

Abstract
This study examined the effects of the peptide CGRP receptor antagonist CGRP8-37 and the newly-developed non-peptide CGRP receptor antagonist BIBN4096BS for their potential to both inhibit the development and reverse tolerance to the antinociceptive action of morphine.
Repeated administration of intrathecal morphine (15 μg), once daily, produced a progressive decline of antinociceptive effect and an increase in the ED50 value in the tailflick and paw pressure tests. Co-administration of CGRP8-37 (4 μg) or BIBN4096BS (0.05, 0.1 μg) with morphine (15 μg) prevented the decline of antinociceptive effect and increase in ED50 value in the tailflick test. Intrathecal administration of the CGRP receptor antagonists did not alter the baseline responses in either tests. Acute CGRP8-37 also did not potentiate the acute actions of spinal morphine.
In animals rendered tolerant to intrathecal morphine, subsequent administration of CGRP8-37 (4 μg) with morphine (15 μg) partially restored the antinociceptive effect and ED50 value of acute morphine, reflecting the reversal of tolerance.
Animals tolerant to intrathecal morphine expressed increased CGRP and substance P-like immunostaining in the dorsal horn of the spinal cord. The increase in CGRP, but not substance P-like immunostaining, was blocked by a co-treatment with CGRP8-37 (4 μg). In animals already tolerant to morphine, the increase in CGRP but not substance P-like immunostaining was partially reversed by CGRP8-37 (4 μg).
These data suggest that activation of spinal CGRP receptors contributes to both the development and expression of spinal opioid tolerance. CGRP receptor antagonists may represent a useful therapeutic approach for preventing as well as reversing opioid tolerance.
Keywords: Morphine tolerance, calcitonin gene-related peptide, immunostaining, spinal cord, antinociception
Introduction
Opioid drugs such as morphine are, to date, the most potent analgesics available for the treatment of acute and chronic pain syndromes. The limiting factor in the use of these drugs, however, is the rapid development of tolerance to their analgesic effects, a factor which reveals itself following repeated opioid exposure (Cox, 1999). On the basis of recent studies investigating the mechanisms responsible for tolerance, it is thought that chronic opioid exposure to spinal or supraspinal areas produces a neuronal adaptation which compromizes opioid activity. However, it has been reported that patients suffering from neuropathic pain syndromes may also exhibit reduced opioid sensitivity without prior exposure to an opioid agonist, and it is thought that common mechanisms may underlie opioid tolerance and neuropathic pain (Mao et al., 1995b). Thus, it is important to determine factors which not only contribute to the development of tolerance, but also to the maintenance of this phenomenon. The ability of an agent to inhibit the development of opioid tolerance as well as reverse established tolerance provides an ideal alternative to opioid morphine treatment alone.
Recent studies have suggested that repeated opioid exposure leads to a sensitization of pathways releasing nociceptive transmitters and/or their second messengers, thus producing a state of tolerance (Mao et al., 1995b). In this respect, considerable focus has been on the role of the excitatory amino acid transmitter L-glutamate. Previous studies have shown that glutamate-mediated activation of the NMDA receptor, a subclass of excitatory amino acid receptor, contributes to both the development and maintenance of morphine tolerance (Mao et al., 1994; Trujillo & Akil, 1991; 1994). Since the neurones utilising L-glutamate as a transmitter also express calcitonin gene-related peptide (CGRP) and substance P (Gibson et al., 1984), the role of these neuropeptides in the genesis of tolerance becomes an important consideration. Several studies have demonstrated that at the cellular level CGRP exerts effects that are functionally opposite to those produced by opioid activity, namely the formation of cyclic AMP (van Rossum et al., 1997; Wimalawansa, 1996) and the augmentation of L-glutamate and substance P release (Kangrga et al., 1990; Le Greves et al., 1985). In behavioural studies, CGRP has been shown to inhibit the antinociceptive effects produced by opioids (Welch et al., 1989). Since opioids produce analgesia, in part, by inhibiting the release of sensory transmitters in the spinal cord (Pohl et al., 1989), an adaptative increase in the release of a neuropeptide such as CGRP could physiologically antagonize opioid action and thus lead to the development of tolerance. Indeed, we have previously shown that morphine tolerant animals express increased levels of CGRP-like immunostaining in the dorsal horn of the spinal cord (Menard et al., 1995; 1996). Additionally, both the chronic spinal morphine-induced increase in CGRP-like immunostaining as well as loss of morphine potency could be prevented by the co-infusion of a CGRP1 peptide receptor antagonist, CGRP8-37, with intrathecal morphine (Menard et al., 1996). Thus, the blockade of CGRP1 receptor activity using CGRP8-37 prevents both the biochemical and behavioural manifestations of opioid tolerance. Considering that CGRP8-37 is a peptide antagonist, its activity following systemic administration is likely to be limited. Therefore, in this study we compared the potential of CGRP8-37 with that of a recently developed non-peptide CGRP receptor antagonist, BIBN4096BS (Doods et al., 2000), to influence the development of spinal morphine tolerance.
Since CGRP plays a significant role in the development of spinal opioid tolerance, we reasoned that increased spinal CGRP might also serve to maintain tolerance. Previous experiments have shown that established tolerance can be reversed by pharmacological manipulation of NMDA receptor activity with antagonists such as ketamine and LY274614 (Tiseo & Inturrisi, 1993; Elliott et al., 1994; Shimoyama et al., 1996). Additionally, the inhibition of the production of intermediaries of NMDA receptor activity (prostaglandins and nitric oxide) also reverses established tolerance (Kolesnikov et al., 1993; Powell et al., 1999). Since CGRP can release L-glutamate (Kangrga et al., 1990) and enhance NMDA receptor-mediated excitation (Murase et al., 1989), it is likely that CGRP also contributes to the maintenance of tolerance. If this is indeed the case, a blockade of the CGRP receptors should restore the effects of morphine in tolerant animals. Thus, in this study we examined whether CGRP receptor antagonism can reverse the behavioural manifestations of tolerance as well as the increases in spinal neuropeptide immunostaining associated with spinal morphine tolerance (Menard et al., 1996).
Methods
Intrathecal catheter implantation
Male Sprague-Dawley rats (Charles River, Quebec) weighing between 225–250 g were used in this study. Animals were given free access to food and water and were maintained according to the guidelines of the Canadian Council on Animal Care. Animals were implanted with intrathecal catheters according to the method described by Yaksh & Rudy (1976). Briefly, under halothane anaesthesia (4%) animals were placed prone in a stereotaxic frame and a small incision was made at the back of the neck. A small puncture was made in the atlanto-occipital membrane of the cisterna magna and a polyethylene catheter (PE-10, 7.5 cm) was inserted such that the caudal tip reached the lumbar enlargement of the spinal cord. The rostral end of the catheter was exteriorized at the top of the head and the wound closed with sutures. The animals were allowed 4–5 days to recover from the surgery and those displaying signs of motor dysfunction (fore limb or hind limb paralysis) were excluded from the study. All drugs were injected through the exteriorized portion of the catheter in a 10 ul volume followed by a flush with 10 ul of 0.9% saline.
