

Abstract
Ventricular arrhythmias are rare but life-threatening side effects of therapy with the second-generation H1 receptor antagonists terfenadine and astemizole. Blockade of the K+ channels encoded by the Human Ether-à-go-go-Related Gene 1 (HERG1) K+ channels, which is the molecular basis of the cardiac repolarizing current IKr, by prolonging cardiac repolarization, has been recognized as the mechanism underlying the cardiac toxicity of these compounds.
In the present study, the potential blocking ability of the novel second-generation H1 receptor antagonist mizolastine of the HERG1 K+ channels heterologously expressed in Xenopus oocytes and in HEK 293 cells or constitutively present in SH-SY5Y human neuroblastoma cells has been examined and compared to that of astemizole.
Mizolastine blocked HERG1 K+ channels expressed in Xenopus oocytes with an estimated IC50 of 3.4 μM. Mizolastine blockade was characterized by a fast dissociation rate when compared to that of astemizole; when fitted to a monoexponential function, the time constants for drug dissociation from the K+ channel were 72.4±11.9 s for 3 μM mizolastine, and 1361±306 s for 1 μM astemizole.
In human embryonic kidney 293 cells (HEK 293 cells) stably transfected with HERG1 cDNA, extracellular application of mizolastine exerted a dose-related inhibitory action on IHERG1, with an IC50 of 350±76 nM. Furthermore, mizolastine dose-dependently inhibited HERG1 K+ channels constitutively expressed in SH-SY5Y human neuroblastoma clonal cells.
The results of the present study suggest that the novel second-generation H1 receptor antagonist mizolastine, in concentrations higher than those achieved in vivo during standard therapy, is able to block in some degree both constitutively and heterologously expressed HERG1 K+ channels, and confirm the heterogeneity of molecules belonging to this therapeutical class with respect to their HERG1-inhibitory action.
Keywords: Potassium channels, mizolastine, antihistamines, drug cardiotoxicity, arrhythmias
Introduction
In the last two decades, second-generation H1 receptor blockers have been developed for the symptomatic treatment of allergic reactions in order to overcome the marked antimuscarinic and sedative properties displayed by first-generation congeners (Sorkin & Heel, 1985). Due to their improved H1 selectivity, absence of sedation, and possibly, antiallergic properties not deriving from their antihistaminic activity, second-generation H1 receptor blockers are among the most prescribed drug families (Slater et al., 1999).
Mizolastine (2-[[[1-[1-[(4-fluorophenyl)methyl)-1H-benzimidazol-2-yl]piperidinyl]methylamino]-4(3H)-pyrimidinone; SL 85.0324) is a new potent and selective second-generation H1 receptor blocker available in Europe, which is effective in relieving the symptoms of allergic rhinitis (Leynadier et al., 1996; Tasman & Weber, 1995) and urticaria (Brostoff et al., 1996). This compound, structurally related to astemizole, is devoid of sedative (Patat et al., 1995; Vuurman et al., 1994) and antimuscarinic (Danjou et al., 1992) properties, and displays a more hydrophilic profile which confers a lower distribution volume, good bioavailability, and limited metabolic conversion by the hepatic CYPs 3A4 and 2A6 (Delauche-Cavallier et al., 1999). These pharmacological properties have represented a distinct advantage over some other antihistamines.
Despite the obvious improvement in the treatment of allergic diseases represented by second-generation antihistamines, concerns over the risk/benefit profile of some members of this pharmacological class have recently been raised (Woosley, 1996). In fact, the rare occurrence of ‘torsade de pointes', a form of polymorphic ventricular arrhythmia, has been described in patients taking intentional overdoses of the second-generation antihistamines astemizole or terfenadine (Craft, 1986; Davies et al., 1989), or suffering from predisposing factors to the development of cardiac arrhythmias and undergoing therapy with these antihistamines (Monahnan et al., 1990). These potentially fatal adverse cardiovascular reactions have increased the level of awareness in the medical community, leading to guidelines established by the European Agency for the Evaluation of Medicinal Products (EMEA) for pre-clinical and clinical studies aimed at reducing the risk for cardiovascular adverse effects by drugs to be introduced in the market (EMEA, 1997).
