

Abstract
The B1 receptor for kinins, stimulated by kinin metabolites without the C-terminal Arg residue (e.g., des-Arg9-bradykinin (BK) and Lys-des-Arg9-BK), is an increasingly recognized molecular target for the development of analgesic and anti-inflammatory drugs. Recently developed antagonists of this receptor were compared to a conventional antagonist, Ac-Lys-[Leu8]-des-Arg9-BK, in pharmacological assays based on the rabbit B1 receptor.
B-9858 (Lys-Lys-[Hyp3, Igl5, D-Igl7, Oic8]des-Arg9-BK) and three other analogues possessing the α-2-indanylglycine5 (Igl5) residue (order of potency B-9858 ≈ B-10146>B-10148>B-10050) were partially insurmountable antagonists of des-Arg9-BK in the contractility assay based on rabbit aortic rings. B-9858-induced depression of the maximal effect was more pronounced in tissues treated with the protein synthesis inhibitor cycloheximide to block the spontaneous increase of response attributed to the post-isolation formation of B1 receptors, and only partly reversible on washing.
By comparison, Ac-Lys-[Leu8]des-Arg9-BK was a surmountable antagonist (pA2 7.5), even in cycloheximide-treated tissues. B-9958 (Lys-[Hyp3, CpG5, D-Tic7, CpG8]des-Arg9-BK) was also surmountable (pA2 8.5).
The binding of [3H]-Lys-des-Arg9-BK to recombinant rabbit B1 receptors expressed in COS-1 cells was influenced by two of the antagonists: while Ac-Lys-[Leu8]des-Arg9-BK competed for the radioligand binding without affecting the Bmax, B-9858 decreased the Bmax in a time-dependent and washout-resistant manner.
B-9858 and analogues possessing Igl5 are the first reported non-competitive, non-equilibrium antagonists of the kinin B1 receptor.
Keywords: Non-competitive antagonism, rabbit aorta, kinin B1 receptor, vascular smooth muscle cell
Introduction
Bradykinin (BK) and related peptides (hereafter termed kinins) are blood-derived peptides that exert pharmacological effects on various tissues and cells, notably the vasculature, sensory neurons and renal and intestinal epithelia (Marceau et al., 1998). Prominent cardiovascular kinin effects support their role as endogenous vasodilator and hypotensive factors (Dendorfer et al., 1999). The effects of kinins are mediated by two types of G protein-coupled receptors, called B1 and B2. These receptors have been identified and defined using a series of pharmacological, molecular and genetic criteria (reviewed by Marceau et al., 1998).
Many successful B2 receptor antagonists have been developed, such as the peptide icatibant (Hock et al., 1991) or the non-peptides FR 173657 (Aramori et al., 1997) and LF 16-0335 (Pruneau et al., 1998). However, fewer B1 antagonists have been reported so far, and they are all des-Arg9-BK-related peptides or pseudopeptides (Galoppini et al., 1999). The therapeutic applications of B1 receptor ligands may have been overlooked, as this receptor subtype does not mediate the effect of exogenous kinins in most experimental systems and is selectively stimulated by des-Arg9-fragments of native kinins, that are produced by one of several metabolic pathways (kininase I; Décarie et al., 1996). However, the B1 receptor gene expression is highly regulated and completely inducible under the effect of several cytokines and mitogen activated protein kinases (Larrivée et al., 1998). Experimental pathologies reveal the anti-inflammatory, analgesic and potential anti-shock effects of the B1 receptor antagonists in animals (Marceau et al., 1998; Ahluwalia & Peretti, 1999; McLean et al., 1999).
Lys-[Leu8]des-Arg9-BK is the optimal B1 receptor antagonist based on natural amino acids for the human and rabbit forms of this receptor (Gobeil et al., 1997; 1999). Recently, metabolically stable B1 and mixed B1+B2 antagonists of high affinity and stability have been synthesized. B-9858 (Lys-Lys-[Hyp3, Igl5, D-Igl7, Oic8]des-Arg9-BK) belongs to this series and has some selectivity for B1 receptors (Gera et al., 1996; Gobeil et al., 1999). This peptide retains the Lys0 residue, a favourable feature for binding to human and rabbit B1 receptors. In binding assays, B-9858 is more potent on the human B1 receptor than on its murine homologue (MacNeil et al., 1997). Potent antagonism of des-Arg9-BK-induced contraction by B-9858 in the rat ileum preparation has also been reported (pA2 8.1; Galoppini et al., 1999), but the antagonism was unexpectedly not reversible in the rabbit aorta preparation (Gobeil et al., 1999). In the anaesthetized dog, representing a haemodynamic system co-expressing B1 and B2 receptors, B-9858 is reportedly 20 times more potent than Lys-[Leu8]des-Arg9-BK in antagonizing des-Arg9-BK-induced hypotension, and the antagonist effect lasts for more than 4 h (versus about 15 min for Lys-[Leu8]des-Arg9-BK; Stewart et al., 1996). The haemodynamic response to des-Arg9-BK in LPS-pretreated rabbits was used to test the in vivo effects of a large series of B1 receptor antagonists (Gobeil et al., 1999). While B-9858 was surpassed by others for in vitro potency or metabolic resistance, it was the most potent compound in vivo, thus supporting the possibility of a distinctive mode of action.
Together these findings suggest that B-9858 may belong to a family of drugs that includes irreversible antagonists of the rabbit B1 receptor, a previously unknown type of drug interaction with this receptor. The present experiments have been undertaken to characterize this behaviour and elucidate its structural basis.
