

Abstract
Ca2+ entry mechanisms underlying spontaneous arteriolar tone and acute myogenic reactivity remain uncertain. These studies aimed to compare the effects of nifedipine and the putative T-channel blocker, mibefradil, on arteriolar myogenic responsiveness and intracellular Ca2+ (Ca2+i).
First order cremaster muscle arterioles (1A) were isolated from rats, cannulated, pressurized to 70 mmHg in the absence of intraluminal flow, and mechanical responses studied by video microscopy. The Ca2+i was measured using fluorescence imaging of Fura 2 loaded arterioles.
Both nifedipine and mibefradil showed dose-dependent inhibition of spontaneous myogenic tone (at 70 mmHg; pEC50 7.04±0.17 vs 6.65±0.20 respectively, n=6 for both, n.s.) and KCl-induced vasoconstriction (at 70 mmHg; pEC50 6.93±0.38 vs 6.45±0.27 respectively, n=6 for both, n.s.).
In arterioles maintained at 50 mmHg, nifedipine (10−7 and 10−5 M) caused a concentration dependent reduction in Ca2+i, however, mibefradil (10−7 and 10−5 M) had no effect. Furthermore nifedipine significantly attenuated the increase in Ca2+i associated with an acute pressure step (50–120 mmHg) whereas mibefradil was considerably less effective.
Mibefradil (10−7 M) significantly attenuated contractile responses to 60 mM KCl without altering the KCl-induced increase in Ca2+i, in contrast to nifedipine (10−7 M) which reduced both Ca2+i and contraction.
Membrane potential of arterioles with spontaneous myogenic tone (70 mmHg) was −41.5±1.0 mV. Nifedipine (10−7 or 10−5 M) had no effect on membrane potential, however mibefradil (10−5 M) caused significant depolarization.
In summary, both mibefradil and nifedipine inhibit arteriolar spontaneous tone and acute myogenic reactivity. While there may be overlap in the mechanisms by which these agents inhibit tone, differences in effects on membrane potential and intracellular Ca2+ levels suggest mibefradil exhibits actions other than blockade of Ca2+ entry in skeletal muscle arterioles.
Keywords: Ca2+ entry, arterioles, myogenic tone, myogenic response, intracellular Ca2+, membrane potential, smooth muscle
Introduction
Arterioles typically exhibit a pressure-dependent vasoconstriction that occurs via a mechanism which is intrinsic to the vascular smooth muscle cells. This property of arteriolar smooth muscle underlies such phenomena as steady-state myogenic tone, acute myogenic reactivity, and rhythmic vasomotion. Previous studies on arterioles isolated from skeletal muscle have shown that the contractile activity is reliant on Ca2+-dependent myosin light chain phosphorylation (for review see Davis & Hill, 1999). While such vasomotor activity has long been recognized to be dependent on the influx of extracellular Ca2+ (for example, Uchida & Bohr, 1969; Duling et al., 1981; Laher et al., 1988; Meininger et al., 1991; Wesselman et al., 1996; Knot & Nelson, 1998) the exact entry mechanisms are uncertain.
The involvement of multiple Ca2+ entry pathways is suggested by several observations including; (1) L-type Ca2+ channel blockers, such as nifedipine and verapamil, do not totally inhibit myogenic tone in all tissues (Hill & Meininger, 1994); (2) persistence of myogenic reactivity in high KCl solutions (Laher & Bevan, 1989; McCarron et al., 1997); and (3) that arteriolar wall intracellular Ca2+ shows an initial increase in response to an acute pressure step in the presence of verapamil (Zou et al., 1996). Further, the increase in cytosolic Ca2+ evoked by stretch of single smooth muscle cells could only be inhibited by approximately 50% in the presence of nifedipine (Davis et al., 1992b). Mechanosensitive channels are typically cation non-selective and a component of the nifedipine-insensitive increase in Ca2+ may be due to direct entry through these channels. However, it is thought that a more important effect of mechanosensitive channel activation is Na+ entry and subsequent membrane depolarization preceding voltage-gated Ca2+ entry (Davis et al., 1992a).
Although a number of studies have demonstrated attenuation of arteriolar tone by L-type Ca2+ channel antagonists, these studies have typically not considered temporal aspects of acute myogenic vasoconstriction and associated mechanisms of Ca2+ entry (Laher & Bevan, 1989; Nelson et al., 1990; Hynes & Duling, 1991; Wesselman et al., 1996). As mentioned above mechanosensitive channels may be involved in the depolarization phase, as may alternate Ca2+ entry pathways (Davis et al., 1992a). A further consideration is that the mechanical state of an arteriole changes during a myogenic constrictor response. In response to an acute increase in intraluminal pressure, for example, an arteriole first distends before constricting to a steady-state diameter typically smaller than that prior to the increase in pressure. While our recent studies, comparing step and ramp pressure increases, have shown that the initial distension phase is not obligatory for attaining the steady state response (Hill et al., 2000) it is not clear whether the same Ca2+ supply mechanisms are utilized. Thus, differing mechanical stimuli such as increased wall tension and overt cell stretch could conceivably activate differing Ca2+ entry mechanisms.
In addition to L-type voltage gated Ca2+ channels a number of electrophysiological studies have demonstrated T-type voltage gated channels in vascular smooth muscle (Loirand et al., 1986; Bean, 1989; Ganitkevich & Isenberg, 1990; Mishra & Hermsmeyer, 1994). The recent development of mibefradil with reported selectivity for T-type voltage gated channels (over L-type channels) (Mishra & Hermsmeyer, 1994; Clozel et al., 1997), has stimulated interest in the functional consequences of these channels and has provided the opportunity to examine the possible involvement of such Ca2+ entry mechanisms in myogenic tone and reactivity. Thus in this study, the effects of mibefradil on myogenic responsiveness, pressure-induced changes in Ca2+i, in skeletal muscle arterioles were compared to those of nifedipine, a dihydropyridine which selectively blocks L-type Ca2+ channels.
Methods
Animals
The studies used male Sprague-Dawley rats weighing 273±8 (170–310) g. Before experiments, rats were housed in pairs in a dedicated animal facility with a 12 : 12 h light dark cycle. During this period rats were allowed free access to a standard rat chow and drinking water. All procedures were approved by the Animal Care and Use Committee at RMIT University.