The QT prolongation and ventricular arrhythmia caused by terfenadine and astemizole have been related to the ability of these drugs to interfere with cardiac potassium channels involved in action potential repolarization (Berul & Morad, 1995), and particularly with the rapid component of the cardiac repolarizing potassium current (IKr) (Woosley et al., 1993; Sanguinetti et al., 1995; Salata et al., 1995). The Human Ether-à-go-go-Related Gene 1 (HERG1) (Warmke & Ganetzky, 1994) seems to represent the molecular basis of IKr, since the K+ currents encoded by HERG1 display biophysical and pharmacological characteristics similar to those of cardiac IKr (Trudeau et al., 1995; Sanguinetti et al., 1995).
The aim of the present study has been to investigate the potential interaction of increasing concentrations of mizolastine with HERG1 K+ channels heterologously expressed in Xenopus oocytes and in human embryonic kidney 293 cells (HEK 293 cells) (Zhou et al., 1998), or endogenously present in SH-SY5Y human neuroblastoma cells (Arcangeli et al., 1995); and to compare the actions of the novel H1 receptor blocker with those of astemizole, one of the second-generation antihistamines with well-known inhibitory actions on the K+ channels encoded by HERG1 (Suessbrich et al., 1996; Taglialatela et al., 1998b).
Methods
Xenopus oocytes isolation
Ovarian lobes were surgically removed from adult female Xenopus frogs (Rettili di Schneider, Varese, Italy) and placed in 100-mm Petri dishes containing a Ca2+-free solution of the following composition (in mM): NaCl 82.5, KCl 2, MgCl2 1, HEPES 5, piruvic acid 2.5, 100 u ml−1 penicillin, and 100 μg ml−1 streptomycin, pH 7.5 with NaOH. After four extensive washes, the oocytes (stage V–VI) were dissociated at room temperature by collagenase treatment (type IA, 45–80 min at a concentration of 2 mg ml−1). At the end of the collagenase treatment, the oocytes were placed in a Ca2+-containing solution of the following composition (in mM): NaCl 100, KCl 2, CaCl2 1.8, MgCl2 1, HEPES 5, piruvic acid 2.5, 100 u ml−1 penicillin, and 100 μg ml−1 streptomycin, pH 7.5 with NaOH. Dissociated oocytes were then placed in a 19°C incubator and used for the experiments on the following day.
Molecular biology and oocyte injection
The cloning of HERG1 (Warmke & Ganetzky, 1994), BEAG (Frings et al., 1998), rKv2.1 (Frech et al., 1989), and rKv3.1 (Taglialatela et al., 1991) has already been described. These cDNAs were cloned into the following vectors: pSp64A+ for HERG1, pCRII for BEAG, and pBluescript for rKv2.1 and rKv3.1. cDNAs were linearized with HindIII for BEAG, EcoRI for HERG1, and NotI for rKv2.1 and rKv3.1. cRNAs were transcribed in vitro from linearized cDNAs by means of a commercially available kit (mCAP, Stratagene), using T7 RNA polymerase for BEAG, rKv2.1, and rKv3.1, and SP6 RNA polymerase for HERG1. RNAs were quantified by the RiboGreen fluorescent kit (Molecular Probes) and stored in a stock solution (250 ng μl−1) at −20°C in 0.1 M KCl. One day after isolation, Xenopus oocytes were microinjected with 46 nl of the respective cRNA stock solution or appropriate dilution.
