrenoceptor
- CABG
coronary artery bypass graft
- DPCPX
1,3-dipropyl-8-cyclopentylxanthine
- GPCR
G protein-coupled receptor
- NHE
Na+/H+ exchanger
- PAR1
protease-activiated receptor 1
References
- AVKIRAN M. Rational basis for use of sodium-hydrogen exchange inhibitors in myocardial ischemia. Am. J. Cardiol. 1999a;83:10G–18G. doi: 10.1016/s0002-9149(99)00215-5. [DOI] [PubMed] [Google Scholar]
- AVKIRAN M. Cardiac sarcolemmal Na+/H+ exchanger activity in ischaemia: potential regulatory factors. Eur. Heart J. Suppl. 1999b;1:K11–K17. [Google Scholar]
- FLIEGEL L., DYCK J.R.B. Molecular biology of the cardiac sodium/hydrogen exchanger. Cardiovasc. Res. 1995;29:155–159. [PubMed] [Google Scholar]
- GUNASEGARAM S., HAWORTH R.S., HEARSE D.J., AVKIRAN M. Regulation of sarcolemmal Na+/H+ exchanger activity by angiotensin II in adult rat ventricular myocytes: opposing actions via AT1 versus AT2 receptors. Circ. Res. 1999;85:919–930. doi: 10.1161/01.res.85.10.919. [DOI] [PubMed] [Google Scholar]
- GURDEN M.F., COATES J., ELLIS F., EVANS B., FOSTER M., HORNBY E., KENNEDY I., MARTIN D.P., STRONG P., VARDEY C.J., WHEELDON A. Functional characterization of three adenosine receptor types. Br. J. Pharmacol. 1993;109:693–698. doi: 10.1111/j.1476-5381.1993.tb13629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LASLEY R.D., MENTZER R.M. Myocardial protection: the adenosine story. Drug Develop. Res. 1996;39:314–318. [Google Scholar]
- LEEM C.H., LAGADIC-GOSSMANN D., VAUGHAN-JONES R.D. Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J. Physiol. 1999;517:159–180. doi: 10.1111/j.1469-7793.1999.0159z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOUTTIT J.B., HUNT A.A.E., MAXWELL M.P., DREW G.M. The time course of cardioprotection induced by GR79236, a selective adenosine A1-receptor agonist, in myocardial ischaemia-reperfusion injury in the pig. J. Cardiovasc. Pharmacol. 1999;33:285–291. doi: 10.1097/00005344-199902000-00016. [DOI] [PubMed] [Google Scholar]
- MAHAFFEY K.W., PUMA J.A., BARBAGELATA A., DICARLI M.F., LEESAR M.A., BROWNE K.F., EISENBERG P.R., BOLLI R., CASAS A.C., MOLINA-VIAMONTE V., ORLANDI C., BLEVINS R., GIBBONS R.J., CALIFF R.M., GRANGER C.B. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial, the acute myocardial infarction study of adenosine (AMISTAD) trial. J. Am. Coll. Cardiol. 1999;34:1711–1720. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- MATHERNE G.P., LINDEN J., BYFORD A.M., GAUTHIER N.S., HEADRICK J.P. Transgenic A1 adenosine receptor overexpression increases myocardial resistance to ischemia. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6541–6546. doi: 10.1073/pnas.94.12.6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENTZER R.M., BIRJINIUK V., KHURI S., LOWE J.E., RAHKO P.S., WEISEL R.D., WELLONS H.A., BARKER M.L., LASLEY R.D. Adenosine myocardial protection: preliminary results of a phase II clinical trial. Ann. Surg. 1999;229:643–650. doi: 10.1097/00000658-199905000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARAYAN P., MENTZER R.M., LASLEY R.D. Phosphatase inhibitor cantharidin blocks adenosine A1 receptor anti-adrenergic effect in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2000;278:H1–H7. doi: 10.1152/ajpheart.2000.278.1.H1. [DOI] [PubMed] [Google Scholar]
- NARAYAN P., VALDIVIA H.H., MENTZER R.M., LASLEY R.D. Adenosine A1 receptor stimulation antagonizes the negative inotropic effects of the PKC activator dioctanoylglycerol. J. Mol. Cell. Cardiol. 1998;30:913–921. doi: 10.1006/jmcc.1998.0648. [DOI] [PubMed] [Google Scholar]
- OBAYASHI K., HORIE M., XIE L.-H., TSUCHIYA K., KUBOTA A., ISHIDA H., SASAYAMA S. Angiotensin II inhibits protein kinase A-dependent chloride conductance in heart via pertussis toxin-sensitive G proteins. Circulation. 1997;95:197–204. doi: 10.1161/01.cir.95.1.197. [DOI] [PubMed] [Google Scholar]
- RUPPRECHT H.-J., VOM DAHL J., TERRES W., SEYFARTH K.M., RICHARDT G., SCHULTHEISS H.-P., BUERKE M., SHEEHAN F.H., DREXLER H. Cardioprotective effects of the Na+/H+ exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation. 2000;101:2902–2908. doi: 10.1161/01.cir.101.25.2902. [DOI] [PubMed] [Google Scholar]
- SNABAITIS A.K., YOKOYAMA H., AVKIRAN M. Roles of mitogen-activated protein kinases and protein kinase C in α1A-adrenoceptor-mediated stimulation of the sarcolemmal Na+/H+ exchanger. Circ. Res. 2000;86:214–220. doi: 10.1161/01.res.86.2.214. [DOI] [PubMed] [Google Scholar]
- THÉROUX P., CHAITMAN B.R., DANCHIN N., ERHARDT L.R.W., MEINERTZ T., SCHRODER J.S., TOGNONI G., WHITE H.D., WILLERSON J.T., JESSEL A.Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations: main results of the GUARDIAN trial Circulation 2000. ‘in press' [DOI] [PubMed]
- YASUTAKE M., HAWORTH R.S., KING A., AVKIRAN M. Thrombin activates the sarcolemmal Na+/H+ exchanger: evidence for a receptor-mediated mechanism involving protein kinase C. Circ. Res. 1996;79:705–715. doi: 10.1161/01.res.79.4.705. [DOI] [PubMed] [Google Scholar]
- YOKOYAMA H., AVKIRAN M.Adenosine A1 receptor stimulation inhibits α1-adrenergic activation of the sarcolemmal Na+/H+ exchanger J. Mol. Cell. Cardiol. 200032A55(Abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOKOYAMA H., YASUTAKE M., AVKIRAN M. α1-Adrenergic stimulation of sarcolemmal Na+/H+ exchanger activity in rat ventricular myocytes: evidence for selective mediation by the α1A-adrenoceptor subtype. Circ. Res. 1998;82:1078–1085. doi: 10.1161/01.res.82.10.1078. [DOI] [PubMed] [Google Scholar]