Nociceptive testing
The antinociceptive potential of all agents was tested using two nociceptive spinal reflex tests: the tailflick and paw pressure tests. The tailflick test (D'Amour & Smith, 1941) was used to evaluate the animal's response to a thermal stimulus. Radiant heat was applied to the bottom section of the animals' tail using an analgesia meter (Owen et al., 1981) and the response latency required for the animal to remove its tail was recorded. The intensity of the heat was adjusted to yield a baseline response of 2–3 s and a cut-off time of 10 s was implemented to minimize tissue damage. The paw pressure test (Loomis et al., 1987) was used to evaluate the animal's response to a mechanical stimulus. Using an inverted air-filled syringe, pressure was applied to the dorsal surface of the animal's hindpaw. The pressure at which the animal withdrew its hindpaw was recorded. A cut-off pressure of 300 mmHg was used.
Induction of morphine tolerance
In order to induce a state of morphine tolerance, animals were injected once daily for 7 days with intrathecal morphine (15 μg). Injections were made each day between 1000 h and midnight. Nociceptive testing was performed before and 30 min after drug administration to determine baseline and drug induced antinociceptive effects, respectively. On the eighth day, cumulative dose-response curves were generated to determine the potency of acute morphine. To generate these curves, animals were given ascending doses of morphine every 30 min until a maximal level of antinociception was reached in both the tailflick and paw pressure test. The morphine ED50 value, an indicator of morphine potency, was derived from the constructed dose-response curves. A state of tolerance was indicated by a progressive decrease in the daily antinociceptive response to morphine, a rightward shift in the acute morphine dose response curve and an increase in the ED50 value of morphine.
Study 1: Effect of CGRP receptor blockade on the development of morphine tolerance
In order to examine the ability of CGRP receptor antagonists to inhibit the development of tolerance, the peptide (CGRP8-37) and non-peptide (BIBN4096BS) antagonists were co-injected with morphine once daily for 7 days. Nociceptive testing was performed daily and cumulative dose response curves were generated on the 8th day, as described above. The ability of these agents to influence the development of tolerance was assessed by examining their effect on (a) the decline in antinociception produced by morphine alone and (b) on morphine potency, as indicated by a shift in the dose-response curve and change in ED50 value. To determine if BIBN4096BS or CGRP8-37 influenced the antinociceptive response to acute intrathecal morphine, the agents under study were co-injected with morphine in opioid naïve animals.
Study 2: To determine the effects of CGRP receptor blockade on established morphine tolerance
In order to examine the ability of CGRP receptor blockade to reverse tolerance, animals were first rendered tolerant to intrathecal morphine. Animals received daily injections of morphine (15 μg) once daily for 5 days. On days 6–10 animals received either the test agent alone or the test agent co-injected with morphine, once daily. On the 11th day cumulative dose-response curves for acute morphine were generated and the ED50 values calculated for these curves. The ability of drug treatments to reverse tolerance was indicated by (a) a recovery of morphine-induced antinociception during chronic treatment and (b) a recovery of morphine potency (ED50) towards control values.
Immunohistochemistry of CGRP and substance P-like immunoreactivity
The spinal cords from the animals treated according to the paradigms outlined above were isolated and examined for CGRP or substance P-like immunostaining (Menard et al., 1995). On the day of tissue removal, rats were anaesthetized with urethane and perfused intracardially with cold phosphate buffered saline and subsequently with 4% paraformaldehyde in 0.1 M phosphate buffer. The L4-5 segments of the spinal cord were removed and postfixed in the same fixative overnight. The spinal cord segments were then transferred to 30% sucrose in 0.1 M phosphate buffer for cryoprotection. The spinal cord segments were cut into 50 um sections using a cryostat. The sections were incubated with 0.3% H2O2, rinsed in PBS and incubated with 10% normal goat serum. Sections were then incubated with rabbit polyclonal anti-CGRP antibody (1 : 4000) or anti-substance P antibody (1 : 8000) diluted in PBS containing 0.3% Triton X-100 and 3% NGS for 36 h at 4°C. The sections were then processed using Vecastain ABC kit (Vector, Burlingame, CA, U.S.A.) according to the instructions of the manufacturer and rinsed with PBS-T between each incubation period. The immunoprecipitates were developed using 3,-3 diaminobenzedine and the chromogen was enhanced using the glucose oxidase-nickel-3,-3 diaminobenzidine method.
The optical density of CGRP and substance P immunoreactivity in the L4 and L5 spinal cord sections from rats with different treatment was measured using image analysis software (SigmaScan, Jandel Scientific Inc., San Rafael, CA, U.S.A.). Five sections were randomly taken from three rats from each of the six treatment groups. The image of each dorsal horn was captured at 79× magnification by using a high-resolution CCD camera. Optical density was measured along three 20 um lines drawn from the marginal border from lamina I toward the deeper dorsal horn in the lateral, middle and medial areas of the dorsal horn area. The optical densities of the three lines were averaged for each treatment group.
Drugs
Morphine sulphate was obtained from BDH Pharmaceuticals (Toronto, Ontario, Canada). α-CGRP8-37 (human) was synthesized at the Institut Nationale de la Recherche Scientifique-Sante (Pointe Claire, Quebec, Canada), and BIBN4096BS was a gift from Dr H. Doods, Boehringer Ingelheim (Germany). Morphine and CGRP8-37 were dissolved in physiological saline (0.9%) and BIBN4096BS was dissolved in 5% cyclodextrin. Polyclonal antiserum to rat CGRP was obtained from Peninsula Laboratories Inc., (Belmont, CA, U.S.A.), and polyclonal antiserum to rat substance P was generously provided by Dr J.M. Polak (Royal Postgraduate Medical School, London, U.K.). 3,3′ diaminobenzidine tetrahydrochloride and glucose oxidase were obtained from Sigma Chemicals (St. Louis, MO, U.S.A.). All other chemicals were obtained from Sigma Chemicals (St. Louis, MO, U.S.A.).
Data analysis
Tailflick and paw pressure values were converted to a maximum percent effect (M.P.E.): M.P.E.=100×[post-drug response−baseline response]/[cut-off value−baseline response]. Data are expressed as mean±s.e.mean in the figures. The ED50 values were determined using a non-linear regressional analysis (Prizm 2, GraphPad Software Inc.). Statistical significance (P<0.05) was determined using a one-way ANOVA followed by a Student Newman-Keuls post hoc test for multiple comparisons between groups.