Methods
Drugs
BK and cycloheximide (CHX) were purchased from Sigma (St. Louis, MO, U.S.A.) and des-Arg9-BK, from Bachem Bioscience Inc. (King of Prussia, PA, U.S.A.). B-9858 was a gift from Laboratoires Fournier S.C.A. (Daix, France). Ac-Lys-[Leu8]des-Arg9-BK and Sar-[D-Phe8]des-Arg9-BK are respectively a competitive antagonist and a metabolically stable agonist of the B1 receptors (Drapeau et al., 1993). The other kinin receptor antagonists B-9958, B-10050, B-10146 and B-10148 were synthesized using general methods described elsewhere (Gera et al., 1996). The structures of the six used B1 receptor antagonists used in this study are given in Table 1.
Table 1.
Structure of the tested B1 receptor antagonists and affinity estimates for their interaction with the rabbit B2 receptors

Contractility studies
Rabbit aortic rings (New Zealand white, 1.5–2 kg, Charles River, St. Constant, Canada) were suspended under a tension of 2 g in 5 ml tissue baths containing oxygenated (95% O2: 5% CO2) and warmed (37°C) Krebs solution as described (Larrivée et al., 1998). Some experiments were also performed on rabbit jugular veins, cut into 2 cm strips and suspended under a tension of 1 g in 5 ml tissue baths containing oxygenated and warmed Krebs solution to which 1 μM captopril was added (Marceau et al., 1994).
Contractility studies in the aortic preparation were based upon the construction of cumulative concentration-responses curves for des-Arg9-BK (a B1 receptor agonist on this tissue). These studies aimed to investigate the potency and surmountability of the B1 receptor antagonists in the vascular smooth muscle preparation. In each tissue, two cumulative concentration-response curves were constructed at times 3.5 and 5.5 h from the beginning of the incubation of the preparation. One of the six antagonists listed in Table 1, or the saline vehicle in control tissues, was introduced at time 5 h and maintained in the bathing fluid during the construction of the second curve (effect of the antagonist at 5.5 h). Contractility results were expressed as a per cent of the maximal response recorded in each tissue when constructing the control curve (3.5 h); this calculation accounts for the fact that the maximal response to this kinin increases as a function of the post-isolation incubation time in this preparation (a behaviour specific for agonists of B1 receptors and attributed to the post-isolation formation of these receptors; Bouthillier et al., 1987; Audet et al., 1994; Larrivée et al., 1998). Additional contractility experiments were designed to monitor the effect of the inhibition of protein synthesis on the depression of the maximal agonist effect induced by B-9858. Tissues were exposed to CHX (71 μM) from the time 4.5 h on, with or without an antagonist (from the time 5 h on).
The contractility experiments in the rabbit jugular vein, which responds to kinins via preformed B2 receptors, were performed to investigate the potency and surmountability of recently produced antagonists against BK (as in Marceau et al., 1994).
Cloning and expression of the rabbit B1 receptor gene
Rabbit genomic DNA was isolated from rabbit liver, as described previously (Bachvarov, et al., 1995). Five hundred micrograms of the rabbit genomic DNA were consecutively used as a template for the PCR amplification of the entire coding region (intronless) of the rabbit B1 receptor, as 5′-ATAAAAGCTTATGGCCTCACAGGGCCCCCTG-3′ and 5′-ATA AGGATCCT TAAT TCCGCCAGAA ACCCCAGAGC-3′ were utilized as PCR sense and anti-sense primers, respectively. These primers were based on the published sequence of the rabbit B1 receptor gene (MacNeil et al., 1995), and contain additional HindIII and BamHI sites (underlined) for the directional cloning of the rabbit B1 receptor coding region in the eukaryotic expression vector pcDNA3 (Invitrogen, Carlsbad, CA, U.S.A.), under the control of the strong cytomegalovirus promoter.
Binding assay to the recombinant rabbit B1 receptor
A binding assay to rabbit B1 receptors was conducted as described (Levesque et al., 1995a), except for the identity of the ligand which was [3H]-Lys-des-Arg9-BK ([3H]-des-Arg10-kallidin, NEN Biosciences, 105 Ci mmol−1). Briefly, COS-1 cells were seeded at a high density in 24-well plates (culture medium DMEM supplemented with 10% foetal bovine serum and antibiotics, Gibco). After 24 h, the cells (70–80% confluent) were transiently transfected with the expression vector described above using the Ex-Gen 500 transfection reagent (MBI Fermentas Inc., Flamborough, Canada) as directed by the manufacturer. Some untransfected cells were used in control experiments. After an additional 48 h culture period, cell wells were washed twice with the binding medium (consisting of Medium 199 supplemented with 0.1% bovine serum albumin, 1 μM amastatin, 1 μM captopril, 1 μM phosphoramidon (Sigma) and sodium azide 0.02% w v−1) and filled with 0.5 ml of prewarmed (37°C) binding medium. The B1 receptor ligand (0.125–4 nM) and cold competing peptides (1 μM of the agonist Lys-des-Arg9-BK for the determination of non-specific binding or nanomolar concentrations of B1 receptor antagonists studied to characterize the effect of these drugs on the receptor population) were added to the wells. Separate protocols dealt with the effect of cell preincubation with the B1 receptor antagonists (30 min) in the culture medium before initiating the binding assay. In one experiment, the binding assay followed immediately the preincubation, and the same antagonist concentrations were maintained during the assay. In another experiment, the preincubation with antagonist was followed by a 1 h washout period at 37°C (wells rinsed twice with the binding buffer) before initiating the binding assay in the absence of competing antagonist. After 60 min of incubation at 37°C, each well was washed three times with 2 ml of ice-cold PBS, pH 7.4. One ml of 0.1 N NaOH was finally added to dissolve the cells. Radioactivity in the resulting suspension was determined by scintillation counting (5 min per vial).
