

Abstract
In a hamster model (genetic symbol dtsz) of primary paroxysmal non-kinesiogenic dystonic choreoathetosis, recent studies have shown beneficial effects of glutamate and dopamine receptor antagonists. Nitric oxide (NO), synthesized from L-arginine by NO synthase in response to glutamate receptor activation, elicits cyclic GMP and modulates glutamate-mediated processes and striatal dopamine release.
Therefore, the effects of NO synthase inhibitors and of L-arginine on severity of dystonia were investigated in dtsz hamsters in which dystonic attacks, characterized by twisting movements and postures, can be induced by stress.
The NO synthase inhibitors NG-nitro-L-arginine (L-NNA), NG-nitro-L-arginine methyl ester (L-NAME) and 7-nitroindazole significantly reduced the severity of dystonia. At antidystonic effective doses neither L-NNA nor L-NAME caused observable side effects, whereas 7-nitroindazole exerted moderate reduction of locomotor activity.
The antidystonic effect of L-NAME was reversed by co-administration of the NO precursor L-arginine. However, L-arginine administered alone did not exert any effect on severity of dystonia.
Cerebellar cyclic GMP levels in brains of mutant hamsters in comparison to non-dystonic control hamsters did not significantly differ, but the cerebellar cyclic GMP levels tended to be increased in dtsz hamsters during a dystonic attack. L-NAME significantly decreased the cerebellar cyclic GMP levels in both dtsz and control hamsters.
Although an overproduction of NO is probably not critically involved in the pathogenesis of paroxysmal dystonia, it may contribute to the manifestation of dystonic attacks, as indicated by the antidystonic effects of NO synthase inhibitors.
Peripheral side effects may limit the clinical use of NO synthase inhibitors, but more selective inhibitors of the neuronal NO synthase should be considered as interesting candidates for the treatment of paroxysmal dystonia.
Keywords: Dystonia, nitric oxide, glutamate, movement disorders
Introduction
Nitric oxide (NO) is an important messenger molecule implicated in multiple physiological and pathophysiological events in the central nervous system (Moncada et al., 1991; Schuman & Madison, 1994). NO is synthesized from L-arginine by NO synthase in response to Ca2+ entry through the activated N-methyl-D-aspartate (NMDA) receptor-gated ion channels. NO diffuses rapidly and, as shown particularly in the cerebellum of rodents, exerts its action in surrounding target cells by increasing cyclic GMP levels (Garthwaite et al., 1988, Schuman & Madison, 1994). It seems likely that NO acts via cyclic GMP dependent mechanisms by modulating the release of various neurotransmitters (Prast et al., 1994; Schuman & Madison, 1994), e.g., NO has been shown to enhance the striatal dopamine release (Bowyer et al., 1995; Stewart et al., 1996). NMDA receptor-mediated neurotransmitter release is regulated, in part, through NO production and can be blocked by NO synthase inhibitors (Dawson & Dawson 1996).
With regard to the close interaction of NMDA-receptor and NO-mediated processes, NO synthase inhibitors have been investigated in experimental models of neurological diseases associated with abnormal motor behaviour, such as parkinsonism and epilepsy, in which enhanced NMDA-receptor activation is thought to be involved in the pathophysiology (e.g. Eblen et al., 1996, Rundfeldt et al., 1995). Primary dystonias, a group of a relatively common movement disorder characterized by sustained muscle contractions causing twisting movements or abnormal postures, have been presumed to be caused by biochemical dysfunctions, such as by dopaminergic overactivity or dopaminergic deficits in the striatum, and altered neuronal activity within the basal ganglia. Apart from the Dopa-responsive dystonia, however, no consistent abnormality has been identified as yet and the benefit of medical treatment is often insufficient (Fahn 1995; McGeer & McGeer, 1995). The knowledge about the pathophysiological relevance of excitatory amino acids in dystonias and efficacy of glutamate receptor antagonists is sparse (McGeer & McGeer, 1995, Richter & Löscher, 1998). To our knowledge, there are no data on the effects of NO synthase inhibitors in dystonias.
Recent pharmacological and neurochemical studies in mutant dystonic hamsters (gene symbol dtsz), a genetic animal model of primary paroxysmal non-kinesiogenic dystonic choreoathetosis, indicated that enhanced activation of NMDA receptors and striatal dopaminergic overactivity is involved in this subtype of dystonia in which dystonic attacks can be provoked by stress and anxiety (Nobrega et al., 1997; Rehders et al., 2000; Richter et al., 1991; Richter & Löscher 1997). Although there is evidence that glutamatergic and dopaminergic overactivity probably does not represent the primary defect, these dysfunctions seem to contribute to the manifestation of dystonic episodes in this animal model (for review see Richter & Löscher, 1998). These recent findings prompted us to examine the role of NO in dystonia in the hamster model.