Results
Study 1: Development of tolerance
The effects of CGRP8-37 on the development of tolerance to spinal morphine in the tailflick and paw pressure tests are represented in Figure 1. Administration of intrathecal saline or CGRP8-37 alone did not produce an antinociceptive response in either the tailflick or the paw pressure test. Administration of intrathecal morphine (15 μg) to rats on day 1 produced a maximal antinociceptive response in both tests. However, once daily administration of morphine (15 μg) resulted in a progressive decline in the analgesic response to baseline levels by day 5, reflecting the development of tolerance. Co-administration of CGRP8-37 (4 μg) with morphine attenuated this decline in both tests. The antinociceptive responses obtained with CGRP8-37/morphine combination treatment were significantly greater than with morphine treatment alone on days 2–7 (P<0.001) in the tailflick test and on days 2–4 (P<0.01) and 6–7 (P<0.05) in the paw pressure test. These results thus confirm our previous observations, using the continuous infusion model of tolerance (Menard et al., 1996), that CGRP receptor blockade with a peptide antagonist indeed inhibits the development of morphine tolerance.
Figure 1.

Time course of the antinociceptive effects of daily administration of intrathecal morphine (15 μg) alone and in combination with CGRP8-37 (4 μg) in the (A) tailflick and (B) paw pressure tests. Morphine and the test agent were administered as a single dose. Nociceptive testing was performed 30 min following each injection. The data are expressed as mean±s.e.mean for 5–7 animals. # Significant differences from the action of morphine # (P<0.05); ## (P<0.01).
The effects of BIBN4096BS, a newly developed non-peptide receptor antagonist, on the development of morphine tolerance in the tailflick and pawpressure test are represented in Figure 2. Co-administration of BIBN4096BS (0.05, 0.1 μg) with morphine decreased the antinociceptive response of morphine alone on day 1 by approximately 20 and 30%, respectively, in both tests. However, despite this the co-administration of BIBN4096BS (0.05, 0.1 μg) with morphine for 7 days attenuated the decline in antinociception in the tailflick test. A 50-fold lower dose (0.001 μg) of BIBN4096BS was also tested and it produced a similar response. The antinociceptive responses obtained on days 4–7 were significantly greater with BIBN4096BS/morphine combination treatment (P<0.05) than with morphine treatment alone in this test. However, treatment with BIBN4096BS unlike CGRP8-37, did not influence the decline in morphine antinociception in the paw pressure test (Figure 2B).
Figure 2.

Time course of the antinociceptive effects of daily administration of intrathecal morphine (15 μg) alone and in combination with BIBN4096BS (0.001, 0.05, 0.1 μg) in the (A) tailflick and (B) paw pressure tests. Morphine and the test agent were administered as a single dose. Nociceptive testing was performed 30 min following each injection. The data are expressed as mean±s.e.mean for 5–7 animals. # Significant differences from the action of morphine # (P<0.05); ## (P<0.01).
At the end of the 7 day treatment period CGRP8-37 maintained the level of morphine induced antinociception at a greater level than did BIBN4096BS in both the tailflick (60 vs 45%) and the paw pressure test (40 vs 20%). Thus, although both agents influenced the development of tolerance in one or both tests, the effects seen with CGRP8-37 were greater.
The ED50 values for the acute antinociceptive potency of morphine determined at the end of the treatment period are represented in Table 1. Chronic 7 day morphine (15 μg) treatment produced a ∼4 fold increase in the acute ED50 values in both tests compared to chronic vehicle treatment, reflecting a significant loss of opioid potency (P<0.001). However, in the group receiving CGRP8-37 (4 μg) with morphine the increase in ED50 value of the opioid agonist was completely prevented in both the tailflick and paw pressure test. Similar treatment with BIBN4096BS (0.001, 0.05, 0.1 μg) also completely prevented the increase in ED50 value in both tests. The ED50 values obtained in both the CGRP8-37/morphine and BIBN4096BS/morphine combination group were not significantly different from those in the saline treated group. Treatment with CGRP8-37 (4 μg) alone for 7 days increased the ED50 values of morphine as compared to saline, but this increase was not statistically significant. Treatment with BIBN4096BS (0.1 μg) alone for seven days decreased the ED50 values of acute morphine from those of vehicle pretreatment, but this effect was also not statistically significant. Thus, CGRP8-37 and BIBN4096BS appeared not to influence significantly morphine potency on their own, but when co-administered with chronic spinal morphine prevented the loss of agonist potency associated with the development of tolerance.
Table 1.
Effect of CGRP8-37 and BIBN4096BS on the development of morphine tolerance

The possibility that CGRP8-37 influenced tolerance by augmenting the actions of morphine was evaluated by combining this agent with a sub-maximal dose of acute morphine and testing its effects in the tailflick and paw pressure tests in drug naïve animals. Co-administration of CGRP8-37 (4 μg) with morphine (7.5 μg) did not influence significantly the effect of morphine in either test (data not shown). Additionally, intrathecal pretreatment with CGRP8-37 (4 μg) did not produce a shift in the dose-response curve for acute morphine in either test. The acute morphine ED50 values in the CGRP8-37 pre-treated animals (8.65±1.4 (tailflick), 12.24±1.4 (paw pressure)) were not different from those in the vehicle pretreated animals (9.74±1.6 (tailflick), 13.09±3.0 (paw pressure)).
The likelihood that BIBN4096BS influenced tolerance by potentiating the acute actions of morphine is unlikely since administration of BIBN4096BS slightly decreased the acute antinociceptive effects of morphine after a single administration (see Figure 2). Intrathecal pretreatment with BIBN4096BS (0.1 μg) shifted the acute dose-response curve for the acute action of morphine to the right in the tailflick but not paw pressure test. The acute morphine ED50 values in the BIBN4096BS pre-treated animals (6.68±0.32 (tailflick), 4.98±0.50 (paw pressure)) were slightly greater than those in the vehicle pre-treated animals animals (4.50±0.27 (tailflick), 4.49±0.20 (paw pressure)) in the tailflick test but not the paw pressure test.
Study 2: Reversal of tolerance
The effects of CGRP8-37 on established morphine tolerance in the tailflick test are presented in Figure 3A,B and Figure 4. In this part of the study, CGRP8-37, but not BIBN4096BS, was tested since CGRP8-37 did not influence the acute effects of morphine. Repeated daily administration of morphine (15 ug) for 10 days resulted in a decay of the antinociceptive response to baseline levels by day 5, reflecting the development of tolerance (Figure 3A). Addition of CGRP8-37 (4 μg) to morphine on days 6–10 partially restored the morphine response to approximately 70% of the original value (Figure 3B). Figure 4 represents the area under the curve values obtained from Figure 3A,B, indicating the degree of restoration in the morphine response for each dose of the 5 CGRP8-37 doses tested. Intervention with CGRP8-37 (0.5, 1, 2, 4, 8 μg) during continued morphine treatment on days 6–10 significantly increased the area under the curve response as compared to morphine treatment alone (P<0.001). The lowest dose of CGRP8-37 tested (0.5 μg) produced the greatest reversal in the morphine effect. Additionally, CGRP8-37 restored the morphine effect to a greater level in the tailflick than in the paw pressure test (paw pressure data not shown). Intervention with CGRP8-37 (4 or 8 μg) without morphine on days 6–10 did not produce a reversal in morphine effect (data not shown).
Figure 3.