Cell culture
Human neuroblastoma SH-SY5Y cells were cultured in Dulbecco's Modified Eagle Medium (DMEM), containing glucose (4.5 g l−1) and 5% foetal calf serum (FCS), and incubated at 37°C in a humidified atmosphere with 10% CO2 in 100-mm plastic Petri dishes. HEK 293 cells were cultured in modified eagle medium (MEM), supplemented with Earle's salts, nonessential amino acids (0.1 mM), penicillin (50 u ml−1), steptomycin (50 μg ml−1), G418 (0.4 mg ml−1) and 10% foetal calf serum (FCS), and incubated at 37°C in a humidified atmosphere with 5% CO2 in 100-mm plastic Petri dishes. For electrophysiological experiments, the cells were seeded on glass coverslips (Fisher) coated with poly-L-lysine (30 μg ml−1). All the experiments were performed 1–4 days after seeding at room temperature (22–23°C).
Electrophysiology
Voltage-clamp with two microelectrodes
The oocytes were voltage-clamped with a commercially available amplifier (Warner OC-725A, Warner Instr. Corp.). Current and voltage electrodes were filled with 3 M KCl, 10 mM HEPES (pH 7.4; ≈amp;1 MΩ resistance). The bath solution contained (in mM): NaCl 88, KCl 10, MgCl2 2.6, CaCl2 0.18, HEPES 5, pH 7.5. This solution was perfused in the recording chamber at a rate of about 0.2 ml min−1. Data were stored on the hard disk of a 486 IBM compatible computer for off-line analysis. The pCLAMP software (version 6.0.2, Axon Instr., CA, U.S.A.) was used for data acquisition and analysis. Currents were recorded at room temperature.
Patch-clamp
Currents from the human neuroblastoma SH-SY5Y and the HERG1-transfected HEK 293 cells were recorded at room temperature using a commercially available amplifier (Axopatch 200A, Axon Instruments, Foster City, CA, U.S.A.). The whole-cell configuration of the patch-clamp technique (Hamill et al., 1981) was adopted using glass micropipettes of 3–7 MΩ resistance. No compensation was performed for pipette resistance and cell capacitance. The cells were perfused with an extracellular solution containing (in mM): KCl 100, EGTA 10 and HEPES 10, pH 7.3 with KOH (for the SH-SY5Y cells), or NaCl 150, KCl 10, CaCl2 3, MgCl2 1, HEPES 10, pH 7.4 with NaOH (for the HERG1-transfected HEK 293 cells). The pipettes were filled with (in mM) CsCl 110, tetraethylammonium-Cl 10 MgCl2 2, EGTA 10, glucose 8, Mg-ATP 2, cyclic AMP 0.25 and HEPES 10, pH 7.3 with NaCl KOH (for the SH-SY5Y cells), or K-Aspartate 130, NaCl 10, CaCl2 4, MgCl2 2, EGTA 10, Mg-ATP 2, cyclic AMP 0.25 and HEPES 10, pH 7.4 with NaOH (for the HERG1-transfected HEK 293 cells; free Ca2+: 51 nM, calculated with MaxChelator Ver. 6.50).
Drugs and statistics
All the reagents were purchased from Sigma Chemicals (Italy). Astemizole was kindly provided by Janssen-Cilag (Rome, Italy). The H1-receptor antagonists were dissolved in dimethylsulphoxide (DMSO) at concentrations between 5 and 50 mM, and stock solutions were kept at −20°C. Appropriate drug dilutions were prepared daily. The maximal DMSO concentration (0.6%) did not affect HERG1 K+ channels recorded in Xenopus oocytes, HEK 293 cells, or SH-SY5Y human neuroblastoma cells. Statistical significance between the data was obtained by means of the Student's t-test or by the ANOVA test followed by the Tukey test. When appropriate, data are expressed as the mean±s.e.mean.