Statistics
Results are expressed as means±s.e.mean and Mann–Whitney statistics were calculated using the InStat 2.0 computer program (GraphPad Software, San Diego, CA, U.S.A.). The parameters of the Schild and Scatchard plots (pA2 determination and binding data treatment, respectively) were obtained using a computer program (Tallarida & Murray, 1987).
Results
Vascular contractility mediated by B1 receptors in the rabbit isolated aorta
Rabbit aortic rings were stimulated with the B1 receptor agonist des-Arg9-BK at 3.5 and 5.5 h. The paired control curves representing the peptide effect in the same preparations at 3.5 h and 5.5 h in the absence of antagonists showed that the potency (EC50) of the peptide remained relatively stable, while the maximal effect (Emax) increased by about 35% during this period (Figure 1; P<10−4, Mann–Whitney test).
Figure 1.
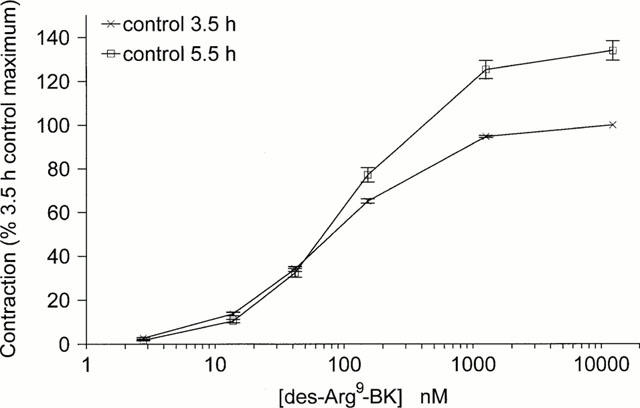
Change in the concentration-effect curve of the B1 receptor agonist des-Arg9-BK as a function of the incubation time in rabbit aortic rings. Responses are expressed as per cent of the maximal effect of the peptide in a control recording established at time 3.5 h in each tissue. Values are the means, and vertical bars the s.e.mean of 36 determinations.
When applied before the construction of the 5.5 h concentration-effect curve, Ac-Lys-[Leu8]des-Arg9-BK shifted the curve to the right, without depressing Emax (Figure 2A). In fact, some apparent increase in des-Arg9-BK Emax was recorded in tissues exposed to the highest concentration of the antagonist (1 μM). The estimated pA2 value was 7.5±0.03, and the slope of the Schild regression close to unity (0.97; calculation of Schild plot parameters based on the EC50 values calculated from the average curves in Figure 2A). The drug B-9858 modified the concentration-effect curve to the B1 receptor agonist (at 5.5 h) as it simultaneously shifted it to the right and decreased the expected Emax observed in the absence of antagonist in control rings sampled from the same animals (Figure 2B). These changes of the curve were consistently observed at drug concentrations of 54 pM and above. The remaining drugs behaved either as surmountable (B-9958, Figure 2C; pA2 estimate 8.5±0.05; regression slope 0.93) or at least partially insurmountable antagonists (B-10050, B-10146, B-10148; Figure 2D–F) considering their action on the agonist Emax. The insurmountable effect of four of the antagonists is not favourable for calculation of an affinity estimate, unless equilibrium is reached during the time frame of the experiment (Kenakin, 1993). The order of potency for the partially insurmountable antagonists was: B-9858 ≈ B-10146>B-10148>B-10150.
Figure 2.
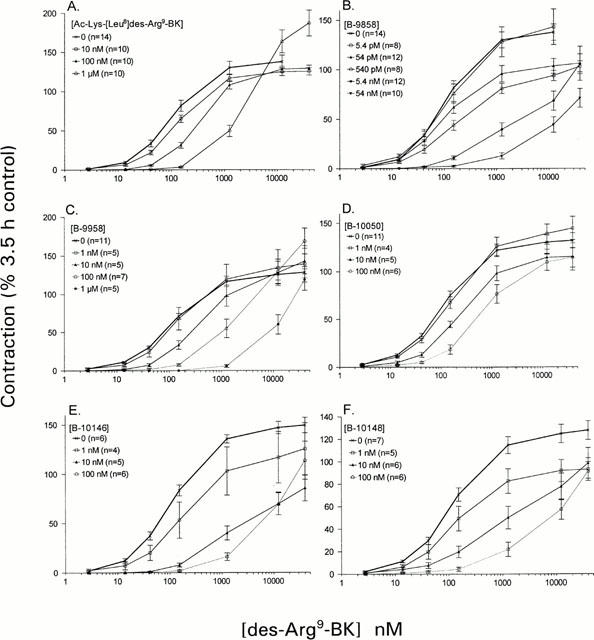
Effect of six B1 receptor antagonists or their saline vehicle on des-Arg9-BK-induced contraction in the rabbit isolated aorta at time 5.5 h. Responses are expressed as a per cent of the maximal effect of des-Arg9-BK in a control recording established at time 3.5 h in each tissue.