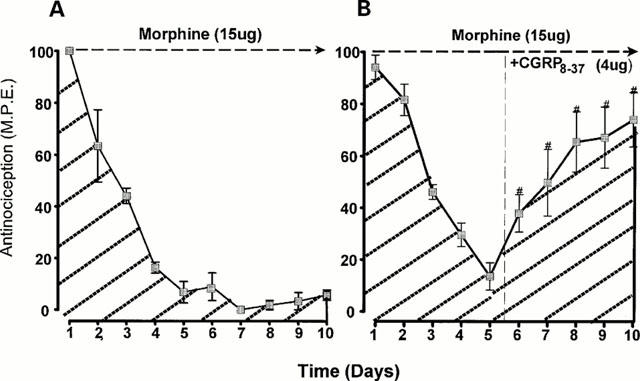
The effects of intrathecal CGRP8-37 on established tolerance to intrathecal morphine in the tailflick test. Tolerance was induced by administration of single morphine (15 μg) injections from days 1–5. CGRP8-37 (4 μg) was administered with morphine from days 6–10. Morphine and the test agent were given as a single injection followed by nocicpetive testing 30 min after each injection. The data are expressed as mean±s.e.mean for 5–7 animals. # Significant differences from the action of morphine (P<0.001).
Figure 4.

The effect of CGRP8-37 on established tolerance to intrathecal morphine expressed as area under the curve. Area under the curve was calculated for the antinociceptive responses obtained in the tailflick test. # Significant differences from morphine days 6–10 (P<0.001).
The ED50 values of acute morphine obtained in the reversal experiments are represented in Table 2. As expected, chronic morphine treatment for 10 days produced an approximate 5 fold increase in the morphine ED50 values as compared to saline treatment. However, co-treatment with CGRP8-37 on days 6–10 reversed the increase in the morphine ED50 values in the tailflick test. Treatment with the 0.5, 2, 4 and 8 μg doses reduced the morphine ED50 values to those obtained with saline treatment reflecting a restoration in morphine potency. Treatment with the 1 μg dose of CGRP8-37 reduced the morphine ED50 value from that obtained with morphine treatment, but it remained significantly greater than the values obtained with saline treatment (P<0.01). In the paw pressure test, the 0.5 and 8 μg doses of CGRP8-37 restored the morphine ED50 values to that obtained with saline treatment. Treatment with 4 μg reduced the ED50 as compared to morphine, but it remained significantly greater than that obtained with saline (P<0.01). Administration of 1 and 2 μg of CGRP8-37 did not reduce the morphine ED50 values from those obtained with morphine treatment. Animals given saline on days 6–10 still showed an increase in the ED50 values suggesting the persistence of tolerance despite discontinuation of morphine treatment for 5 days. Treatment with CGRP8-37 (4 or 8 μg) alone on days 6–10 prevented the increase in ED50 value in both tests. Thus, CGRP8-37 was found to have the potential of reversing established tolerance with and without co-administration of morphine, although its actions tend to be test specific.
Table 2.
Effect of CGRP8-37 and BIBN4096BS on the reversal of morphine tolerance

Immunohistochemical studies
CGRP-like Immunostaining
Figure 5 shows the photomicrographs of CGRP immunoreactive axons in the dorsal horn of the spinal cord from rats chronically treated with morphine. The corresponding mean optical density (OD) values of immunostaining in the dorsal horn are presented in Figure 6. In the saline-treated animals, CGRP-like immunostaining was abundant in the superficial laminae of the dorsal horn (Figure 5A). Chronic treatment with morphine for 7 days increased CGRP-like immunostaining in this area (Figure 5C). The mean optical density of CGRP-like immunostaining in morphine treated animals was significantly greater than in saline treated animals (P<0.001). Co-treatment with CGRP8-37 (4 μg) partially prevented the increase in CGRP-like immunostaining (Figure 5D). The mean optical density in this group was significantly less than that in the morphine treated group (P<0.001), but was still significantly greater than that in the saline treated group (P<0.001). In animals already tolerant to spinal morphine, the increase in CGRP-like immunostaining could be partially reversed by subsequent treatment with CGRP8-37 (4 μg) (Figure 5E). The mean optical density in this group was significantly less than that in the morphine treated group (P<0.001), but also significantly greater than that in the saline treated group (P<0.01). Subsequent treatment with CGRP8-37 and morphine co-injected in tolerant animals completely reversed the increase in CGRP-like immunostaining (Figure 5F). Thus, CGRP8-37 demonstrated the potential to prevent and to reverse partially the increase in CGRP-like immunostaining associated with spinal morphine tolerance. These effects are not a consequence of the antagonist alone, since CGRP8-37 treatment by itself did not decrease CGRP-like immunoreactivity as compared to saline treatment (Figure 5B, Figure 6).
Figure 5.

Photomicrographs of CGRP immunoreactive axons in the dorsal horn of L4–L5 spinal cords of rats following daily intrathecal injection of (A) saline 7 days, (B) CGRP8-37 (4 μg) 7 days, (C) morphine (15 μg) 7 days, (D) morphine +CGRP8-37 7 days, (E) morphine 5 days, then CGRP8-37 5 days, (F) morphine 5 days, then morphine +CGRP8-37 5 days. Scale bar 200 um.
Figure 6.

Mean optical density of CGRP and substance P immunoreactive axons in the dorsal horn of L4–L5 spinal cords of rats. *Significant from saline *(P<0.01), **(P<0.001). # Significant from morphine (P<0.001).
Substance P-like immunostaining
Figure 7 shows the photomicrographs of substance P immunoreactive axons in the dorsal horn of the spinal cord from rats chronically treated with morphine. The corresponding mean optical density (OD) values of substance P-like immunostaining in the dorsal horn are presented in Figure 6. In saline treated animals, substance P-like immunoreactivity was also abundant in the superficial laminae of the dorsal horn (Figure 7A). However, the mean optical density of the substance P-like immunostaining was less than the CGRP-like immunostaining in saline-treated animals (Figure 6). Chronic treatment with morphine for 7 days increased substance P-like immunostaining in the dorsal horn (Figure 7B). However, co-treatment with CGRP8-37 (4 μg) did not prevent the increase in substance P-like immunostaining (Figure 7D). Subsequent treatment with CGRP8-37 (4 μg) alone or co-injected with morphine also did not reverse the increase in substance P-like immunostaining induced by chronic morphine (Figure 7E,F). The mean optical density of substance P-like immunostaining in animals treated with CGRP8-37 was not significantly different from that in morphine treated animals. Thus, unlike CGRP-like immunostaining, substance P-like immunostaining was not significantly influenced by the CGRP8-37 treatment.
Figure 7.

Photomicrographs of substance P immunoreactive axons in the dorsal horn of L4–L5 spinal cords of rats following daily intrathecal injection of (A) saline 7 days, (B) CGRP8-37 (4 μg) 7 days, (C) morphine (15 μg) 7 days, (D) morphine +CGRP8-37 7 days, (E) morphine 5 days, then CGRP8-37 5 days, (F) morphine 5 days, then morphine +CGRP8-37 5 days. Scale bar 200 um.