Four distinct isoforms of NO synthase have been identified (Schuman & Madison, 1994). NG-nitro-L-arginine (L-NNA) and NG-nitro-L-arginine methyl ester (L-NAME) and 7-nitroindazole inhibit endothelial and neuronal NO synthase isoforms (Moore et al., 1991; Moore & Handy, 1997). Depending on the used compound, dose and pretreatment time, there are conflicting data on effects of NO synthase inhibitors in animal models of epilepsy, parkinsonism or cerebral ischaemia (Eblen et al., 1996; Rundfeldt et al., 1995; Schuman & Madison, 1994). Therefore, in the present study, we examined the effects of different NO synthase inhibitors, i.e. L-NNA, L-NAME and 7-nitroindazole, as well as the effects of the NO precursor L-arginine on the severity of dystonia. Furthermore, cyclic GMP levels were determined in brains of dtsz hamsters and non-dystonic control hamsters before and after treatment with L-NAME.
Methods
Animals
The present experiments were carried out in groups of dtsz hamsters which were obtained by selective breeding (for detailed descriptions see Löscher et al., 1989; Richter & Löscher, 1998). For measurements of cyclic GMP additional groups of age-matched non-dystonic hamsters of an outbred line were used. Dystonic attacks in dtsz mutant hamsters, characterized by generalized twisting movements and abnormal postures of limbs and trunk, can be induced by handling and mild environmental stimuli (Löscher et al., 1989; Richter & Löscher, 1998).
As shown by previous clinical, electrophysiological and pharmacological studies, the dtsz hamster shows all characteristics of primary paroxysmal non-kinesiogenic dystonia (for review see Richter & Löscher, 1998). Similar to primary dystonia in humans, dystonia in mutant hamsters occurs in the absence of morphological alterations in the brain or spinal cord (Wahnschaffe et al., 1990). Brain regions with abnormal neuronal activity are the basal ganglia, thalamic and deep cerebellar nuclei and the red nucleus (Richter & Löscher, 1998).
In mutant hamsters the severity of dystonia is age-dependent with maximum severity between the 30th and 40th day of life (max-period, suitable to study antidystonic effects of drugs). Thereafter, the severity of dystonia slowly declines (post-max period, suitable to examine prodystonic drug-effects) until complete remission of dystonia appears with the age of about 10 weeks (Richter & Löscher, 1993).
Drug testing
In dtsz hamsters dystonic attacks can be induced by the procedure of triple stimulation (Löscher et al., 1989), i.e. (1) taking the animal from its home cage and placing it on a balance, (2) i.p. injection of vehicle (pre- and post-drug control) or of the drug (i.e. in the present study of NG-nitro-L-arginine methyl ester (L-NAME), NG-nitro-L-arginine (L-NNA), 7-nitroindazole or L-arginine), (3) placement of the hamster in a new environment. The severity of dystonia was rated by the following score-system (Löscher et al., 1989): stage 1, flattened ears and flattened posture; stage 2, facial contortions, rearing with forelimbs crossing, disturbed gait with retarded setting of the forepaws; stage 3, stiffened hindlimbs so that the animals appear to walk on tiptoes; stage 4, twisting movements and loss of balance; stage 5, hindlimbs hyperextended caudally; stage 6, immobilization in a twisted, hunched posture with hind- and forelimbs tonically extended forward and opisthotonus. The examiner of the severity of dystonia was unaware of the drugs used in the present investigations. The individual maximum stage of dystonia is usually reached within 3 h after the hamsters were placed in the new cage. Therefore the hamsters were observed for 3 h. During this period the severity of dystonia, the latencies to the different stages and, in case of drug trials, the side effects (not quantified) were noticed. After reaching the maximum stage the hamsters usually recover within 2–5 h. The control trials were done 2 days before and 2 days after drug testing.
L-NAME (used as HCL), L-NNA and L-arginine, purchased from Sigma (Munich, Germany), were freshly dissolved in water (L-NNA with the aid of dilute HCL). 7-nitroindazole (RBI Biotrend, Cologne, Germany) was freshly suspended in 1% Tween 80. All drugs were administered intraperitoneally. Injection volume was 5 ml kg−1. For control trials (pre- and post-drug recordings) the hamsters received the same volume of vehicle.
The significance of differences in severity of dystonia and latencies to onset of the first unequivocal signs of dystonia (stage 2) and to maximum severity (stage 6) were calculated by the Wilcoxon signed test for paired replicates.
Measurements of cyclic GMP
Determinations of cyclic GMP concentrations were undertaken to examine if antidystonic effects of 50 mg kg−1 L-NAME are accompanied by decreases of cyclic GMP. For the determinations of brain cyclic GMP levels two groups of dtsz mutant hamsters and two groups of control hamsters were decapitated (at the age of 34 days) 3 h after triple stimulation procedure. One group of dtsz and control hamsters was decapitated 3 h after administration of vehicle (basal) and a second group 3 h after administration of L-NAME (50 mg kg−1 i.p.). When the animals were decapitated, dtsz hamsters exhibited severe (basal) or moderate (after L-NAME) dystonia, while no motor disturbances occurred in both groups of control hamsters.