Protein synthesis blockade was applied after the recording of the first concentration-effect curve in some tissues in order to determine whether the stabilization of des-Arg9-BK Emax with CHX (Deblois et al., 1989) could modify antagonist surmountability. The curve obtained at 5.5 h in the presence of CHX alone was similar to the control curve constructed at 3.5 h, as the maximal response at 5.5 h amounted to 95.7% of that recorded at 3.5 h in the same tissues (Figure 3). The depression of the maximal response induced by B-9858 (54 nM) was more profound in CHX-treated tissues (11.5%, relative to the response recorded without the antagonist, Figure 3, versus 52% in tissues not treated with CHX, Figure 2B). Treatment with Ac-Lys-[Leu8]des-Arg9-BK (1 μM) shifted the agonist concentration-effect curve to the right, somewhat more in the presence (Figure 3) than in the absence (Figure 2A) of CHX, but the surmountability of the drug was preserved in the presence of CHX.
Figure 3.
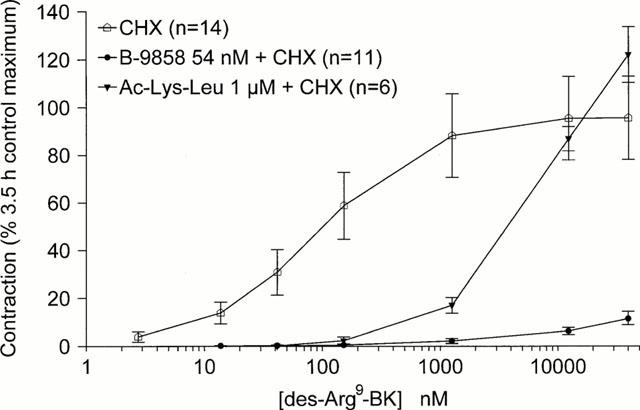
Effect of protein synthesis inhibition applied to rabbit aortic rings after the construction of the initial concentration-effect curve of des-Arg9-BK, but before the subsequent curve recorded in the presence or absence of the antagonists B-9858 or Ac-Lys-[Leu8]des-Arg9-BK. Responses are expressed as in Figures 1 and 2.
A special protocol was designed to evaluate the reversibility of the depressing effect of B-9858 on des-Arg9-BK Emax (Figure 4). The agonist peptide was applied at the single, nearly maximal concentration of 11.1 μM at 3.5, 5.5 and 8.5 h. B-9858 (54 nM) was present only in the time window 5–6 h and the tissues were abundantly washed thereafter. The protein synthesis inhibitor CHX was present in some tissues from the time point 4.5 h and on. As reported above, B-9858 depressed the effect of a large concentration of des-Arg9-BK, and significantly more in tissues exposed to CHX (Figure 4, P=0.04, Mann–Whitney test). An extensive recovery of the agonist effect in the following period was observed in the absence of CHX, but not in its presence (74.3±13.2% of the control contraction in the absence of CHX, 40.2±4.0% in the presence of CHX, P=0.04).
Figure 4.
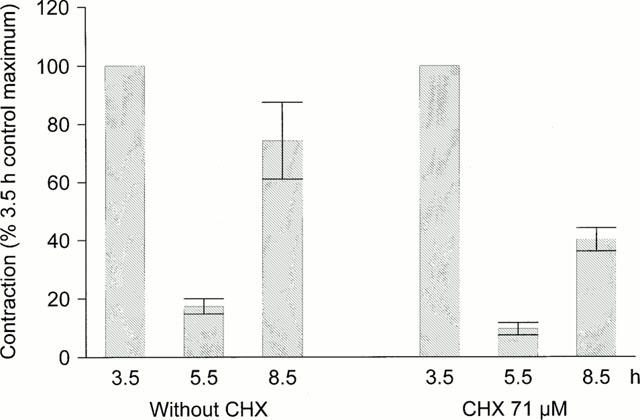
Contractile effect of des-Arg9-BK (11.1 μM) on rabbit aortic rings, as modified by incubation time (control response recorded at 3.5 h), the presence of the antagonist B-9858 (54 nM in the time window 5–6 h in all tissues) and by the presence of cycloheximide (CHX, 71 μM, from the time point 4.5 h on in some tissues only). Values are expressed as per cent of the control contraction and are the means±s.e.mean of nine determinations. See text for statistics.
Effect of B-9858 and analogues on the B2 receptors of the rabbit jugular vein
The recently developed B1 receptor antagonists were investigated for receptor subtype specificity by testing them as antagonists of the B2 receptor-mediated effect of BK on the jugular vein. The five compounds exhibited apparently surmountable antagonist actions with variable potencies; pA2 estimates based on Schild plot construction (Marceau et al., 1994) are reported in Table 1.
Radioligand binding to B1 receptors
Untransfected COS-1 cells bound very little [3H]-Lys-des-Arg9-BK in a specific manner (Figure 5A). In initial studies, the specific binding of [3H]-Lys-des-Arg9-BK to recombinant rabbit B1 receptors transiently expressed by COS-1 cells was found to be saturable (Figure 5A). The calculated Scatchard regression parameters were a KD of 0.37 nM and a Bmax of 89±8 fmol well−1 for this particular transfection. When antagonists were introduced simultaneously with the ligand in the binding reaction, the parameters were modified (Figure 5A): Ac-Lys-[Leu8]des-Arg9-BK or B-9858 did not modify the Bmax (85±14 or 88±9 fmol well−1, respectively), but decreased the apparent affinity (apparent KD increased to 0.69 or 0.72 nM, respectively), as expected for competitive antagonists (Scatchard plots not shown). A separate experiment involved a 30 min preincubation of the cells with the B1 receptor antagonists in complete culture medium before performing the binding assay (Figure 5B, corresponding Scatchard plots 5C). Aside from a lower ligand Bmax in these particular cells, the results of the saturation curves without antagonist or with 10 nM of Ac-Lys-[Leu8]des-Arg9-BK were comparable to previous findings (control Bmax=36±3 fmol well−1, control KD=0.24 nM; in the presence of the antagonist, Bmax=39±5 fmol well−1, KD=0.77 nM). However, preincubation of the cells with B-9858 resulted in a loss of binding sites (Bmax=26±3 fmol well−1, KD=1.02 nM).