Discussion
We have previously reported that the activity of the nociceptive sensory transmitter CGRP contributes to the development of tolerance to the antinociceptive actions of spinally infused morphine (Menard et al., 1995; 1996). In the present study, we sought to characterize further the role of CGRP in opioid tolerance, using both a peptide and a newly developed non-peptide CGRP receptor antagonist known as BIBN4096BS. Since an important goal of this study was to determine if the activity of CGRP also underlies the maintenance of tolerance to spinal morphine, we examined the potential of CGRP receptor blockade to reverse established tolerance. To meet this objective the present study, unlike our earlier studies involving continuous opioid infusion, utilised a daily morphine injection model. This approach allowed for drug intervention immediately following the establishment of tolerance. The results demonstrated that in this model, both CGRP receptor antagonists (CGRP8-37 and BIBN4096BS) effectively reduce the development of spinal morphine tolerance and, additionally, that CGRP8-37 restores the potency of morphine in animals already tolerant to this opiate. This study also revealed that CGRP8-37 both prevents and reverses the increase in CGRP-like immunostaining observed in morphine-tolerant animals. Taken together, these results suggest that CGRP related systems contribute to both the development and maintenance of tolerance to spinal morphine.
The CGRP receptor antagonist, CGRP8-37, is well recognized as a competitive antagonist for the CGRP1 receptor subtype (Dennis et al., 1990; Quirion et al., 1992). BIBN4096BS is a novel competitive, non-peptide CGRP1 receptor antagonist (Doods et al., 2000). Although Yu et al. (1994) previously reported that CGRP8-37 alone produces an antinociceptive effect, we used a dose of CGRP8-37 that was lower and that did not produce an antinociceptive response in either the tailflick or paw pressure test. Based on previous studies it has been shown that BIBN4096BS has a Ki for the rat receptor of 3.4±0.5 nM (Doods et al, 2000) and that CGRP8-37 has a Ki for the rodent receptor of 4.0±1.6 nM (Taylor et al, 1996). Thus, in the rat CGRP8-37 and BIBN4096BS have comparable affinities, although the latter has greater affinity for the human CGRP receptor (Doods et al, 2000). On a molar level, the concentrations of CGRP8-37 and BIBN4096BS used in the development study were 1.25 nM and 1–114 pM, respectively. The doses of BIBN4096BS used did not produce analgesic effects on their own. As was observed in our previous studies (Mendard et al., 1996), CGRP8-37 inhibited the development of morphine tolerance, and this effect was mimicked by BIBN4096BS. The action of CGRP receptor blockade on the development of tolerance to spinal morphine was not due to a potentiation of acute morphine action, or a sensitization to morphine as was previously observed with the NMDA receptor antagonist MK801 (Dunbar & Yaksh, 1996) or inhibitors of cyclooxygenase activity like ketorolac and ibuprofen (Powell et al., 1999). In fact, BIBN4096BS slightly inhibited the acute antinociceptive action of morphine in the tail-flick test but still attenuated the loss of morphine potency associated with chronic treatment. While both antagonists completely prevented the loss of morphine potency in tolerant animals, examination of the time response curve showed that CGRP8-37 reduced the decline in morphine effect to a greater extent than BIBN4096BS. This could be partly related to the ability of BIBN4096BS to attenuate the morphine effect. The latter apparently does not involve an interaction with CGRP1 receptors, as the peptide antagonist did not produce this effect. Both antagonists appeared to be more effective against the development of tolerance to thermal antinociception than mechanical antinociception, suggesting that CGRP activity may have a more prominent role in mediating thermal nociception. Although information regarding such a role of CGRP is lacking, there is evidence that different types of nociceptive stimuli can exert differential actions on the release of sensory neuropeptides in the dorsal horn (Kuraishi et al., 1985; 1990; Salt & Hill, 1983). Recent evidence suggests a prominent role of substance P in mechanical nociception. Trafton et al, (1999) showed that NK-1 receptor internalization was greater in response to a mechanical rather than a thermal noxious stimulus. Our preliminary studies show that a substance P receptor antagonist is more effective than the CGRP receptor antagonists in influencing tolerance to morphine action against mechanical stimuli (unpublished data). Thus, CGRP may preferentially affect peptide containing primary afferent neurones that are involved in thermal nociception.
The fact that CGRP receptor blockade inhibited and reversed both the behavioural and biochemical manifestations of tolerance indicates that spinal CGRP innervation and receptors play a role in both the initiation and maintenance of tolerance. How this activity influences morphine tolerance is not known but both direct and indirect mechanisms may be involved. CGRP receptors are located throughout the dorsal horn and evidence from autoradiographic studies suggest the likely pre-synaptic (Ye et al., 1999) and post-synaptic localization of CGRP receptors in this area (Yashpal et al., 1992; Menard et al., 1995; van Rossum et al., 1997). At the intracellular level, CGRP receptor activation increases cyclic AMP formation via a G-protein coupled mechanism (van Rossum et al., 1997; Wimalawansa, 1996), whereas opioid receptor activation decreases cyclic AMP formation (Sharma et al., 1975). Similarly, CGRP also facilitates the release of L-glutamate (Kangrga et al., 1990) and substance P (Oku et al., 1987) whereas opioids inhibit this release (Yaksh et al., 1980; Nicol et al., 1996). Thus, increased CGRP activity, likely due to augmented release of the neuropeptide in response to chronic opioid treatment, could attenuate opioid receptor-mediated decreases in adenylyl cyclase activity, impair opioid inhibition of neurotransmitter release, and thus reduce the antinociceptive response to morphine. Alternatively, CGRP may influence opioid tolerance indirectly by influencing excitatory amino acid receptor activity. Considerable evidence supports the notion that neuronal adaptations increasing NMDA receptor-mediated events underlies opioid tolerance (Mao et al., 1994; Trujillo & Akil, 1991). An important locus of this adaptive process has been identified in the spinal cord, since intrathecal co-administration of NMDA receptor antagonists with morphine blocks the development of tolerance (Dunbar & Yaksh, 1996; Shimoyama et al., 1996). Previous work on the isolated spinal cord has shown that CGRP releases L-glutamate (Kangrga et al., 1990) and enhances the depolarization of spinal neurones produced by the activation of NMDA receptors (Murase et al., 1989). As CGRP is co-localized with L-glutamate, it is strategically placed to modulate the activity of this transmitter and its receptors. Thus, an increase in CGRP, under the influence of morphine, may result in the amplification of NMDA receptor function and a physiological antagonism of the opioid-induced antinociception. Interestingly, it has been reported that the tolerance to systemic or spinal morphine can be reversed by NMDA receptor antagonists (Elliott et al., 1994; Shimoyama et al., 1996; Tiseo & Inturrisi, 1993) and the CGRP1 receptor antagonist CGRP8-37 shares this property. Thus, CGRP antagonists may reverse opioid tolerance by decreasing the positive modulatory action of CGRP on NMDA receptors and reducing the physiological antagonism of opioid action. Alternately, CGRP may inhibit morphine tolerance through interactions with substance P. In the spinal cord, CGRP is co-localized and co-released with substance P (Wiesenfeld-Hallin et al., 1984), a potent modulator of nociceptive transmission in the dorsal horn which contributes to pain-related behaviours. Previous studies have demonstrated that the nociceptive potential of CGRP may be attributed to substance P since CGRP: (a) potentiates spinal substance P release (Oku et al., 1987), (b) inhibits substance P degradation by proteases (Le Greves et al., 1985), and (c) augments the nociception produced by substance P (Oku et al., 1987). An increase in substance P release may inhibit opioid activity.