The brains were quickly dissected (frontal cortex, striatum, cerebellum) and homogenized in an ice-cold 6% TCA. The homogenates were centrifuged at 2500×g for 15 min and the supernatants were extracted three times with ether. The remaining homogenates were used for protein determinations. The extracts were vacuum-dried overnight. Dried samples were kept at −80°C until analysis. For cyclic GMP detection, a commercial enzymimmunoassay kit (Biotrak, Amersham) was used. Samples were redissolved in 1 ml assay buffer and 50 μl aliquots were used in the assay. cyclic GMP values were expressed as pmol/mg protein. Protein determinations were done using the method of Lowry et al. (1951) using a microplate reader.
The significance of differences in cyclic GMP levels were calculated with ANOVA and post hoc by the Tukey test.
Results
As shown in Figure 1, L-NAME significantly reduced the severity of dystonia in mutant hamsters at the dose of 50 mg kg−1 during the 2nd and 3rd hour of observation. L-NAME did not exert significant effects on the latency to onset of dystonic symptoms. At 5 or 10 mg kg−1, no significant effects on severity or latency to onset of dystonia were recorded. At all doses tested, L-NAME did not cause any observable adverse effects.
Figure 1.
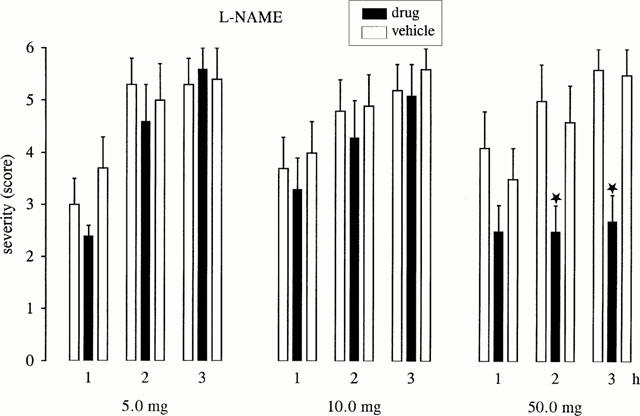
Effect of L-NAME on severity of dystonia in mutant hamsters at the age of maximum severity (max period). Usually, the individual maximum severity of dystonia is reached within 3 h after induction of dystonia by triple stimulation including the i.p. injection of drugs (black bars) or vehicle for pre- and post-drug controls (open bars). The figure shows the average of the maximum individual severity scores of dystonia reached within the 1st, 2nd and 3rd hour after i.p. administration, reflecting the progression of dystonia in mutant hamsters after treatment with the compounds and without drug-treatment (vehicle controls). Control recordings were undertaken 2 days before (pre-drug control) and 2 days after (post-drug control) the drug trial. Asterisks indicate significant reduction of severity in comparison to the pre- and post-drug control (*P<0.05). Data are shown as mean±s.e.mean of seven (5.0 mg kg−1), nine (10.0 mg kg−1) or eight (50.0 mg kg−1) dystonic hamsters.
L-NNA retarded the progression of dystonia in mutant hamsters (Figure 2). The severity of dystonia was decreased during the 1st (75 mg) or 2nd (50 mg) hour of observation. At both doses tested, L-NNA did not exert significant effects on latency to onset of dystonia, but the latency to the maximum severity (stage 6) was significantly increased (P<0.05) from 89.4±22.7 (pre-drug) or 74.0±23.8 (post-drug) to 163.8±4.7 min (50 mg) and from 69.4±6.5 (pre-drug) or 87.0±16.9 (post-drug) to 129.9±16.4 min (75 mg). At both doses no behavioural side effects were observed.
Figure 2.
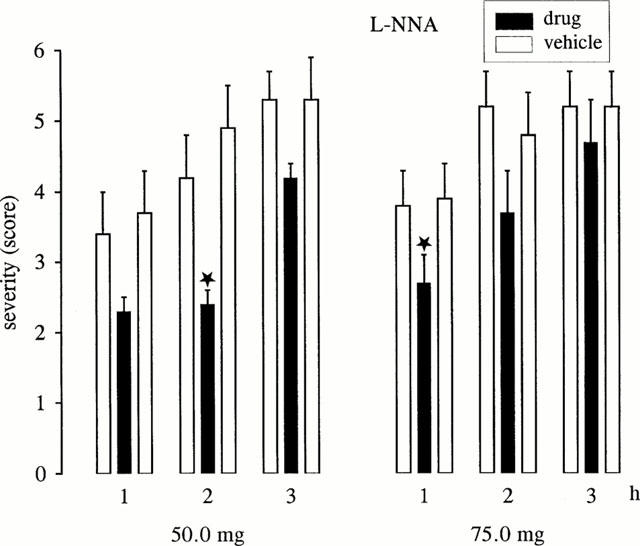
Effect of L-NNA on severity of dystonia in mutant hamsters at the age of maximum severity (max period). The figure shows the average of the maximum individual severity scores of dystonia reached within the 1st, 2nd and 3rd hour after i.p. administration of 50.0 or 75.0 mg kg−1. Control recordings were taken 2 days before (pre-drug control) and 2 days after (post-drug control) the drug trial. Asterisks indicate significant reduction of severity in comparison to the pre-drug and post-drug control (*P<0.05; **P<0.01). Data are shown as mean±s.e.mean of nine (50.0 mg kg−1) or 10 (75.0 mg kg−1) dystonic hamsters.