Figure 5.
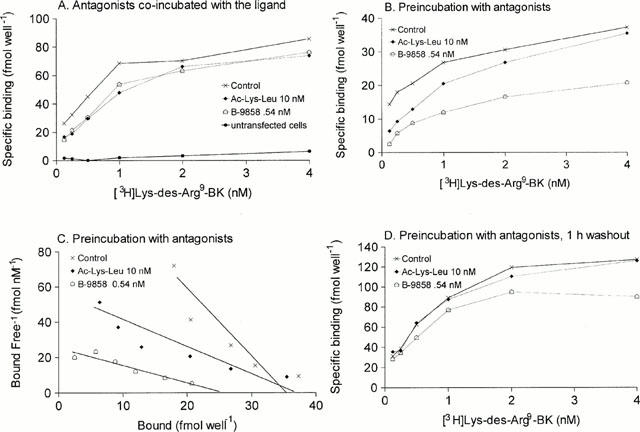
Binding of [3H]-Lys-des-Arg9-BK to recombinant rabbit B1 receptors expressed in COS-1 cells as influenced by antagonist drugs (A). Saturation curves obtained without antagonist (Control) or in the presence of Ac-Lys-[Leu8]des-Arg9-BK (10 nM) or of B-9858 (0.54 nM). Antagonist drugs were introduced at the same time as the radioligand. In panel A, the ligand binding to untransfected cells is also shown (B). Saturation curves obtained in a different transfection with the same drugs; the cells were incubated with the antagonists in the culture medium for 30 min before initiating the binding assay. (C) Scatchard plots derived from the saturation data from panel B. (D) Effect of preincubation with antagonists followed by drug washout on the radioligand binding. See text for details and data analysis.
A 1 h drug washout period was applied after the 30 min preincubation period with antagonists in a separate experiment to test for antagonist binding reversibility (Figure 5D). In these experiments, the antagonists were not present in the binding buffer when the radioligand was introduced. Results from wells pretreated with B-9858 suggest a low reversibility of the antagonism exerted by this compound, as the observed Bmax (106±12 fmol well−1) is lower than the control value (144±15 fmol well−1). Apparent KD values in these experiments were similar (0.44 and 0.56 nM, respectively). Ac-Lys-[Leu8]des-Arg9-BK binding seems to be completely reversible under the conditions applied, as the calculated Scatchard plot parameters (Bmax=140±15 fmol well−1; KD=0.55 nM,) coincide with those of the control curve.
Discussion
B-9858 is a member of a recently described, structurally constrained family of peptide antagonists for the kinin receptors (Gera et al., 1996: Stewart et al., 1996). These antagonists exhibited resistance to peptidases, high potency and a relative pharmacological ‘promiscuity', as several of them were active on both B1 and B2 receptors. Using rabbit vascular smooth muscle preparations, we showed that B-9858 is a surmountable antagonist of moderate potency of the B2 receptor (pA2 7.0), while it is a very potent and insurmountable B1 receptor antagonist (Figure 2B). By comparison, the antagonist of classical design Ac-Lys-[Leu8]des-Arg9-BK did not depress the estimated 5.5 h Emax of the agonist des-Arg9-BK (Figure 2A). The tested series of B-9858 analogues (Table 1, Figure 2) allows formulation of several hypothesis on the effect of structure on affinity for B1 receptors: the D-pentafluorophenylalanine (F5F) residue in position 7 confers a particularly high affinity, when two compounds that differ only at this position are compared (B-10146 and B-10050); however D-2-indanylglycine7 (D-Igl7), also confers a high potency, as seen by comparing B-9858 with B-10148. Diverse residues with a cyclic side-chain can be located in position 8 without important affinity changes (e.g., compare B-10146 to B-10148). Finally, the compounds suspected of insurmountable antagonism possess the Igl5 residue. This residue is absent in Ac-Lys-[Leu8]des-Arg9-BK and B-9958, and both of these compounds are surmountable. Our pA2 estimate for the latter drug is close to the one previously reported on the rabbit isolated aorta using another experimental approach (8.4, Gobeil et al., 1997). That cumulative concentration-effect curves reach a higher Emax in the presence of increasing concentrations of the two surmountable antagonists is of obscure significance, but has also been observed in other systems (e.g., see Pruneau et al., 1998; 1999).