In the present study, chronic morphine treatment increased both CGRP and substance P-like immunostaining in the dorsal horn of the spinal cord. Although substance P is present in smaller amounts, the relative increase in both peptides compared to that of naïve animals was comparable. The administration of a CGRP receptor antagonist clearly inhibited the increase in CGRP-like immunostaining suggesting that it is a receptor-mediated event. The influence of CGRP receptor activation on CGRP levels may be expressed at the level of the primary afferent neurone via pre-synaptically located receptors or through activation of post-synaptic receptors on projection neurones. The former possibility is favoured by a recent observation with dorsal root ganglia cells maintained in culture, a preparation in which only the pre-synaptic neurones are represented, that CGRP-immunostaining is increased following chronic morphine exposure (Quirion et al., 1999). Alternatively, the increase in CGRP may be mediated by activation of the post-synaptic CGRP receptor, leading to production of a retrograde messenger that influences pre-synaptic CGRP levels. Although there is currently no evidence that CGRP activity in the spinal cord produces such a messenger, a recent study has demonstrated that in smooth muscle cells, CGRP activates cyclic AMP and nitric oxide pathways to induce relaxation (Rekik et al., 1997). Nitric oxide is a transmitter molecule produced following NMDA receptor activation and has been shown to increase the release of neurotransmitters pre-synaptically. Since CGRP augments NMDA receptor activity, CGRP may influence NMDA induced nitric production and increase pre-synaptic neurotransmitter release.
The action of chronic morphine treatment on substance P in this model was surprising, as this was not observed in previous studies involving chronic morphine infusion (Menard et al., 1995; 1996). The basis for this difference is not clear but it may be related to the fact that spinal exposure to morphine in this model was lower and substance P-like immunostaining in the dorsal horn region was quantitated. The increase in substance P-like immunostaining, however, appears to occur via a CGRP1 receptor-independent mechanism since the peptide antagonist did not influence this response. The reasons for these differential effects are not known, but could be related to differential action of CGRP8-37 on the CGRP and substance P containing neurones in the dorsal horn. Currently, the role of substance P in opioid tolerance is largely undefined although previous studies have implicated this peptide in the development of physical dependence (Johnston & Chahl, 1991; Kreeger & Larson, 1993). An increase in the release of substance P could lead to the release of retrograde messengers and the attenuation of opioid receptor activity. In this respect, it has been reported that substance P is a very potent releaser of prostanoids at the spinal level (Marriott et al., 1991; Hua et al., 1999). Prostanoids augment the activity of primary afferents releasing neurotransmitters via presynaptic receptors (Matsumura et al., 1992; Vasko et al., 1994). Thus, an increase in substance P activity could reduce the ability of opioids to inhibit transmitter release and produce antinociception. We have recently shown that intrathecal application of prostanoid synthesis inhibitors effectively blocks and reverses the development of opioid tolerance (Powell et al., 1999). Our preliminary findings with a substance P receptor antagonist (SR140333) have also revealed its potential to inhibit spinal morphine tolerance (Powell et al., 1999, unpublished results).
We observed that the development of tolerance to spinal morphine was characterized by both a decline in morphine-induced antinociception and a reduction in morphine potency. Although CGRP receptor blockade influenced both indices of tolerance, the decrement in morphine antinociception was attenuated but not completely prevented. This suggests that the agents under study produced a partial inhibition of the CGRP receptor, or that additional factors contribute to tolerance. Despite the use of several doses, CGRP8-37 failed to reverse completely the increases in CGRP-like immunostaining or the response to morphine. Studies on the combinations of antagonists against various nociceptive transmitters or messengers could provide a more definitive answer.
The ability of CGRP8-37 to restore the antinociceptive effects of morphine in tolerant animals has important clinical implications. Recent studies have demonstrated that in models of inflammation, hyperalgesia and nerve injury, animals not previously exposed to opioids exhibit a reduced response to these agents (Dickenson & Sullivan, 1986; Xu & Wiesenfeld-Hallin, 1991; Mao et al., 1995a), an effect that is also seen in patients suffering from neuropathic pain. Moreover, the excitatory amino acid receptor-based neuronal adaptations that contribute to these pain states have also been implicated in tolerance (Mao et al., 1995b). Activation of spinal NMDA receptors appears to play a crucial role in the development and maintenance of hyperalgesia associated with nerve injury (Mao et al., 1995b). Since NMDA receptors are also implicated in morphine tolerance and there are reciprocal interactions between CGRP and L-glutamate, it is likely that adaptations in spinal CGRP systems contribute to the hyperalgesia associated with nerve injury. Thus, CGRP receptor blockade may have the potential to reverse reduced opioid sensitivity due to neuronal adaptations resulting from nerve injury.
In conclusion, the results of the present study demonstrate that CGRP receptor activity contributes to both the development and maintenance of spinal morphine tolerance. The combination of a CGRP receptor antagonist with morphine presents an option for the inhibition of clinical tolerance and may provide a new avenue for restoring opioid responsiveness in neuropathic pain states. Thus, the development of a potent, selective, non-peptide CGRP receptor antagonists, which would be systemically active, may provide novel methods of modulating opioid tolerance. BIBN4096BS (Doods et al, 2000) is a first exciting step in that regard.
Acknowledgments
The authors acknowledge grant support from the Medical Research Council of Canada and travel support from the Ontario-Quebec Exchange Program.