7-Nitroindazole significantly decreased the severity of dystonia during the 1st and 2nd hour of observation at a dose of 75 mg kg−1, while no effects on severity or latencies were found after administration of 50 mg kg−1 (see Figure 3). At a dose of 75 mg kg−1, 7-nitroindazole significantly (P<0.05) increased the latency to onset of dystonia and the latency to the maximum severity (stage 6) from 74.6±12.5 (pre-drug) or 74.4±4.8 (post-drug) to 121.8±19.6 min. At the antidystonic effective dose, 7-nitroindazole caused a moderate decrease of spontaneous locomotion and in four animals moderate ataxia during the first hour, i.e. 20–60 min after administration, while no side effects were observed at the lower dose of 50 mg. The disappearance of side effects caused by 75 mg kg−1 7-nitroindazole already within the 1st hour after administration and the lack of antidystonic effects during the 3rd hour of observations suggested a short duration of action of 7-nitroindazole. Therefore, the NO synthase inhibitor L-NAME was used for co-administrations with L-arginine and for examinations of the effects on cyclic GMP levels.
Figure 3.
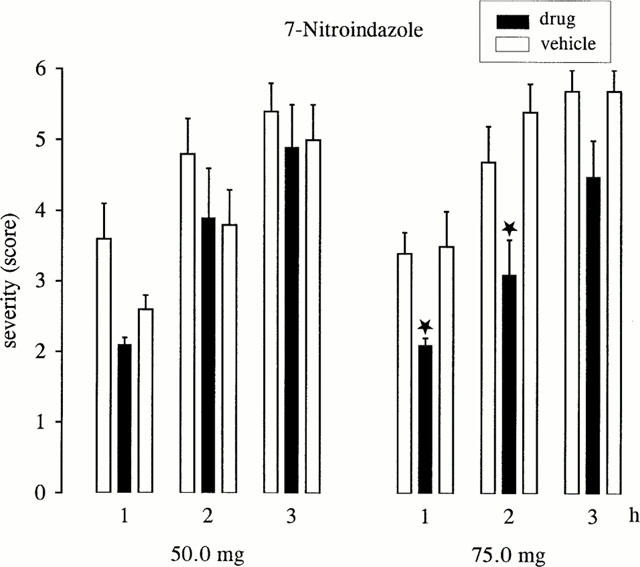
Effect of 7-nitro indazole on severity of dystonia in mutant hamsters at the age of maximum severity of dystonia (max period). The figure shows the average of the maximum individual severity scores of dystonia reached within the 1st, 2nd and 3rd h after i.p. administration of 50.0 or 75.0 mg kg−1. Control recordings were taken 2 days before (pre-drug control) and 2 days after (post-drug control) the drug trial. Data are shown as mean±s.e.mean of nine (50.0 mg kg−1) or 11 (75.0 mg kg−1) dystonic hamsters. Asterisks indicate significant reduction of severity in comparison to the pre- and post-drug control (*P<0.05).
The NO precursor L-arginine did not exert any effects on the severity of dystonia and on latencies to onset or to maximum severity (Figure 4). Even high doses of L-arginine did not cause observable behavioural side effects. As shown in Figure 4, the antidystonic efficacy of L-NAME was reversed by co-administration of L-arginine.
Figure 4.
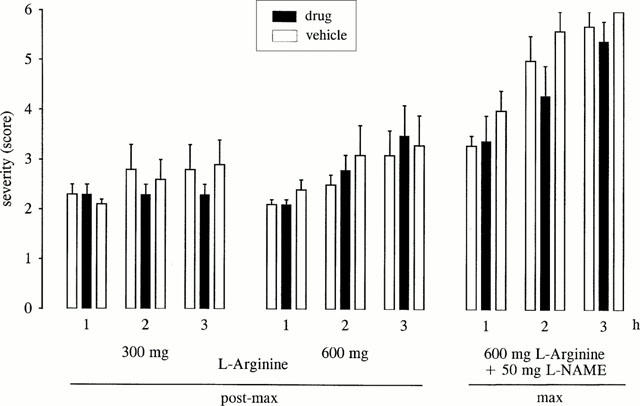
Effect of the NO precursor L-arginine on severity of dystonia in mutant hamsters administered alone (300 or 600 mg kg−1) at an age of 50–55 days (at which the severity of dystonia is decreased, post-max period) or in co-administration with L-NAME (600 mg L-arginine+50 mg L-NAME) at the age of maximum severity. The figure shows the average of the maximum individual severity scores of dystonia reached within the 1st, 2nd and 3rd hour after i.p. administrations. Control recordings were taken 2 days before (pre-drug control) and 2 days after (post-drug control) the drug trial. Data are shown as mean±s.e.mean of eight dystonic hamsters.