A practical problem when studying the B1 receptors based on the rabbit isolated aorta is to separate drug effect from the effect of time on the system. The progressive increase of B1 receptor agonist Emax in the rabbit aorta has been found to be sizeable during the time period 3.5–5.5 h (Figure 1). Thus, it was hypothesized that some of the residual effects of des-Arg9-BK in the presence of a representative insurmountable antagonist, B-9858, may be partially dependent on the renewal of receptors at the cell surface. The use of protein synthesis inhibitors such as CHX supports the concept of tissue injury-induced de novo formation of B1 receptors (Bouthillier et al., 1987; Audet et al., 1994). Notably, when CHX is applied from the beginning of the in vitro incubation, rabbit aortic tissues remain unresponsive to des-Arg9-BK, but respond to the agonists of other receptor types in a stable manner. Another documented use of CHX on this preparation served to stabilize the response to des-Arg9-BK when it had reached a certain level (Deblois et al., 1989), suggesting that a definite receptor population can be stabilized and repeatedly stimulated in the system. Application of CHX after the construction of the 3.5 h concentration-effect curve effectively stabilized the subsequent response to des-Arg9-BK (Figure 3). This procedure further depressed the residual effect of the agonist in the presence of B-9858 (Figure 3), establishing more clearly the insurmountable nature of the antagonism exerted by this drug. Ac-Lys-[Leu8]des-Arg9-BK did not reduce des-Arg9-BK Emax in CHX-treated tissues (Figure 3), suggesting that this compound behaves as a competitive antagonist on the stabilized B1 receptor population. However, Ac-Lys-[Leu8]des-Arg9-BK was about half as potent in CHX-treated tissues, suggesting another form of distortion by the unstable behaviour of the preparation.
Non-competitive antagonism can be further classified into equilibrium or non-equilibrium types (Kenakin, 1993), the latter category being defined as a time-dependent removal of receptors from the equilibrium by the action of an irreversible or slowly reversible antagonist. A 3 h washout period allowed a certain recovery of the des-Arg9-BK Emax in B-9858-treated tissues (Figure 4), but the effect of adding CHX to some preparations suggests that the recovery may be due in part to the formation of new B1 receptors, and that the reversibility of B-9858 antagonism from the preformed receptor is actually low. The lack of equilibrium invalidates the use of pA2 or pKB scales to measure the potency of antagonist (Kenakin, 1993).
We conducted our radioligand binding assay at 37°C in the presence of sodium azide; these conditions allow reaching an effective equilibrium without ligand internalization in this system (Levesque et al., 1995a). The observed control KD values were in the same range than those obtained using rabbit smooth muscle cells and the same tritiated ligand (Schneck et al., 1994; Galizzi et al., 1994). Preincubation of living cells with antagonists was employed in some experiments; this was a valid approach, notably because N-acetylation in Ac-Lys-[Leu8]des-Arg9-BK confers a complete resistance to degradation in serum, relative to the more potent but fragile sequence Lys-[Leu8]des-Arg9-BK (Drapeau et al., 1993). While Ac-Lys-[Leu8]des-Arg9-BK retained its surmountable effect and reached equilibrium in the binding essay, B-9858 exerted a complex effect, consisting of a shift to the right of the saturation curve and, only in cells pretreated with the drug, of a time-dependent loss of binding sites (Figure 5). A variation of the assay also evidenced the low reversibility of B-9858 binding (Figure 5D). Binding assays based on rabbit aortic smooth muscle cells, cultured and characterized as previously described (Levesque et al., 1993; 1995b), also revealed that B-9858, but not Ac-Lys-[Leu8]des-Arg9-BK, reduced [3H]-Lys-des-Arg9-BK Bmax (data not shown). Thus, B-9858 is a prototype of a non-competitive, non-equilibrium antagonist for the kinin B1 receptor. This type of interaction was heretofore unknown for this receptor type, but several examples involving other related receptor types are known (e.g. the peptide icatibant at the rabbit BK B2 receptor; Marceau et al., 1994; Bachvarov et al., 1995; Houle et al., 2000; the clinically used non-peptide angiotensin antagonists at the human AT1 receptor, Vanderheyden et al., 1999).
Non-equilibrium antagonism at the rabbit B1 receptor exhibits several interesting features that should orient future molecular investigations: the drugs are charged peptides of relatively high molecular weight, making unlikely the interference with non-receptor intracellular sites; unlike the B2 receptor, the B1 receptor is not believed to undergo important agonist-induced phosphorylation and internalization (Austin et al., 1997; Faussner et al., 1998). Finally, the irreversible or slowly reversible binding of B-9858 can be exploited to demonstrate receptor up-regulation by inactivating a pre-existing receptor population, thus supporting studies of the dynamic regulation of this receptor type.
Acknowledgments
Supported by the Medical Research Council of Canada (MRCC; grant MOP-14077). J.-F. Larrivée and S. Houle have been the Recipients of Studentships from the FCAR-FRSQ program, Quebec, and the MRCC, respectively. D.R. Bachvarov is a Scholar of the FRSQ. Synthetic work was supported by U.S. NIH grant HL-26284.