Isolated arteriole preparation
Rats were anaesthetized with sodium thiopentone (100 mg.kg−1, Pentothal, Abbott Australasia), after which the right or left cremaster muscle was exteriorized, excised from the animal and placed in a cooled (4°C) chamber containing dissection buffer ((mM): 3-N-morpholino propanesulphonic acid (MOPS) 3; NaCl 145; KCl 5; CaCl2 2.5; MgSO4 1; NaH2PO4 1; EDTA 0.02; pyruvate 2; glucose 5 and 1% albumin) (Duling et al., 1981). Segments of the main intramuscular arteriole (1A) were dissected from the muscle as previously described (Meininger et al., 1991). Individual vessel segments were then cannulated with glass micropipettes, secured using 10-0 monofilament suture and mounted in a 5 ml volume tissue chamber. The vessel preparation was positioned on the stage of an inverted microscope and the arterioles continuously superfused (2–4 ml.min−1) with a physiologic salt solution (PSS), containing (mM): NaCl 111, NaHCO3 25.7, KCl 4.9, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, glucose 11.5 and 2-N-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) 10. Arteriole segments were gradually pressurized to 70 mmHg, using either a height-adjustable reservoir or a pressure servo control system (Living Systems Instrumentation, Burlington, Vermont, U.S.A.). Pressure was monitored constantly using a pressure transducer at the level of the inflow pipette. Vessels were warmed to 34°C during a 60 min equilibration period, checked for pressure leaks and allowed to develop spontaneous basal tone. Vessel length was adjusted prior to the development of spontaneous tone by increasing segment length such that pressure steps to 120 mmHg did not cause a lateral bowing of the vessel. This procedure has been verified, in preliminary studies, to result in optimal myogenic responsiveness.
In experiments requiring measurement of changes in Ca2+i, vessels were incubated (60 min, room temperature) with 1 μM fura 2-acetoxy methylester (fura 2-AM; Molecular Probes, Eugene, OR, U.S.A.) in buffer containing 0.5% DMSO and 0.01% pluronic. Only the abluminal surface of the vessel segment was exposed to the fura 2-AM solution to restrict dye loading to the vascular smooth muscle layer (Meininger et al., 1991; Zou et al., 1995). The dye loading procedure was followed by a 30 min wash period with PSS. Fura 2-AM loaded arterioles were exposed to epi-illumination (75W Xenon source) with light of alternating excitation wavelengths (340 and 380 nm) using a spinning filter wheel (divided into 340 and 380 nm sections) operating at 30 Hz. Fluorescence emission at 510 nm was measured using a photomultiplier tube (Hamematsu, Bridgewater, NJ, U.S.A.) in combination with a signal conditioner (Texas A&M University, TX, U.S.A.). Simultaneous transillumination with wavelengths greater than 610 nm provided a non-fluorescent image which enabled measurement of internal arteriolar diameter while fluorescence intensities were measured. The high wavelength image was directed, by a beam splitter, to a CCD camera. This procedure did not interfere with measurements of Ca2+-related fluorescence. Fluorescent image intensities were expressed as the 340 : 380 nm ratio to allow quantitative estimates of changes in arteriolar wall Ca2+i. Details of these procedures have been presented in previous publications (Meininger et al., 1991; Zou et al., 1995; Hill et al., 2000). Diameter of the arterioles was measured on the video image using electronic calipers and the data stored using a MacLab A-D system.
Protocols
Effect of mibefradil and nifedipine on spontaneous myogenic tone and responsiveness to 60 mM potassium
Arterioles equilibrated at an intraluminal pressure of 70 mmHg, were superfused (2 ml.min−1) with either of the Ca2+ channel antagonists, mibefradil and nifedipine, over the range 10−8 M to 3×10−5 M for 15 min prior to measuring vasoactive responses. The responsiveness to a depolarizing stimulus of 60 mM K+ (KCl iso-osmotically substituted for NaCl; KPSS) was recorded after rapidly changing the superfusate to KPSS. This protocol for responsiveness to 60 mM KPSS was repeated on additional arterioles maintained at an intraluminal pressure of 10 mmHg; at this pressure arterioles lack spontaneous myogenic tone. At the completion of the protocol arterioles were rendered passive by superfusion (10 min) with PSS containing 2 mM EGTA and 0 mM Ca2+. Diameter values (in μm) were normalized for each arteriole as the ratio of active: passive diameter at 70 mmHg (d/d70) or in the case of the arterioles at an intraluminal pressure of 10 mmHg the passive diameter at 10 mmHg, designated d/d10.
In a separate set of arterioles the effect of increasing concentrations of mibefradil on arteriolar myogenic tone was examined in the presence of a maximally effective concentration of nifedipine (10 μM).
Effect of mibefradil and nifedipine on Ca2+ and vasomotor responses following an acute 50–120 mmHg pressure step
In an additional group of arterioles the intraluminal pressure was reduced to 50 mmHg, for at least 5 min to achieve a steady state diameter after which the pressure was increased rapidly to 120 mmHg for a period of 10 min. The step change in intraluminal pressure was performed 3–4 times on each arteriole; i.e. without drug or after exposure (15 min) to either nifedipine (10−7 and 10−5 M) or mibefradil (10−7 and 10−5 M). To avoid interactions or additive effects mibefradil and nifedipine were studied in separate vessels.
A further 11 arterioles were loaded with 1 μM fura 2-AM and the protocol repeated with simultaneous measurement of diameter and arteriolar wall Ca2+. Experiments were performed both in the absence and presence of Fura loading to ensure that the presence of the Ca2+ indicator did not affect either the mechanical response or the actions of the inhibitors. The passive diameter and wall Ca2+ of these arterioles was measured at intraluminal pressures of 50, 70 and 120 mmHg following 10 min superfusion with 0 mM Ca2+, 2 mM EGTA PSS. The arteriole diameter and Ca2+ responses associated with these step changes in pressure were compared at three time points; (i) mean values over 1 min at 50 mmHg prior to the pressure step (basal); (ii) mean values occurring during the first minute of the pressure step (max.) and (iii) the steady state value at 120 mmHg defined as the mean of minute 8–9 of the 10 min period.