As shown in Figure 5, no significant differences of cerebellar cyclic GMP levels could be detected between groups of mutant hamsters and non-dystonic control animals (P=0.092) which were decapitated 3 h after triple stimulation procedure including the injection of vehicle (basal). The cyclic GMP concentration only tended to be increased in mutant hamsters which exhibited severe dystonia (mean 5.1±0.6) after vehicle injections. Antidystonic effective doses of L-NAME (50 mg kg−1) significantly decreased cyclic GMP concentrations in the cerebellum in both mutant hamsters and control animals. The dtsz hamsters showed only moderate dystonia (mean 2.6±0.5) before decapitation, i.e. 3 h after administration of L-NAME, supporting the marked antidystonic efficacy of this NO synthase inhibitor. The cyclic GMP levels in the striatum and frontal cortex of dtsz and control hamsters (not illustrated) were too low to allow any comparisons between the groups, possibly because higher volumes of aliquots of the samples were necessary for analyses of cyclic GMP levels in these regions of hamster brains than those used in the present study, which have been shown to be suitable for determinations in rat brains (Eblen et al., 1996).
Figure 5.
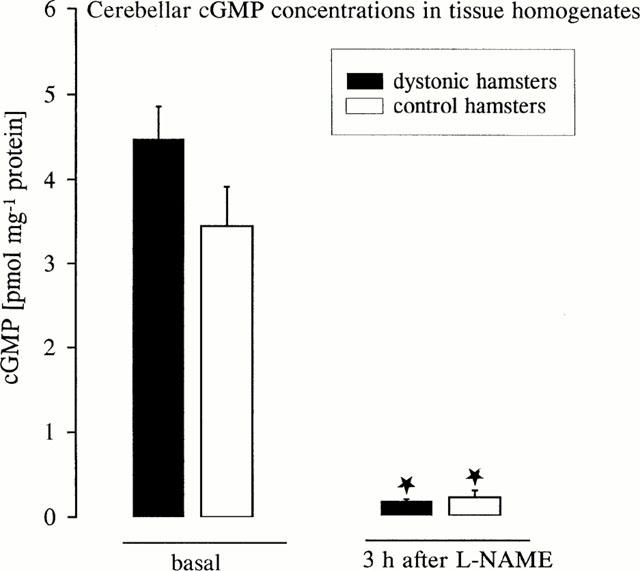
Levels of cyclic GMP in the cerebellum of mutant dtsz hamster and control hamsters 3 h after administration of vehicle (basal) or after treatment with L-NAME (50 mg kg−1). Asterisks indicate significant reduction of cyclic GMP after treatment with L-NAME in comparison to basal levels (*P<0.001). Data are shown as mean±s.e.mean of 7–9 animals.
Discussion
The present data demonstrate for the first time antidystonic activity of NO synthase inhibitors at doses which did not cause marked central side effects in dtsz mutant hamsters. As reported for various other effects of NO synthase inhibitors in rats (Connop et al., 1995; Rundfeldt et al., 1995), the different NO synthase inhibitors showed a different time-course of antidystonic efficacy in mutant hamsters. In contrast to L-NNA and 7-nitroindazole, L-NAME did not only retard the progression of dystonia, but significantly reduced the severity of dystonia during the whole period of observation. In rodents, the duration of action of 7-nitroindazole, which shows a trend towards higher inhibition of neuronal NO synthase (Moore et al., 1991; Moore & Handy, 1997), is known to be shorter than that of L-NAME which inhibits endothelial and neuronal NO synthase (Iadecola et al., 1994; MacKenzie et al., 1994). Therefore, the more marked antidystonic effects of L-NAME in comparison with 7-nitroindazole is probably based on its longer action but not to the different affinity to the NO synthase isoforms, also indicated by the low antidystonic activity of L-NNA. The measurements of cerebellar cyclic GMP levels 3 h after administration of the antidystonic effective dose of L-NAME indicate that the beneficial effect was accompanied by a pronounced inhibition of neuronal NO synthase.