Abbreviations
- Ac-Lys
N-acetyl-L-lysine
- BK
bradykinin
- ChG
α-cyclohexylglycine
- CHX
cycloheximide
- CpG
α-cyclopentylglycine
- F5F
pentafluorophenylalanine
- Hyp
trans-4-hydroxyproline
- Igl
α-2-indanylglycine
- Oic
octahydroindole-2-carboxylic acid
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
References
- AHLUWALIA A., PERETTI M. B1 receptors as a new inflammatory target: Could this B the 1. Trends Pharmacol. Sci. 1999;20:100–104. doi: 10.1016/s0165-6147(99)01321-8. [DOI] [PubMed] [Google Scholar]
- ARAMORI I., ZENKOH J., MORIKAWA N., O'DONNELL N., ASANO M., NAKAMURA K., IWAMI M., KOJO H., NOTSU Y. Novel subtype selective nonpeptide bradykinin receptor antagonists FR167344 and FR173657. Mol. Pharmacol. 1997;51:171–176. doi: 10.1124/mol.51.2.171. [DOI] [PubMed] [Google Scholar]
- AUDET R., PETITCLERC E., DRAPEAU G., RIOUX F., MARCEAU F. Further analysis of the upregulation of bradykinin B1 receptors in isolated rabbit aorta by using metabolic inhibitors. Eur. J. Pharmacol. 1994;271:551–555. doi: 10.1016/0014-2999(94)90819-2. [DOI] [PubMed] [Google Scholar]
- AUSTIN K.E., FAUSSNER A., ROBINSON H.E., CHAKRAVARTY S., KYLE D.J., BATHON J.M., PROUD D. Stable expression of the human kinin B1 receptor in Chinese hamster ovary cells. J. Biol. Chem. 1997;272:11420–11425. doi: 10.1074/jbc.272.17.11420. [DOI] [PubMed] [Google Scholar]
- BACHVAROV D.R., SAINT-JACQUES E., LARRIVÉE J.-F., LEVESQUE L., RIOUX F., DRAPEAU G., MARCEAU F. Cloning and pharmacological characterization of the rabbit bradykinin B2 receptor. J. Pharmacol. Exp. Ther. 1995;275:1623–1630. [PubMed] [Google Scholar]
- BOUTHILLIER J., DEBLOIS D., MARCEAU F. Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vitro and in vivo. Br. J. Pharmacol. 1987;92:257–264. doi: 10.1111/j.1476-5381.1987.tb11319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEBLOIS D., BOUTHILLIER J., MARCEAU F. Pharmacological modulation of the up-regulated responses to des-Arg9-bradykinin in vivo and in vitro. Immunopharmacology. 1989;17:187–198. doi: 10.1016/0162-3109(89)90047-7. [DOI] [PubMed] [Google Scholar]
- DÉCARIE A., RAYMOND P., GERVAIS N., COUTURE R., ADAM A. Serum interspecies differences in metabolic pathways of bradykinin and [des-Arg9]BK: Influence of enalaprilat. Am. J. Physiol. 1996;270:H1340–H1347. doi: 10.1152/ajpheart.1996.271.4.H1340. [DOI] [PubMed] [Google Scholar]
- DENDORFER A., WOLFRUM S., DOMINIAK P. Pharmacology and cardiovascular implications of the kinin-kallikrein system. Jpn. J. Pharmacol. 1999;79:403–426. doi: 10.1254/jjp.79.403. [DOI] [PubMed] [Google Scholar]
- DRAPEAU G., AUDET R., LEVESQUE L., GODIN D., MARCEAU F. Development and in vivo evaluation of metabolically resistant antagonists of B1 receptors for kinins. J. Pharmacol. Exp. Ther. 1993;266:192–199. [PubMed] [Google Scholar]
- FAUSSNER A., PROUD D., TOWNS M., BATHON J.M. Influence of the cytosolic carboxyl termini of human B1 and B2 kinin receptors on receptor sequestration, ligand internalization, and signal transduction. J. Biol. Chem. 1998;273:2617–2623. doi: 10.1074/jbc.273.5.2617. [DOI] [PubMed] [Google Scholar]
- GALIZZI J.P., BODINIER M.C., CHAPELAIN B., LY S.M., COUSSY L., GIREAUD S., NELIAT G., JEAN T. Up-regulation of [3H]-des-Arg10-kallidin binding to the bradykinin B1 receptor by interleukin-1β in isolated smooth muscle cells: correlation with B1 agonist-induced PGI2 production. Br. J. Pharmacol. 1994;113:389–394. doi: 10.1111/j.1476-5381.1994.tb17001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALOPPINI C., MEINI S., TANCREDI M., DI FENZA A., TRIOLO A., QUARTARA L., MAGGI C.A., FORMAGGIO F., TONOLIO C., MAZZUCCO S., PAPINI A., ROVERO P. A new class of pseudopeptide antagonists of the kinin B1 receptor containing alkyl spacers. J. Med. Chem. 1999;42:409–414. doi: 10.1021/jm980495r. [DOI] [PubMed] [Google Scholar]
- GERA L., STEWART J.M., WHALLEY E.T., BURKARD M., ZUZACK J.S. New bradykinin antagonists having very high potency at B1 receptors. Immunopharmacology. 1996;33:183–185. doi: 10.1016/0162-3109(96)00100-2. [DOI] [PubMed] [Google Scholar]
- GOBEIL F., CHARLAND S., FILTEAU C., PERRON S.I., NEUGEBAUER W., REGOLI D. Kinin B1 receptor antagonists containing α-methyl-L-phenylalanine: In vitro and in vivo antagonist activities. Hypertension. 1999;33:823–829. doi: 10.1161/01.hyp.33.3.823. [DOI] [PubMed] [Google Scholar]
- GOBEIL F., NEUGEBAUER W., NGUYEN-LE X.K., NSA ALLOGHO S., PHENG L.H., BLOUIN D., WHALLEY E.T., REGOLI D. Pharmacological profiles of the human and rabbit B1 receptors. Can. J. Physiol. Pharmacol. 1997;75:591–595. [PubMed] [Google Scholar]