Effect of mibefradil and nifedipine on Ca2+ and vasomotor responses to 60 mM K+
Two groups of arterioles (n=5 per group) loaded with 1 μM fura 2 (AM) and pressurised to 70 mmHg were depolarized by rapidly changing the bathing solution from PSS to 60 mM KCl-PSS (KPSS) under control conditions or in the presence of either mibefradil (10−7 and 10−5 M) or nifedipine (10−7 and 10−5 M). The passive diameter and wall Ca2+i of these arterioles was measured following 10 min superfusion with 0 mM Ca2+, 2 mM EGTA PSS. The arteriole diameter and Ca2+ responses associated with the depolarizing stimulus were compared at two time points; (i) mean values over a 1 min period prior to addition of KPSS (basal) and (ii) maximum responses occurring during the first minute of exposure to the stimulus.
Determination of resting membrane potential in cannulated arterioles
As membrane potential is known to influence the opening state of the various populations of voltage gated Ca2+ channels, measurements of resting potential were made in arterioles in the absence and presence of spontaneous myogenic tone. To remove myogenic tone intraluminal pressure was set to 10 mmHg while spontaneous tone was studied at an intraluminal pressure of 70 mmHg. To determine the effects of the Ca2+ channel blockers on membrane potential, measurements were made (in separate groups of vessels) in the presence of either nifedipine (10−7 and 10−5 M) or mibefradil (10−7 and 10−5 M). Intracellular recordings of smooth muscle membrane potential were made with glass microelectrodes filled with 2 M KCl and with tip resistances of 100–200 MΩ.
Drugs and chemicals
Nifedipine (Sigma) was prepared daily as a stock solution at 10−2 M in dimethylsulphoxide and further diluted in PSS. Mibefradil (obtained as a gift from La Roche) was dissolved daily as a stock solution in water at 10−2 M and further diluted in PSS. Buffer salts and chemicals were obtained from Sigma unless otherwise stated.
Statistics
Analysis of the concentration response curves was made using the line fitting function in Graphpad Prism (version 2.0, Graphpad Software Inc, San Diego). Results are presented as the pEC50, (negative log of the EC50) and were considered different at P<0.05 as calculated by the Student t-test. The significance of changes in group data, that represent the response to step changes in pressure were determined using analysis of variance (ANOVA) and the SPSS statistical package (SPSS version 6.01). When the ANOVA indicated an overall significant changes, differences between individual groups were determined using the t-test. Results are presented as the mean±standard error of the mean (s.e.mean) and were considered different when P<0.05, n represents both the number of arteriole segments and rats studied.
Results
Effect of mibefradil and nifedipine on spontaneous myogenic tone and associated Ca2+i levels, and responsiveness to 60 mM potassium
To avoid possible interactions or additive effects, the actions of mibefradil and nifedipine were studied in separate vessel preparations. Arterioles in each group were comparable in dimension; passive diameters being 168±8 μm and 160±5 μm in the mibefradil (n=6) and nifedipine (n=6) groups respectively. Similarly, arterioles showed a comparable level of spontaneous tone, d/d70 values being 0.53±0.03 and 0.61±0.05 respectively (Figure 1). In time-control studies the level of spontaneous tone was maintained constant for the period of the experiment; similarly the response to 60 mM K+ was consistent for the 10 responses required for each experiment (Figure 1).
Figure 1.
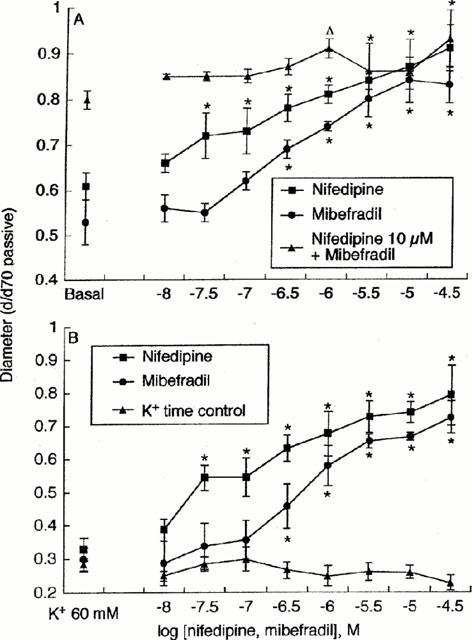
Concentration-dependent effects of mibefradil (n=6) and nifedipine (n=6) on spontaneous arteriolar tone (A) and KCl (60 mM)–induced vasoconstriction (B). Vessel diameters are plotted relative to the passive diameter at an intraluminal pressure of 70 mmHg. Basal indicates the level of spontaneous tone at 70 mmHg in the absence of Ca2+ channel antagonists (A); in (B) responses to 60 mM KCl in the absence of antagonist are shown to the left of the figure. (A) includes studies of the effect of increasing concentrations of mibefradil on myogenic tone in the presence of a maximally effective concentration of nifedipine (n=3). (B) includes a time control for KCl-induced vasoconstriction performed in the absence of Ca2+ channel antagonists (n=3). Results are presented as mean±s.e.mean.
The effect of increasing concentrations of mibefradil and nifedipine on spontaneous arteriolar tone at an intraluminal pressure of 70 mmHg is shown in Figure 1A. Both Ca2+ channel antagonists reduced the level of myogenic tone in a concentration-dependent manner and with similar potencies; the pEC50 for nifedipine being 7.04±0.17 and for mibefradil 6.65±0.20 (P>0.05, n.s.). Addition of increasing concentrations of mibefradil in the presence of a maximally effective concentration of nifedipine (10 μM) did not cause a further loss in tone as compared to the effect of the L-type blocker alone (Figure 1A).
The effects of the inhibitors on steady-state Ca2+i levels, at intraluminal pressures of 50, 70 and 120 mmHg, are detailed in Figure 2; results are shown normalized with respect to Ca2+i levels under passive conditions. Normalization was performed to account for any variation in baseline ratio values as it was necessary to use separate vessel groups for the mibefradil and nifedipine studies. Consistent with our previous studies (Zou et al, 1995), in the absence of Ca2+ channel antagonists Ca2+i increased with increasing intraluminal pressure. Nifedipine caused a dose-dependent reduction in Ca2+i levels at each pressure studied (Figure 2B). In contrast, mibefradil, at either concentration, failed to significantly reduce Ca2+i at any of the pressures studied (Figure 2A).
Figure 2.