Since NO synthase inhibitors share the antidystonic action of NMDA receptor antagonists, previously described in mutant hamsters (Löscher & Richter, 1993; Richter & Löscher, 1997; Richter et al., 1991), NO could serve as a mediator of pathophysiologically increased glutamatergic transmission involved in the manifestation of a dystonic attack. Recent determinations of excitatory amino acid concentrations in discrete brain regions of dtsz hamsters, examined in the absence of dystonia, failed to show significant changes (Löscher & Hörstermann, 1992). Furthermore, autoradiographic analysis of NMDA receptor density in 67 brain regions, using the ligand [3H]N-(1-[2-thienyl]cyclohexyl)3,4-piperidine ([3H]-TCP), which binds to the phencyclidine site in the ion channel of the NMDA receptor channel, demonstrated that NMDA receptor binding is not substantially altered in mutant hamsters in the absence of dystonic attacks compared to age-matched controls (Nobrega et al., 1997). However, during a dystonic episode there was a tendency towards enhanced binding in most regions, including a significant increase in the ventrolateral thalamic nucleus (Nobrega et al., 1997). The consequence of enhanced glutamatergic transmission during a dystonic attack, as suggested by the increased [3H]-TCP binding in dystonic brains during manifestation of dystonia (Nobrega et al., 1997) and antidystonic effects of competitive and non-competitive NMDA receptor antagonists (Richter et al., 1991; Richter & Löscher, 1997), could be an enhanced NO synthesis, leading by increasing cyclic GMP levels to enhanced release of glutamate and dopamine, as shown by several in vivo and in vitro studies (e.g. Bowyer et al., 1995; Prast et al., 1994; Stewart et al., 1996). These considerations are not clearly supported by the measurements of cyclic GMP levels, but there was, at least, a tendency of enhanced cyclic GMP levels in brains of mutant hamsters which exhibited severe dystonia. In contrast, in the dt rat, a genetic animal model of permanent dystonia in which cerebellar dysfunctions play a critical role, decreased cyclic GMP levels were found in the cerebellum (Lorden et al., 1985). It should be noted that the cerebellum seems not to be the site of the primary defect in mutant hamsters and that cerebellar cyclic GMP levels are probably not important for dystonia in this animal model. The present study does not exclude significant increases of cyclic GMP levels in pathophysiologically more important brain regions, such as thalamic nuclei, or in subregions, such as the dorsal striatum (Richter & Löscher, 1998). Furthermore, it remains unclear whether the inhibition of endothelial NO synthase and effects on vascular tone are involved the antidystonic action of NO synthase inhibitors. Thus, the present pharmacological data give rise to further investigations of the role of NO in the pathophysiology of dystonia. Immunhistochemical examinations of endothelial and neuronal NO synthase in different brain regions of dtsz hamsters are under way.
With regard to antidystonic effects of the NO synthase inhibitors in mutant hamsters, aggravation of dystonia would have been expected to be caused by L-arginine, the biological precursor for NO synthesis (Moncada et al., 1991). By itself, L-arginine, however, failed to exert prodystonic effects even at a high dose of 600 mg kg−1 which reversed the beneficial effect of L-NAME. This result, which is difficult to explain, is consistent with observed effects on locomotor activity in normal mice (Starr & Starr, 1995). While L-arginine (500 mg kg−1), administered alone, had no effect on locomotor activity in mice, the amino acid reversed the reduction of locomotor activity induced by a high dose (125 mg kg−1) of L-NAME (Starr & Starr, 1995). Nevertheless, the lack of prodystonic effects of L-arginine in dtsz hamsters argues against a primary role of enhanced glutamatergic activity and NO formation in the pathogenesis of primary dystonia, which is in line with the previous neuro-chemical findings (Löscher & Hörstermann, 1992; Nobrega et al., 1997), as described above. Actually, several lines of evidence suggest that dystonia in mutant hamsters is primarily caused by disturbed GABAergic inhibition (Richter & Löscher, 1998), leading to overactivity of the glutamatergic and dopaminergic system which both seem to contribute to the manifestation of a dystonic attack.
Since recent dopamine receptor autoradiographic studies and pharmacological examinations indicated that an enhanced striatal activity of dopamine is involved in the dystonic syndrome in mutant hamsters (Rehders et al., 2000; Richter & Löscher, 1998), the inhibitory effects of NO synthase inhibitors on striatal dopamine release (Bowyer et al., 1995) may be relevant for their antidystonic activity and for the reduction of locomotor activity, observed after administration of high doses of 7-nitroindazole in the present study and also described in mice (Starr & Starr, 1995). Furthermore, the anxiolytic effect of 7-nitroindazole is possibly related to antidopaminergic activity (Dunn et al., 1998). With regard to induction of dystonic attacks by stress and beneficial effects of benzodiazepines in dtsz hamsters and in patients with paroxysmal non-kinesiogenic dystonic choreoathetosis, anxiolytic effects of NO synthase inhibitors may be an important component for the antidystonic efficacy.
In contrast to NMDA receptor antagonists (Richter et al., 1991; Richter & Löscher, 1997), NO synthase inhibitors exerted antidystonic effects at doses which did not cause marked side effects in mutant hamsters. Although L-NAME and L-NNA did not exert observable central adverse effects, these compounds are known to inhibit the activity of endothelial NO synthase and cause pronounced increases of arterial blood pressure at doses which exerted antidystonic effects in the hamster model (Dwyer et al., 1991) which limit the suitability for clinical treatment of movement disorders. However, more selective inhibitors of the neuronal NO synthase might be interesting candidates for treatment of dystonia.