- HOCK F.J., WIRTH K.J., ALBUS U., LINZ W., GERHARDS H.J., WIEMER G., HENKE S., BREIPOHL G., KÖNIG W., KNOLLE J., SCHÖLKENS B.A. Hoe 140 a new potent and long acting bradykinin antagonist: In vitro studies. Br. J. Pharmacol. 1991;102:769–773. doi: 10.1111/j.1476-5381.1991.tb12248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOULE S., LARRIVÉE J.-F., BACHVAROVA M., BOUTHILLIER J., BACHVAROV D.R., MARCEAU F. Antagonist-induced intracellular sequestration of the rabbit bradykinin B2 receptor. Hypertension. 2000;35:1319–1325. doi: 10.1161/01.hyp.35.6.1319. [DOI] [PubMed] [Google Scholar]
- KENAKIN T.P. Pharmacologic analysis of drug-receptor interaction. New York: Raven Press Ltd; 1993. [Google Scholar]
- LARRIVÉE J.-F., BACHVAROV D.R., HOULE F., LANDRY J., HUOT J., MARCEAU F. Role of the mitogen-activated protein kinases in the expression of the kinin B1 receptors induced by tissue injury. J. Immunol. 1998;160:1419–1426. [PubMed] [Google Scholar]
- LEVESQUE L., DRAPEAU G., GROSE J.H., RIOUX F., MARCEAU F. Vascular mode of action of kinin B1 receptors and development of a cellular model for the investigation of these receptors. Br. J. Pharmacol. 1993;109:1254–1262. doi: 10.1111/j.1476-5381.1993.tb13757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVESQUE L., HARVEY N., RIOUX F., DRAPEAU G., MARCEAU F. Development of a binding assay for the B1 receptors for kinins. Immunopharmacology. 1995a;29:141–147. doi: 10.1016/0162-3109(94)00053-i. [DOI] [PubMed] [Google Scholar]
- LEVESQUE L., LARRIVÉE J.-F., BACHVAROV D.R., RIOUX F., DRAPEAU G., MARCEAU F. Regulation of kinin-induced contraction and DNA synthesis by inflammatory cytokines in the smooth muscle of the rabbit aorta. Br. J. Pharmacol. 1995b;116:1673–1679. doi: 10.1111/j.1476-5381.1995.tb16390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACNEIL T., BIERILO K.K., MENKE J.G., HESS J.F. Cloning and pharmacological characterization of a rabbit bradykinin B1 receptor. Biochim. Biophys. Acta. 1995;1264:223–228. doi: 10.1016/0167-4781(95)00152-7. [DOI] [PubMed] [Google Scholar]
- MACNEIL T., FEIGHER S., HRENIUK D.L., HESS J.F., VAN DER PLOEG L.H.T. Partial agonists and full antagonists at the human and murine bradykinin B1 receptors. Can. J. Physiol. Pharmacol. 1997;75:735–740. [PubMed] [Google Scholar]
- MARCEAU F., HESS J.F., BACHVAROV D.R. The B1 receptors for kinins. Pharmacol. Rev. 1998;50:357–386. [PubMed] [Google Scholar]
- MARCEAU F., LEVESQUE L., DRAPEAU G., RIOUX F., SALVINO J.M., WOLFE H.R., SEOANE P.R., SAWUTZ D.G. Effects of peptide and nonpeptide antagonists of bradykinin B2 receptors on the venoconstrictor action of bradykinin. J. Pharmacol. Exp. Ther. 1994;269:1136–1143. [PubMed] [Google Scholar]
- MCLEAN P.G., PERRETTI M., AHLUWALIA A. Inducible expression of the kinin B1 receptor in the endotoxemic heart: mechanisms of des-Arg9 bradykinin-induced coronary vasodilation. Br. J. Pharmacol. 1999;128:275–282. doi: 10.1038/sj.bjp.0702743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRUNEAU D., LUCCARINI J.M., FOUCHET C., DEFRÊNE E., FRANCK R.M., LOILLIER B., DUCLOS H., ROBERT C., CREMERS B., BÉLICHARD P., PAQUET J.-L. LF 16.0335, a novel potent and selective nonpeptide antagonist of the human bradykinin B2 receptor. Br. J. Pharmacol. 1998;125:365–372. doi: 10.1038/sj.bjp.0702083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRUNEAU D., PAQUET J.L., LUCCARINI J.M., DEFRÊNE E., FOUCHET C., FRANK R.-M, , LOILLIER B., ROBERT C., BÉLICHARD P., DUCLOS H., CREMERS B., DODEY P. Pharmacological profile of LF 16-0687, a new potent non-peptide bradykinin B2 receptor antagonist. Immunopharmacology. 1999;43:187–194. doi: 10.1016/s0162-3109(99)00128-9. [DOI] [PubMed] [Google Scholar]
- SCHNECK K.A., HESS J.F., STONESIFER G.Y., RANSOM R.W. Bradykinin B1 receptors in rabbit aorta smooth muscle cells in culture. Eur. J. Pharmacol. 1994;266:277–282. doi: 10.1016/0922-4106(94)90137-6. [DOI] [PubMed] [Google Scholar]
- STEWART J.M., GERA L., HANSON W., ZUZAK J.S., BURKARD M., MCCULLOUGH R., WHALLEY E.T. A new generation of bradykinin antagonists. Immunopharmacology. 1996;31:51–60. doi: 10.1016/0162-3109(96)00084-7. [DOI] [PubMed] [Google Scholar]
- TALLARIDA R.J., MURRAY R.B. Manual of Pharmacologic Calculations with Computer Programs. New York: Springer-Verlag; 1987. [Google Scholar]
- VANDERHEYDEN P.M.L., FIERENS F.L.P., DE BACKER J.P., FRAEYMAN N., VAUQUELIN G. Distinction between surmountable and insurmountable selective AT1 receptor antagonists by use of CHO-K1 cells expressing human angiotensin II AT1 receptors. Br. J. Pharmacol. 1999;126:1057–1065. doi: 10.1038/sj.bjp.0702398. [DOI] [PMC free article] [PubMed] [Google Scholar]