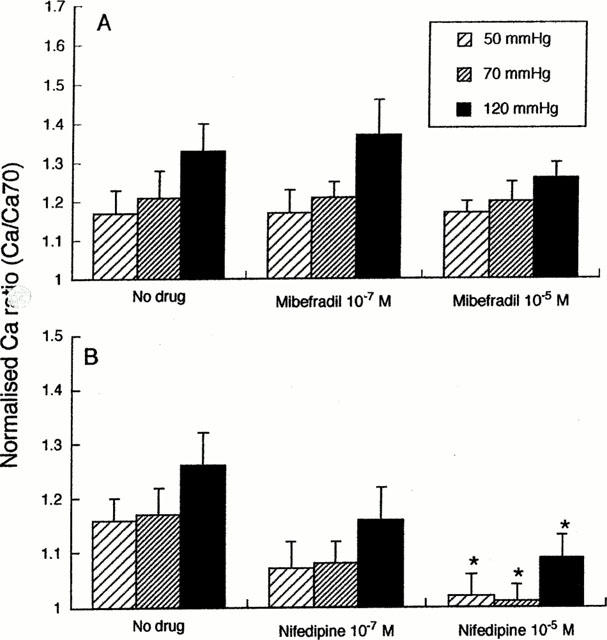
Concentration-dependent effects of mibefradil (A; n=6) and nifedipine (B; n=5) on Ca2+i at steady-state intraluminal pressures of 50, 70 and 120 mmHg. Fluorescence ratios (340 : 380 nm) are normalized to the ratio obtained under passive conditions at 70 mmHg. Results are presented as mean±s.e.mean; *P<0.05.
The constrictor response to 60 mM K+ (intraluminal pressure 70 mmHg) was inhibited by increasing concentrations of nifedipine and mibefradil (Figure 1B), with each antagonist again showing a similar potency for inhibition of the constrictor response; pEC50 for nifedipine being 6.93±0.38 and for mibefradil 6.45±0.27 (n=6 for both; P>0.05, n.s.). While arterioles at an intraluminal pressure of 10 mmHg did not exhibit active myogenic tone, vasoconstriction to KCl (60 mM) was inhibited by both mibefradil and nifedipine in similar manner to that at 70 mmHg. At 10 mmHg calculated pEC50 for nifedipine was 6.50±0.59 and for mibefradil 6.67±0.17 (n=4; P>0.05, n.s.).
Effect of mibefradil and nifedipine on Ca2+ and vasomotor responses following an acute 50–120 mmHg pressure step
In addition to examining the effects of the Ca2+ channel antagonists on arterioles in a steady state, studies were conducted to examine the effects on the temporal response to an acute 50–120 mmHg step increase in intraluminal pressure. Group data representing the mean values at 50 mmHg (basal); peak responses during the rapid pressure increase to 120 mmHg (corresponding to the maximal stretch or tension stimulus), and after reaching steady state conditions at 120 mmHg are shown in Figure 3. In the absence of antagonists the acute pressure step caused an initial distension from basal levels of 0.63±0.03 and 0.65±0.04 (expressed as a percentage of passive diameter at 70 mmHg) to 0.77±0.03 and 0.82±0.05 for the mibefradil and nifedipine groups, respectively. These values are equivalent to a distension of 124±4% and 126±3% relative to basal for the mibefradil and nifedipine groups respectively. Following the distension phase arterioles constricted to 0.59±0.03 (95±4% of basal; mibefradil Figure 3A) and 0.61±0.07 (93±8% of basal; nifedipine Figure 3B). As stated above both mibefradil and nifedipine caused dose-dependent vasodilatation at 50 mmHg and, in addition, attenuated active myogenic vasoconstriction such that in the presence of either 10−5 M mibefradil or nifedipine vessels failed to constrict significantly following the pressure-induced distension (Figure 3A,B).
Figure 3.
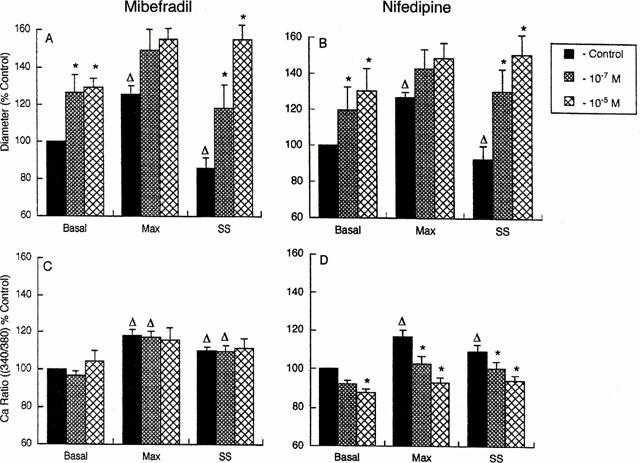
Group data from experiments summarizing the effects of mibefradil (n=6) and nifedipine (n=5) on basal, minimum and steady-state diameter (A and B), and basal, maximum and steady-state Ca2+i (C and D) responses occurring during a pressure step from 50 to 120 mmHg. See text for methods of determining maximum and steady-state values. Diameters are expressed as per cent of the basal diameter (50 mmHg) in the absence of Ca2+ channel antagonists. Changes in intracellular Ca2+ are presented as a percent of the pre-stimulation ratio value independent of the effect of the Ca2+ channel antagonists (C and D). Results are presented as mean±s.e.mean; *P<0.05 compared to corresponding value in the absence of antagonist; ΔP<0.05 compared to basal value at a given antagonist concentration.
Results for changes in Ca2+i are shown with data normalized as a percentage of the pre-step levels (basal) in the absence of antagonists (Figure 3C,D). In the absence of antagonists the acute 50–120 mmHg pressure step, and subsequent vessel distension, resulted in a 128.1±9.0% and 116.8±3.5% maximal increase in Ca2+i for the mibefradil and nifedipine groups of vessels respectively (Figure 3C). This was followed by a decline to a steady-state level of 114.0±4.2% (mibefradil) and 109.2±3.5% (nifedipine) (Figure 3C,D). Prior to the pressure step (i.e. at 50 mmHg) nifedipine caused a dose-dependent decrease in Ca2+i while mibefradil had no measurable effect on Ca2+i (see above and Figure 3C,D). Nifedipine dose-dependently inhibited the Ca2+i response to both phases of the pressure step while mibefradil had little effect on pressure-induced increases in Ca2+i (Figure 3C,D).