Abbreviations
- CGRP
Calcitonin gene-related peptide
- NMDA
N-methyl-D-aspartate
References
- COX B.M.Molecular and cellular mechanisms in opioid tolerance Towards a New Pharmacotherapy of Pain 1999John Wiley, Chichester, UK; 137–156.eds. Bausbaum A.I. & Besson J.M. pp [Google Scholar]
- D'AMOUR F.E., SMITH D.L. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- DENNIS T., FOURNIER A., CADIEUX A., POMERLEAU F., JOLICOEUR F.B., , ST, QUIRION R. hCGRP8-37, a calcitonin gene-related peptide antagonist revealing calcitonin gene-related peptide receptor heterogeneity in brain and periphery. J. Pharmacol. Exp. Ther. 1990;254:123–128. [PubMed] [Google Scholar]
- DICKENSON A.H., SULLIVAN A.F. Electrophysiological studies on the effects of intrathecal morphine on nociceptive neurones in the rat dorsal horn. Pain. 1986;24:211–222. doi: 10.1016/0304-3959(86)90044-8. [DOI] [PubMed] [Google Scholar]
- DOODS H., HALLERMAYER G., WU D., ENTZEROTH M., RUDOLF K, , ENGEL W., EBERLEIN W. Pharmacological profile of BIBN4096BS, the first selective small molecule CGRP antagonist. Br. J. Pharmacol. 2000;129:420–423. doi: 10.1038/sj.bjp.0703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNBAR S., YAKSH T.L.Concurrent spinal infusion of MK801 blocks spinal tolerance and dependence induced by chronic intrathecal morphine in the ra Anesthesiology 1996841177–1188.[published erratum appears in Anesthesiology 1996 Sep; 85(3):695] [DOI] [PubMed] [Google Scholar]
- ELLIOTT K., HYNANSKY A., INTURRISI C.E. Dextromethorphan attenuates and reverses analgesic tolerance to morphine. Pain. 1994;59:361–368. doi: 10.1016/0304-3959(94)90022-1. [DOI] [PubMed] [Google Scholar]
- GIBSON S.J., POLAK J.M., BLOOM S.R., SABATE I.M., MULDERRY P.M., GHATEI M.A., MCGREGOR G.P., MORRISON J.F., KELLY J.S., EVANS R.M. Calcitonin gene-related peptide immunoreactivity in the spinal cord of man and of eight other species. J. Neurosci. 1984;4:3101–3111. doi: 10.1523/JNEUROSCI.04-12-03101.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUA X.Y., CHEN P., MARSALA M., YAKSH T.L. Intrathecal substance P-induced thermal hyperalgesia and spinal release of prostaglandin E2 and amino acids. Neurosci. 1999;89:525–534. doi: 10.1016/s0306-4522(98)00488-6. [DOI] [PubMed] [Google Scholar]
- JOHNSTON P.A., CHAHL L.A. Tachykinin antagonists inhibit the morphine withdrawal response in guinea-pigs. N.S. Arch. Pharmacol. 1991;343:283–288. doi: 10.1007/BF00251127. [DOI] [PubMed] [Google Scholar]
- KANGRGA I., LAREW J.S., RANDIC M. The effects of substance P and calcitonin gene-related peptide on the efflux of endogenous glutamate and aspartate from the rat spinal dorsal horn in vitro. Neurosci. Lett. 1990;108:155–160. doi: 10.1016/0304-3940(90)90723-m. [DOI] [PubMed] [Google Scholar]
- KOLESNIKOV Y.A., PICK C.G., CISZEWSKA G., PASTERNAK G.W. Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5162–5166. doi: 10.1073/pnas.90.11.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KREEGER J.S., LARSON A.A. Substance P-(1–7), a substance P metabolite, inhibits withdrawal jumping in morphine-dependent mice. Eur. J. Pharmacol. 1993;238:111–115. doi: 10.1016/0014-2999(93)90513-h. [DOI] [PubMed] [Google Scholar]
- KURAISHI Y. [Neuropeptide-mediated transmission of nociceptive information and its regulation. Novel mechanisms of analgesics]. [Japanese] Yakugaku Zasshi. 1990;110:711–726. doi: 10.1248/yakushi1947.110.10_711. [DOI] [PubMed] [Google Scholar]
- KURAISHI Y., HIROTA N., SATO Y., HINO Y., SATOH M., TAKAGI H. Evidence that substance P and somatostatin transmit separate information related to pain in the spinal dorsal horn. Brain Res. 1985;325:294–298. doi: 10.1016/0006-8993(85)90326-9. [DOI] [PubMed] [Google Scholar]
- LE REVES P., NYBERG F., TERENIUS L., HOKFELT T. Calcitonin gene-related peptide is a potent inhibitor of substance P degradation. Eur. J. Pharmacol. 1985;115:309–311. doi: 10.1016/0014-2999(85)90706-x. [DOI] [PubMed] [Google Scholar]
- LOOMIS C.W., MILNE B., CERVENKO F.W. Determination of cross tolerance in rat spinal cord using intrathecal infusion via sequential mini-osmotic pumps. Pharmacol. Biochem. Behav. 1987;26:131–139. doi: 10.1016/0091-3057(87)90545-4. [DOI] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., MAYER D.J. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J. Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., MAYER D.J. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995a;61:353–364. doi: 10.1016/0304-3959(95)00022-K. [DOI] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., MAYER D.J. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995b;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- MARRIOTT D., WILKIN G.P., COOTE P.R., WOOD J.N. Eicosanoid synthesis by spinal cord astrocytes is evoked by substance P; possible implications for nociception and pain. Adv. Prostaglandin Thromboxane Leukotriene Res. 1991;21B:739–741. [PubMed] [Google Scholar]
- MATSUMURA K., WATANABE Y., IMAI-MATSUMURA K., CONNOLLY M., KOYAMA Y., ONOE H. Mapping of prostaglandin E2 binding sites in rat brain using quantitative autoradiography. Brain Res. 1992;581:292–298. doi: 10.1016/0006-8993(92)90720-t. [DOI] [PubMed] [Google Scholar]
- MENARD D.P., VAN ROSSUM D., KAR S., JOLICOEUR F.B., JHAMANDAS K., QUIRION R. Tolerance to the antinociceptive properties of morphine in the rat spinal cord: alteration of calcitonin gene-related peptide-like immunostaining and receptor binding sites. J. Pharmacol. Exp. Ther. 1995;273:887–894. [PubMed] [Google Scholar]
- MENARD D.P., VAN ROSSUM D., KAR S., ST PIERRE S., SUTAK M., JHAMANDAS K., QUIRION R. A calcitonin gene-related peptide receptor antagonist prevents the development of tolerance to spinal morphine analgesia. J. Neurosci. 1996;16:2342–2351. doi: 10.1523/JNEUROSCI.16-07-02342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURASE K., RYU P.D., RANDIC M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely isolated rat spinal dorsal horn neurons. Neurosci. Lett. 1989;103:56–63. doi: 10.1016/0304-3940(89)90485-0. [DOI] [PubMed] [Google Scholar]
- NICOL B., ROWBOTHAM D.J., LAMBERT D.G. mu- and kappa-opioids inhibit K+ evoked glutamate release from rat cerebrocortical slices. Neurosci. Lett. 1996;218:79–82. doi: 10.1016/s0304-3940(96)13104-9. [DOI] [PubMed] [Google Scholar]