Acknowledgments
This study was supported by grants from the Deutsche Forschungsgemeinschaft (Lo 271/4-3). We thank Christiane Bartling for technical assistance.
Abbreviations
- L-NAME
NG-nitro-L-arginine methyl ester
- L-NNA
NG-nitro-L-arginine
References
- BOWYER J.F., CLAUSING P., GOUGH B., SLIKKER W., HOLSON R.R. Nitric oxide regulation of methamphetamine-induced dopamine release in caudate putamen. Brain Res. 1995;699:62–70. doi: 10.1016/0006-8993(95)00877-s. [DOI] [PubMed] [Google Scholar]
- CONNOP B.P., KALISCH B.E., BOEGMAN R.J., JHAMANDAS K., BENINGER R.J. Enhancement of 7-nitro indazole-induced inhibition of brain nitric oxide synthase by norharmane. Neurosci. Lett. 1995;190:69–72. doi: 10.1016/0304-3940(95)11502-n. [DOI] [PubMed] [Google Scholar]
- DAWSON T.M., DAWSON V.L. Nitric oxide synthase: role as a transmitter/mediator in the brain and endocrine system. Annu. Rev. Med. 1996;47:219–227. doi: 10.1146/annurev.med.47.1.219. [DOI] [PubMed] [Google Scholar]
- DUNN R.W., REED T.A.W., COPELAND P.D., FRYE C.A. The nitric oxide synthase inhibitor 7-nitroindazole displays enhanced anxiolytic efficacy without tolerance in rats following subchronic administration. Neuropharmacology. 1998;37:899–904. doi: 10.1016/s0028-3908(98)00076-8. [DOI] [PubMed] [Google Scholar]
- DWYER M.A., BREDT D.S., SNYDER S.H. Nitric oxide synthase: irreversible inhibition by L-NG-nitroarginine in brain in vitro and in vivo. Biochem. Biophys. Res. Commun. 1991;176:1136. doi: 10.1016/0006-291x(91)90403-t. [DOI] [PubMed] [Google Scholar]
- EBLEN F., LÖSCHMANN P.-A., WÜLLNER U., TURSKI L., KLOCKGETHER T. Effects of 7-nitroindazole, NG-nitro-L-arginine, and D-CPPene on harmaline-induced postural tremor, N-methyl-D-aspartate-induced seizures, and lisuride-induced rotations in rats with nigral 6-hydroxydopamine lesions. Eur. J. Pharmacol. 1996;299:9–16. doi: 10.1016/0014-2999(95)00795-4. [DOI] [PubMed] [Google Scholar]
- FAHN S. Medical treatment of dystonia Handbook of dystonia 1995New York: Marcel Dekker; 317–328.ed. Tsui, J.K.C., Calne, D.B. pp [Google Scholar]
- GARTHWAITE J., CHARLES S.L., CHESS-WILIAMS R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- IADECOLA C., XU X., ZHANG F., HU J., EL-FAKAHANY E.E. Prolonged inhibition of brain nitric oxide synthase by short-term systemic administration of nitro-L-arginine methyl ester. Neurochem. Res. 1994;19:501–505. doi: 10.1007/BF00967330. [DOI] [PubMed] [Google Scholar]
- LORDEN J.F., OLTMANS G.A., McKEON T.W., LUTES J., BEALES M. Decreased cerebellar 3′5′-cyclic guanosine monophosphate levels and insensitivity to harmaline in the genetically dystonic rat (dt) J. Neurosci. 1985;5:2618–2625. doi: 10.1523/JNEUROSCI.05-10-02618.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LÖSCHER W., FISHER J.E. , Jr, SCHMIDT D., FREDOW G., HÖNACK D., ITURRIAN W.B. The sz mutant hamster: a genetic model of epilepsy or of paroxysmal dystonia. Movement Dis. 1989;4:219–232. doi: 10.1002/mds.870040304. [DOI] [PubMed] [Google Scholar]
- LÖSCHER W., HÖRSTERMANN D. Abnormalities in amino acid neurotransmitters in discrete brain regions of genetically dystonic hamsters. J. Neurochem. 1992;59:689–694. doi: 10.1111/j.1471-4159.1992.tb09423.x. [DOI] [PubMed] [Google Scholar]
- LÖSCHER W., RICHTER A. The glycine/NMDA receptor ligand (+)-HA-966 but not D-cycloserine exert antidystonic efficacy in a genetic animal model of dystonia. Europ. J. Pharmacol. 1993;239:245–247. doi: 10.1016/0014-2999(93)91004-7. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDELL R.J. Protein measurement with Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MACKENZIE G.M., ROSE R., BLAND-WARD P.A., MOORE P.K., JENNER P., MARSDEN C.D. Time course of inhibition of brain nitric oxide synthase by 7-nitro indazole. NeuroReport. 1994;5:1993–1996. doi: 10.1097/00001756-199410000-00039. [DOI] [PubMed] [Google Scholar]