Effect of mibefradil and nifedipine on Ca2+i and vasomotor responses to 60 mM potassium
Arterioles in these studies exhibited comparable passive diameters ((d70) 147±6 μm and 159±6 μm) and levels of myogenic tone (0.51±0.02 and 0.59±0.03 (d/d70)) for the mibefradil and nifedipine groups respectively. As in the previous sections both mibefradil (10−7 and 10−5 M) and nifedipine (10−7 and 10−5 M caused marked vasodilation (Figure 4A,B). Rapid exchange of the superfusate from PSS to KPSS in the absence of antagonists caused an initial increase in Ca2+i, to 138±7 and 158±13% of control associated with rapid constriction to 40±6 and 45±4% of control in the mibefradil and nifedipine groups respectively (Figure 4). Mibefradil at 10−7 and 10−5 M significantly decreased basal Ca2+i to 92±1 and 91±3% of control (Figure 4C); however, a concentration-dependent effect was not apparent. Mibefradil (10−7 M) did not inhibit the KPSS-induced increase in Ca2+i (138±12%; not significantly different to control) while 10−5 M mibefradil significantly reduced the Ca2+i increase to 108±4% control (Figure 4C). Exposure of arterioles to nifedipine (10−7 and 10−5 M) caused both a concentration-dependent reduction of basal Ca2+i (91±2 and 87±2% of control respectively) and a significant reduction of the maximum Ca2+i values (123±5 and 108±4% of control) following superfusion with KPSS (Figure 4D).
Figure 4.
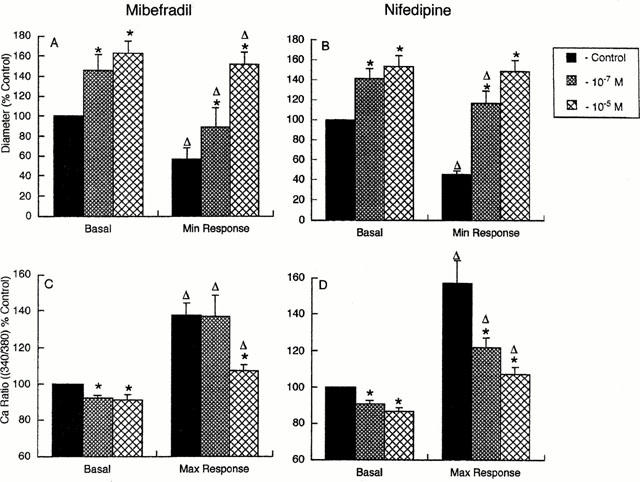
Group data from experiments summarizing the effects of mibefradil (n=5) and nifedipine (n=5) on diameter (A and B) and Ca2+i (C and D) responses to 60 mM KPSS. Diameters are expressed as per cent of the basal diameter (70 mmHg) in the absence of Ca2+ channel antagonists. Changes in intracellular Ca2+ are presented as a per cent of the basal ratio value. Results are presented as mean±s.e.mean; *P<0.05 compared to corresponding value in the absence of antagonist; ΔP<0.05 compared to basal value at a given antagonist concentration.
The maximum constriction in response to KPSS superfusion was inhibited by both mibefradil (10−7 and 10−5 M) and nifedipine (10−7 and 10−5 M) (Figure 4A,B). At 10−7 M mibefradil the KCl-induced contractile response was significantly (P<0.05) attenuated despite maximum Ca2+i levels being comparable to those in the absence of the antagonist (Figure 4A,C). In contrast the lower dose of nifedipine significantly inhibited both the KCl-induced increase in Ca2+i and the resultant vasoconstriction.
Determination of resting membrane potential in cannulated arterioles
Measurements of resting membrane potential were performed in arterioles prepared under identical conditions to the above experiments. In the absence of myogenic tone (intraluminal pressure 10 mmHg) vessels were 86.8±11.5 or 87.1±8.2% of passive diameter in the mibefradil or nifedipine groups respectively and resting membrane potentials were −59.8±1.6 and −56.5±2.1 mV (Figure 5). Arterioles pressurized to 70 mmHg constricted to 48.6±4.2 and 50.1±4.2% of passive diameter for the respective mibefradil and nifedipine groups and exhibited significantly depolarized membrane potentials of −37.6±1.6 and −38.7±1.3 mV (n=4, P<0.01) (Figure 5). As demonstrated previously mibefradil (10−5 M) dilated vessels maintained at 70 mmHg despite this concentration of mibefradil further depolarizing vessels to −28.8±3.2 mV (Figure 5). Mibefradil (10−5 M) also caused significant depolarization of arterioles maintained at 10 mmHg, without any effect on vessel diameter (Figure 5). Nifedipine (10−7 and 10−5 M) significantly dilated vessels maintained at 70 mmHg only and did not alter membrane potential at either pressure (Figure 5).
Figure 5.
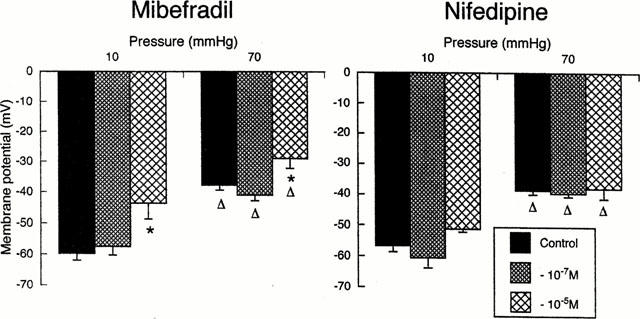
Comparison of the effects of mibefradil and nifedipine on arteriolar membrane potential (n=4 per group). At 10 mmHg vessels lack myogenic tone due to insufficient distending pressure while 70 mmHg approximates the in vivo pressure within the cremaster muscle 1A (Hill et al., 1992). Effects in the presence of mibefradil are shown on the left and in the presence of nifedipine in panels on the right. Results are presented as mean±s.e.mean; *P<0.05 compared to corresponding value in the absence of antagonist; ΔP<0.05 compared to value at 10 mmHg at a given antagonist concentration.