- OKU R., SATOH M., FUJII N., OTAKA A., YAJIMA H., TAKAGI H. Calcitonin gene-related peptide promotes mechanical nociception by potentiating release of substance P from the spinal dorsal horn in rats. Brain Res. 1987;403:350–354. doi: 10.1016/0006-8993(87)90074-6. [DOI] [PubMed] [Google Scholar]
- OWEN J.A., MILNE B., JHAMANDAS K., NAKATSU K. Assembly of an inexpensive tail flick analgesia meter. J. Pharmacol. Meth. 1981;6:33–37. doi: 10.1016/0160-5402(81)90081-4. [DOI] [PubMed] [Google Scholar]
- POHL M., LOMBARD M.C., BOURGOIN S., CARAYON A., BENOLIEL J.J., MAUBORGNE A., BESSON J.M., HAMON M., CESSELIN F. Opioid control of the in vitro release of calcitonin gene-related peptide from primary afferent fibres projecting in the rat cervical cord. Neuropeptides. 1989;14:151–159. doi: 10.1016/0143-4179(89)90039-5. [DOI] [PubMed] [Google Scholar]
- POWELL K.J., HOSOKAWA A., BELL A., SUTAK M., MILNE B., QUIRION R., JHAMANDAS K. Comparative effects of cyclo-oxygenase and nitric oxide synthase inhibition on the development and reversal of spinal opioid tolerance. Br. J. Pharmacol. 1999;127:631–644. doi: 10.1038/sj.bjp.0702587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUIRION R., MA W., ZHENG W.-H., KAR S. Morphine induced calcitonin gene-related peptide increase in cultured dorsal root ganglion neurons. Soc. Neurosci. 1999;25:1434. doi: 10.1016/s0306-4522(00)00226-8. [DOI] [PubMed] [Google Scholar]
- QUIRION R., VAN ROSSUM D., DUMONT Y., ST-PIERRE S., FOURNIER A. Characterization of CGRP1 and CGRP2 receptor subtypes. Ann. N. Y. Acad. Sci. 1992;657:88–105. doi: 10.1111/j.1749-6632.1992.tb22759.x. [DOI] [PubMed] [Google Scholar]
- REKIK M., DELVAUX M., FREXINOS J., BUENO L. The calcitonin gene-related peptide activates both cAMP and NO pathways to induce relaxation of circular smooth muscle cells of guinea-pig ileum. Peptides. 1997;18:1517–1522. doi: 10.1016/s0196-9781(97)00246-5. [DOI] [PubMed] [Google Scholar]
- SALT T.E., HILL R.G. Pharmacological differentiation between responses of rat medullary dorsal horn neurons to noxious mechanical and noxious thermal cutaneous stimuli. Brain Res. 1983;263:167–171. doi: 10.1016/0006-8993(83)91216-7. [DOI] [PubMed] [Google Scholar]
- SHARMA S.K., KLEE W.A., NIRENBERG M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc. Natl. Acad. Sci. U.S.A. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMOYAMA N., SHIMOYAMA M., INTURRISI C.E., ELLIOTT K.J. Ketamine attenuates and reverses morphine tolerance in rodents. Anesthesiology. 1996;85:1357–1366. doi: 10.1097/00000542-199612000-00017. [DOI] [PubMed] [Google Scholar]
- TAYLOR G.M., MEERAN K., O'SHEA D., SMITH D.M., GHATEI M.A., BLOOM S.R. Adrenomedullin inhibits feeding in the rat by a mechanism involving calcitonin gene-related peptide receptors. Endocrinology. 1996;137:3260–3264. doi: 10.1210/endo.137.8.8754748. [DOI] [PubMed] [Google Scholar]
- TISEO P.J., INTURRISI C.E. Attenuation and reversal of morphine tolerance by the competitive N-methyl-D-aspartate receptor antagonist, LY274614. J. Pharmacol. Exp. Ther. 1993;264:1090–1096. [PubMed] [Google Scholar]
- TRAFTON J.A., ABBADIE C., MARCHAND S., MANTYH P.W., BASBAUM A.I. Spinal opioid analgesia: How critical is the regulation of substance P signalling. J. Neurosci. 1999;19:9642–9653. doi: 10.1523/JNEUROSCI.19-21-09642.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRUJILLO K.A., AKIL H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- TRUJILLO K.A., AKIL H. Inhibition of opiate tolerance by non-competitive N-methyl-D-aspartate receptor antagonists. Brain Res. 1994;633:178–188. doi: 10.1016/0006-8993(94)91538-5. [DOI] [PubMed] [Google Scholar]
- VAN ROSSUM D., HANISCH U.K., QUIRION R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci. Biobehav. Rev. 1997;21:649–678. doi: 10.1016/s0149-7634(96)00023-1. [DOI] [PubMed] [Google Scholar]
- VASKO M.R., CAMPBELL W.B., WAITE K.J. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J. Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WELCH S.P., SINGHA A.K., DEWEY W.L. The antinociception produced by intrathecal morphine, calcium, A23187, U50, 488H, [D-Ala2, N-Me-Phe4, Glyol]enkephalin and [D-Pen2, D-Pen5]enkephalin after intrathecal administration of calcitonin gene-related peptide in mice. J. Pharmacol. Exp. Ther. 1989;251:1–8. [PubMed] [Google Scholar]
- WIESENFELD-HALLIN Z., HOKFELT T., LUNDBERG J.M., FORSSMANN W.G., REINECKE M., TSCHOPP F.A., FISCHER J.A. Immunoreactive calcitonin gene-related peptide and substance P coexist in sensory neurons to the spinal cord and interact in spinal behavioral responses of the rat. Neurosci. Lett. 1984;52:199–204. doi: 10.1016/0304-3940(84)90374-4. [DOI] [PubMed] [Google Scholar]
- WIMALAWANSA S.J. Calcitonin gene-related peptide and its receptors: molecular genetics, physiology, pathophysiology, and therapeutic potentials. Endocrine Rev. 1996;17:533–585. doi: 10.1210/edrv-17-5-533. [DOI] [PubMed] [Google Scholar]
- XU X.J., WIESENFELD-HALLIN Z. The threshold for the depressive effect of intrathecal morphine on the spinal nociceptive flexor reflex is increased during autotomy after sciatic nerve section in rats. Pain. 1991;46:223–229. doi: 10.1016/0304-3959(91)90079-D. [DOI] [PubMed] [Google Scholar]
- YASKH T.L., RUDY T.A. Chronic catheterization of the subarachnoid space. Physiol. Behav. 1976;7:1032–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- YAKSH T.L., JESSELL T.M., GAMSE R., MUDGE A.W., LEEMAN S.E. Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature. 1980;286:155–157. doi: 10.1038/286155a0. [DOI] [PubMed] [Google Scholar]
- YASHPAL K., KAR S., DENNIS T., QUIRION R. Quantitative autoradiographic distribution of calcitonin gene-related peptide (hCGRP alpha) binding sites in the rat and monkey spinal cord. J. Comp. Neurol. 1992;322:224–232. doi: 10.1002/cne.903220208. [DOI] [PubMed] [Google Scholar]
- YE Z., WIMALAWANSA S.J., WESTLUND K.N. Receptor for calcitonin gene-related peptide: localization in the dorsal and ventral spinal cord. Neurosci. 1999;92:1389–1397. doi: 10.1016/s0306-4522(99)00088-3. [DOI] [PubMed] [Google Scholar]
- YU L.C., HANSSON P., LUNDEBERG T.The calcitonin gene-related peptide antagonist CGRP8-37 increases the latency to withdrawal responses in rat Brain Res. 1994653223–230.[published erratum appears in Brain Res 1994 Dec 15;666(2):295] [DOI] [PubMed] [Google Scholar]