- MCGEER E.G., MCGEER P.L. Pathology of dystonias Handbook of dystonia 1995New York: Marcel Dekker; 77–102.eds. Tsui J.K.C. & Calne, D.B. pp [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- MOORE P.K., BADDEDGE R.C., WALLACE P., GAFFEN Z.A., HART S.L. 7-Nitro indazole, an inhibitor of nitric oxide synthase, exhibits anti-nociceptive activity in the mouse without increasing blood pressure. Br. J. Pharmacol. 1991;108:296–297. doi: 10.1111/j.1476-5381.1993.tb12798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOORE P.K., HANDY R.L.C. Selective inhibitors of neuronal nitric oxide synthase - is no NOS really good NOS for the nervous system. Trends Pharmacol. Sci. 1997;18:204–206. doi: 10.1016/s0165-6147(97)01064-x. [DOI] [PubMed] [Google Scholar]
- NOBREGA J.N., RICHTER A., JIWA D., RAYMOND R., LÖSCHER W. Alterations in N-methyl-D-aspartate receptor binding in dystonic hamster brains. Brain Res. 1997;744:161–165. doi: 10.1016/s0006-8993(96)01209-7. [DOI] [PubMed] [Google Scholar]
- PRAST H., FISCHER H., GRASS K., PHILIPPU A. Nitric oxide is a modulator of acetylcholine and glutamate release in the ventral striatum Monitoring molecules in neuroscience 1994261–262.Proceedings of the 6th International Conference on in vivo Methods. eds. Louillot, A., Durkin T., Spampinato, U. & Cador, M. pp
- REHDERS J.H., LÖSCHER W., RICHTER A. Evidence for striatal dopaminergic overactivity in paroxysmal dystonia indicated by microinjections in a genetic rodent model. Neuroscience. 2000;97:267–277. doi: 10.1016/s0306-4522(00)00073-7. [DOI] [PubMed] [Google Scholar]
- RICHTER A., FREDOW G., LÖSCHER W. Antidystonic effects of the NMDA receptor antagonists memantine, MK-801 and CGP 37849 in a hamster model of paroxysmal dystonia. Neurosci. Lett. 1991;133:57–60. doi: 10.1016/0304-3940(91)90056-y. [DOI] [PubMed] [Google Scholar]
- RICHTER A., LÖSCHER W. Alterations in pharmacological sensitivity of GABAergic but not dopaminergic and glutamatergic systems during ontogenesis in dystonic mutant hamsters. Europ. J. Pharmacol. 1993;231:111–119. doi: 10.1016/0014-2999(93)90691-a. [DOI] [PubMed] [Google Scholar]
- RICHTER A., LÖSCHER W. Dextrorphan, but not dextromethorphan, exerts antidystonic effects in mutant dystonic hamsters. Brain Res. 1997;745:336–338. doi: 10.1016/s0006-8993(96)01254-1. [DOI] [PubMed] [Google Scholar]
- RICHTER A., LÖSCHER W. Pathophysiology of idiopathic dystonia: findings from genetic animal models. Progress Neurobiol. 1998;54:1–45. doi: 10.1016/s0301-0082(97)00089-0. [DOI] [PubMed] [Google Scholar]
- RUNDFELDT C., KOCH R., RICHTER A., MEVISSEN M., GERECKE U., LÖSCHER W. Dose-dependent anticonvulsant and proconvulsant effects of nitric oxide synthase inhibitors on seizure threshold in a cortical stimulation model in rats. Eur. J. Pharmacol. 1995;274:73–81. doi: 10.1016/0014-2999(94)00711-f. [DOI] [PubMed] [Google Scholar]
- SCHUMAN E.M., MADISON D.V. Nitric oxide and synaptic function. Ann. Rev. Neurosci. 1994;17:153–183. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- STARR M.S., STARR B.S. Do NMDA receptor-mediated changes in motor behaviour involve nitric oxide. Eur. J. Pharmacol. 1995;272:211–217. doi: 10.1016/0014-2999(94)00644-m. [DOI] [PubMed] [Google Scholar]
- STEWART T.L., MICHEL A.D., BLACK M.D., HUMPHREY P.P.A. Evidence that nitric oxide causes calcium-independent release of [3H]dopamine from rat striatum in vitro. J. Neurochem. 1996;66:131–137. doi: 10.1046/j.1471-4159.1996.66010131.x. [DOI] [PubMed] [Google Scholar]
- WAHNSCHAFFE U., FREDOW G., HEINTZ P., LÖSCHER W. Neuropathological studies in a mutant hamster model of paroxysmal dystonia. Movement Dis. 1990;5:286–293. doi: 10.1002/mds.870050405. [DOI] [PubMed] [Google Scholar]