Discussion
The results of the present studies demonstrate that both mibefradil, a putative selective T-channel inhibitor, and the L-type Ca2+ channel blocker, nifedipine, dose-dependently attenuate spontaneous arteriolar myogenic tone. Consistent with the findings of Lam et al. (1998) mibefradil also effectively inhibited arteriolar contractile responses to 60 mM KCl independently of co-existing myogenic tone. Relative selectivity of either inhibitor for myogenic or KCl-induced tone was not apparent in our cannulated isolated skeletal muscle arteriole preparation. Differences do appear, however, to exist between the mode of action of the two antagonists as a reduction in Ca2+i did not appear to play as significant a role in mibefradil-induced inhibition of constrictor responses as was apparent for nifedipine. Furthermore, mibefradil (10−5 M) caused significant smooth muscle membrane depolarization which was not observed following exposure to nifedipine.
Analysis of concentration-response relationships showed no significant differences between the potencies of mibefradil and nifedipine in attenuating either steady-state spontaneous myogenic tone or KCl-induced contractions. The calculated pEC50 for mibefradil was approximately 6.5, some two orders of magnitude less than that reported for selective inhibition of T-channels in isolated vascular smooth muscle cells but in the range for mibefradil acting on a mixed population of T- and L-type channels (Mishra & Hermsmeyer, 1994). The effectiveness of mibefradil in attenuating myogenic tone was similar to that reported for inhibition of agonist and K+-induced responses in rat mesenteric (Sarsero et al., 1998) and cerebral (Lam et al., 1998) arteries. Given that mibefradil was no more effective than the selective L-type channel antagonist, nifedipine, it seems that T-channel blockade is, at best, a minimal component of the dilator action of mibefradil on myogenic tone or K+-induced constriction in rat skeletal muscle arterioles.
The effects of mibefradil and nifedipine on KCl-induced vasoconstriction were examined at two distinct intraluminal pressures, 10 and 70 mmHg. These studies were performed to examine the effectiveness of these agents on the response to K+ in the absence (10 mmHg) and presence (70 mmHg) of myogenic tone. Experiments were not confined to 10 mmHg as at this pressure vessels may not only lack myogenic tone (and therefore interaction with other constrictor mechanisms) but may also be compromised in terms of optimal mechanical responsiveness (for example, being on the ascending limb of the length–tension relationship). The results indicated that regardless of the level of intraluminal pressure both agents dose-dependently inhibited the depolarization-induced constrictor response with similar potency.
In the present studies, blockade of L-type Ca2+ channels with high concentrations of nifedipine abolished approximately 85% of spontaneous arteriolar tone as compared to the totally passive state obtained after superfusion with buffer containing 0 mM Ca2+ and 2 mM EGTA. Addition of increasing concentrations of mibefradil in the presence of a maximally effective concentration of nifedipine did not cause a further loss in tone as compared to the effect of the L-type blocker alone. In apparent contrast Mishra & Hermsmeyer (1994) demonstrated that in cells isolated from the azygos vein, T channel activity persisted in the presence of concentrations of nisoldipine that totally blocked L-channel current. If such electrophysiological studies can be extrapolated to the current functional studies it suggests that the effects of mibefradil may be explained by a nifedipine-like effect (i.e. an action on L-type Ca2+ entry) or that the high concentration of nifedipine blocked any available T-channel entry. Further, as suggested above the arteriolar tone remaining in the presence of nifedipine does not appear to be a function of T-type Ca2+ entry.
A number of studies of cannulated arterioles (Harder, 1984; Knott & Nelson, 1998), including the present, have shown that steady-state vascular smooth muscle membrane potential shows an inverse relationship with intraluminal pressure. From these data it would appear that at physiological pressures, approximately 70 mmHg for the cremaster muscle 1A in vivo (Hill et al., 1992), membrane potential would be in the range of approximately −35 to −45 mV. Based on the single cell electrophysiological studies of Mishra & Hermsmeyer (1994) it would be expected that if T-channels were present in 1A arteriolar smooth muscle then these channels would be voltage-inactivated at these membrane potentials. Thus, these investigators showed that transient Ca2+ currents were not evident when voltage stepped from a holding potential of −30 mV as compared to a holding potential of −80 mV. Alternatively, it is conceivable that a change in membrane stretch or tension could alter the voltage gating properties of such a channel. Modulation of the voltage sensitivity of T-channels by calcium/calmodulin dependent protein kinase II (CaMKII) has been demonstrated in adrenal glomerulosa cells (Chen et al., 1999). No data are available, however, to directly support such modulation in response to the mechanical forces which occur during an increase in arteriolar intraluminal pressure although McCarron has suggested such modulation of the voltage-dependency of L-type channels (McCarron et al., 1997).
An additional observation in the present study was the depolarizing effect of mibefradil at the higher concentration examined (10−5 M). This would be expected to further decrease the probability of T-channel activation and would conceivably enhance Ca2+ entry into smooth muscle by opening more voltage-dependent channels, possibly opposing any vasodilatory effect of Ca2+-channel blockade. Interestingly higher concentrations of mibefradil (3×10−5 M) tended to cause vasoconstriction in our arteriole preparation (data not shown). Consistent with an effect on membrane potential, mibefradil (3×10−5 M) has been reported to depolarize macrovascular endothelial cells, although 10−5 M mibefradil was found to cause transient membrane hyperpolarization (Nilius et al., 1997). These authors suggested that mibefradil inhibited populations of Cl− channels including those activated by Ca2+ and volume changes. Although a role for vascular smooth muscle Cl− channels in arteriolar myogenic responses is controversial inhibition of these channels could conceivably impair the mechanically-induced vasoconstriction (Nelson et al, 1997; Davis & Hill, 1999). However, such a mechanism would be expected to be accompanied by hyperpolarization rather than the depolarization affected by the higher concentration of mibefradil.
An additional aim of the current studies was to examine the effect of mibefradil on arteriolar smooth muscle intracellular Ca2+ levels; both at steady-state and during an acute myogenic stimulus (50–120 mmHg pressure step). The rationale for these experiments was based on previous observations showing that an acute pressure step results in a biphasic Ca2+ response with an initial peak corresponding to the pressure-induced distension followed by a decline to a steady-state level as stable constriction is achieved (Hill et al., 2000). Thus it was considered conceivable that distinct Ca2+ entry mechanisms contribute to different phases of the contractile response. Mibefradil and nifedipine showed similar inhibition of the contractile response to a 50–120 mmHg pressure step, with a 10−7 M dose of the inhibitors attenuating the mechanical response and 10−5 M rendering the vessels passive. In contrast, while nifedipine caused a dose-dependent reduction of Ca2+i levels at each stage of the pressure-step mibefradil did not affect Ca2+i levels at 10−7 M while 10−5 M caused only a modest reduction in Ca2+i. On the basis of these observations it would appear that there are differences in the mechanism(s) by which these two agents inhibit the myogenic response.
A possible explanation for mibefradil inhibiting myogenic contraction by mechanisms in addition to decreasing Ca2+ entry lies in reports of it having effects on other signal transduction pathways which are utilised in myogenic vasoconstriction. For example, Hermsmeyer & Miyagawa (1996) have suggested that mibefradil inhibits protein kinase C activation associated with endothelin stimulation of vascular smooth muscle. Protein kinase C has been implicated in myogenic constriction of arterioles (Hill et al., 1990; Osol et al., 1991; Miller et al., 1997) and as such its inhibition by mibefradil could remove a Ca2+-independent component of myogenic tone. In addition, mibefradil has been suggested to act as an opener of KATP channels (Mocanu et al., 1999) the action of which would be expected to cause both hyperpolarization and vasodilation. Given the involvement of L-type Ca2+ channels in myogenic tone this would be expected, however, to add to the decrease in intracellular Ca2+ levels. Mibefradil has been shown to inhibit certain cytochrome P450 isozymes (Prueksaritanont et al., 1999), however not those implicated in the pressure-dependent generation of vasoactive metabolites of arachidonic acid (Campbell & Harder, 1999). Alternatively, given the fact that mibefradil (10−7 M) attenuated KCl-induced contraction with less effect on Ca2+ entry (relative to nifedipine) it could be argued that mibefradil may exert inhibitory effects at the level of the contractile proteins.
In summary, both mibefradil and nifedipine inhibit arteriolar spontaneous tone and acute myogenic reactivity. While there appears to be overlap in the mechanisms by which these agents inhibit tone, differences in effects on intracellular Ca2+ levels suggest mibefradil exhibits actions other than blockade of Ca2+ entry. No evidence was found to support the notion that mechanisms of Ca2+ entry, underlying spontaneous arteriolar myogenic tone or acute myogenic reactivity, involve a component specifically inhibited by mibefradil.
Acknowledgments
Studies described in this manuscript were supported by grants from the National Health and Medical Research Council of Australia. and the Ramacciotti Foundation. Thanks are extended to Dr Eric Ertel of F. Hoffman–La Roche (Basel, Switzerland) for the kind gift of mibefradil and to Dr Sharmini Rajanayagam for constructive criticism prior to submission of the manuscript.
Abbreviations
- Ca2+i
intracellular Ca2+
- HEPES
10 mM 2-N-hydroxyethylpiperazine-N′-2-ethanesulphonic acid
- KPSS
potassium-substituted physiological salt solution
- MOPS
3-N-morpholino propanesulphonic acid
- PSS
physiological salt solution
References
- BEAN B.P. Classes of calcium channels in vertebrate cells. Ann. Rev. Physiol. 1989;51:367–384. doi: 10.1146/annurev.ph.51.030189.002055. [DOI] [PubMed] [Google Scholar]
- CAMPBELL W.B., HARDER D.R. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circ. Res. 1999;84:484–488. doi: 10.1161/01.res.84.4.484. [DOI] [PubMed] [Google Scholar]
- CHEN X-L., BAYLISS D.A., FERN R.J., BARRETT P.Q. A role for T-type Ca2+ channels in the synergistic control of aldosterone production by ANG II and K+ Am. J. Physiol. 1999;276:F674–F683. doi: 10.1152/ajprenal.1999.276.5.F674. [DOI] [PubMed] [Google Scholar]
- CLOZEL J.P., ERTEL E.A., ERTEL S.I. Discovery and main pharmacological properties of mibefradil (Ro 40-5967), the first selective T-type calcium channel blocker. J. Hypertens. 1997;1 Suppl. 5:S17–S25. doi: 10.1097/00004872-199715055-00004. [DOI] [PubMed] [Google Scholar]
- DAVIS M.J., HILL M.A. Signaling mechanisms underlying the vascular myogenic response. Physiological Revs. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- DAVIS M.J., DONOVITZ J.A., HOOD J.D. Stretch-activated single-channel and whole-cell currents in vascular smooth muscle cells. Am. J. Physiol. 1992a;262:C1083–C1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- DAVIS M.J., MEININGER G.A., ZAWIEJA D.C. Stretch-induced increases in intracellular calcium in isolated vascular smooth muscle cells. Am. J. Physiol. 1992b;263:H1292–H1299. doi: 10.1152/ajpheart.1992.263.4.H1292. [DOI] [PubMed] [Google Scholar]
- DULING B.R., GORE R.W., DACEY R.G., JR, DAMON D.N. Methods for isolation, cannulation, and in vitro study of single microvessels. Am. J. Physiol. 1981;241:H108–H116. doi: 10.1152/ajpheart.1981.241.1.H108. [DOI] [PubMed] [Google Scholar]
- GANITKEVICH V.Y., ISENBERG G. Contribution of two types of calcium channels to membrane conductance of single myocytes from guinea-pig coronary artery. J. Physiol. 1990;426:19–42. doi: 10.1113/jphysiol.1990.sp018125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDER D.R. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ. Res. 1984;55:197–202. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- HERMSMEYER K., MIYAGAWA K. Protein kinase C mechanism enhances vascular muscle relaxation by the Ca2+ antagonist, Ro 40-5967. J. Vasc. Res. 1996;33:71–77. doi: 10.1159/000159134. [DOI] [PubMed] [Google Scholar]
- HILL M.A., MEININGER G.A. Calcium entry and myogenic phenomena in skeletal muscle arterioles. Am. J. Physiol. 1994;267:H1085–H1092. doi: 10.1152/ajpheart.1994.267.3.H1085. [DOI] [PubMed] [Google Scholar]
- HILL M.A., FALCONE J.C., MEININGER G.A. Evidence for protein kinase C involvement in arteriolar myogenic reactivity. Am. J. Physiol. 1990;259:H1586–H1594. doi: 10.1152/ajpheart.1990.259.5.H1586. [DOI] [PubMed] [Google Scholar]
- HILL M.A., TRIPPE K.M., LI Q-X., MEININGER G.A. Arteriolar arcades and pressure distribution in cremaster muscle microcirculation. Microvasc. Res. 1992;44:117–124. doi: 10.1016/0026-2862(92)90106-y. [DOI] [PubMed] [Google Scholar]
- HILL M.A., ZOU H., DAVIS M.J., POTOCNIK S.J., PRICE S. Transient increases in diameter and Ca2+i following acute pressure increases are not obligatory for myogenic constriction. Am. J. Physiol. 2000;278:H345–H352. doi: 10.1152/ajpheart.2000.278.2.H345. [DOI] [PubMed] [Google Scholar]
- HYNES M.R., DULING B. R. Ca2+ sensitivity of isolated arterioles from the hamster cheek pouch. Am. J. Physiol. 1991;260:H355–H361. doi: 10.1152/ajpheart.1991.260.2.H355. [DOI] [PubMed] [Google Scholar]
- KNOT H.J., NELSON M.T. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J. Physiol. 1998;508:199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAHER I., BEVAN J.A. Stretch of vascular smooth muscle activates tone and 45Ca2+ influx. J. Hypertens. 1989;7 Suppl. 4:S17–S20. [PubMed] [Google Scholar]
- LAHER I., VAN BREEMEN C., BEVAN J.A. Stretch-dependent calcium uptake associated with myogenic tone in rabbit facial vein. Circ. Res. 1988;63:669–672. doi: 10.1161/01.res.63.3.669. [DOI] [PubMed] [Google Scholar]
- LAM E., SKARSGARD P., LAHER I. Inhibition of myogenic tone by mibefradil in rat cerebral arteries. Eur. J. Pharmacol. 1998;358:165–168. doi: 10.1016/s0014-2999(98)00611-6. [DOI] [PubMed] [Google Scholar]
- LOIRAND G., PACAUD P., MIRONNEAU C., MIRONNEAU J. Evidence for two distinct calcium channels in rat vascular smooth muscle cells in short-term primary culture. Pflügers Arch. 1986;407:566–568. doi: 10.1007/BF00657519. [DOI] [PubMed] [Google Scholar]
- MCCARRON J.G., CRICHTON C.A., LANGTON P.D., MACKENZIE A., SMITH G.L. Myogenic contraction by modulation of voltage-dependent calcium currents in isolated rat cerebral arteries. J. Physiol. 1997;498:371–379. doi: 10.1113/jphysiol.1997.sp021864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEININGER G.A., ZAWIEJA D.C., FALCONE J.C., HILL M.A., DAVEY J.P. Calcium measurement in isolated arterioles during myogenic and agonist stimulation. Am. J. Physiol. 1991;261:H950–H959. doi: 10.1152/ajpheart.1991.261.3.H950. [DOI] [PubMed] [Google Scholar]
- MILLER F.J., DELLSPERGER K.C., GUTTERMANN D.D. Myogenic constriction of human coronary arterioles. Am. J. Physiol. 1997;273:H257–H264. doi: 10.1152/ajpheart.1997.273.1.H257. [DOI] [PubMed] [Google Scholar]
- MISHRA S., HERMSMEYER K. Selective inhibition of T-type Ca2+ channels by RO 40-5967. Circ. Res. 1994;75:144–148. doi: 10.1161/01.res.75.1.144. [DOI] [PubMed] [Google Scholar]
- MOCANU M.M., GADGIL S., YELLON D.M., BAXTER G.F. Mibefradil, a T-type and L-type calcium channel blocker, limits infarct size through a glibenclamide-sensitive mechanism. Cardiovasc. Drugs Ther. 1999;13:115–122. doi: 10.1023/a:1007732025184. [DOI] [PubMed] [Google Scholar]
- NELSON M.T., CONWAY M.A., KNOT H.J., BRAYDEN J.E. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J. Physiol. (Lond). 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON M.T., PATLAK J.B., WORLEY J.F., STANDEN N.B. Calcium channels, potassium channels and voltage dependence of arterial smooth muscle tone. Am. J. Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- NILIUS B., PRENEN J., KAMOUCHI M., VIANA F., VOETS T., DROOGMANS G. Inhibition by mibefradil, a novel calcium channel antagonist, of Ca2+–and volume-activated Cl− channels in macrovascular endothelial cells. Br. J. Pharmacol. 1997;121:547–555. doi: 10.1038/sj.bjp.0701140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSOL G., LAHER I., CIPOLLA M. Protein kinase C modulates basal myogenic tone in resistance arteries from the cerebral circulation. Circ. Res. 1991;68:359–367. doi: 10.1161/01.res.68.2.359. [DOI] [PubMed] [Google Scholar]
- PRUEKSARITANONT T., MA B., TANG C., MENG Y., ASSANG C., LU P., REIDER P.J., LIN J.H., BAILLE T.A. Metabolic interactions between mibefradil and HMG-CoA reductase inhibitors: an in vitro investigation with human liver preparations. Br. J. Clin. Pharmacol. 1999;47:291–298. doi: 10.1046/j.1365-2125.1999.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARSERO D., FUJIWARA T., MOLENAAR P., ANGUS J.A. Human vascular to cardiac tissue selectivity of L- and T-type calcium channel antagonists. Brit. J. Pharmacol. 1998;125:109–119. doi: 10.1038/sj.bjp.0702045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCHIDA E., BOHR D.F. Myogenic tone in isolated perfused resistance vessels from rats. Am. J. Physiol. 1969;216:1343–1350. doi: 10.1152/ajplegacy.1969.216.6.1343. [DOI] [PubMed] [Google Scholar]
- WESSELMAN J.P., SCHUBERT M.R., VANBAVEL E., PFAFFENDORF M., SPAAN J.A.E. Voltage-operated calcium channels are essential for the myogenic responsiveness of cannulated rat mesenteric small arteries. J. Vasc. Res. 1996;33:32–41. doi: 10.1159/000159129. [DOI] [PubMed] [Google Scholar]
- ZOU H. , RATZ P.H., HILL M.A. Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. Am. J. Physiol. 1995;269:H1590–H1596. doi: 10.1152/ajpheart.1995.269.5.H1590. [DOI] [PubMed] [Google Scholar]
- ZOU H. , RATZ P.H., HILL M.A. Regulation of [Ca2+]i during myogenic and agonist-induced contraction. FASEB J. 1996;10:A57. [Google Scholar]