Results
Differential effect of mizolastine on the K+ currents carried by four cloned K+ channels expressed in Xenopus oocytes
Upon microinjection with HERG1 cRNA, Xenopus oocytes expressed a K+ current having biophysical properties which resembled those of the IKr current (Trudeau et al., 1995; Sanguinetti et al., 1995). This K+ current is activated by depolarization yet displays a pronounced inward rectification of the current-voltage relationship at positive potentials (>0 mV); furthermore, it shows rather slow kinetics of activation, and exhibits a large inward component upon repolarization to −100 mV, a membrane potential value below the equilibrium potential for K+ ions. Extracellular perfusion with mizolastine (10 μM) inhibited HERG1 K+ channels in Xenopus oocytes (Figure 1); the per cent of inhibition (≈70%), was similar whether measured at the peak of the inward current elicited at −100 mV following long (1–2 s) depolarizing pulses to 0 mV or at the peak of the outward current during the depolarizing pulse. Analogously, 10 μM mizolastine also blocked by ≈amp;70% the outward K+ currents carried by the channels encoded by the bovine isoform of the Ether-à-go-go Gene (BEAG; Frings et al., 1998), when heterologously expressed in Xenopus oocytes. By contrast, the K+ currents flowing through the channels encoded by Kv2.1 were insensitive to blockade by 10 μM mizolastine; whereas those carried by Kv3.1 were only slightly affected by the same concentration of the second-generation antihistamine (Figure 1).
Figure 1.
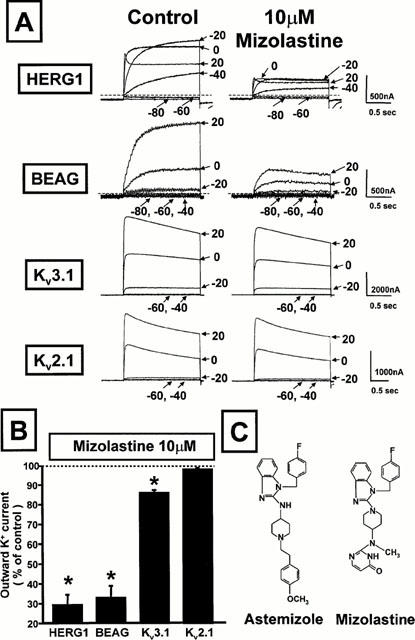
Effect of mizolastine on the K+ currents carried by four cloned K+ channels expressed in Xenopus oocytes. (A) Representative current traces recorded with the two-microelectrode voltage-clamp technique from four different oocytes expressing HERG1, BEAG, Kv3.1, and Kv2.1 recorded in control condition and after 5 min perfusion with 10 μM mizolastine. Holding potential: −90 mV (HERG1 and BEAG), −70 mV (Kv3.1 and Kv2.1); test potentials from −80 mV to +20 mV (HERG1 and BEAG) or from −60 mV to + 20 mV (Kv3.1 and Kv2.1) in 20 mV steps; return potential: −100 mV (HERG1 and BEAG) or −70 mV (Kv3.1 and Kv2.1). When reported, the dashed lines in the panel indicate the zero current level. (B) The per cent variation of the outward K+ current induced by 5 min perfusion with 10 μM mizolastine (Idrug/Icontrol) at voltages which maximally activated the conductance for each K+ channel (0 mV for HERG1, +20 mV for BEAG, Kv3.1 and Kv2.1). Each experimental point is the mean±s.e.mean of 3–8 separate experiments. Asterisks denote values significantly different from control values (P<0.05). (C) Chemical structures of mizolastine and astemizole.
Figure 2A shows the effect of different concentrations of mizolastine (1, 10 and 30 μM) on the inward K+ currents carried by heterologously expressed HERG1 expressed in Xenopus oocytes recorded upon cell hyperpolarization to −100 mV following depolarization to 0 mV to activate the current. In Figure 2B, the per cent of control HERG1 K+ tail currents blockade exerted by the different mizolastine concentrations (0.3–30 μM), calculated from the ratio Idrug/Icontrol of the peak inward K+ currents elicited at −100 mV following depolarization to 0 mV (without subtraction of the steady-state inward current measured at the end of the hyperpolarizing pulse), were normalized and fit to the following binding isotherm: y=max/(1+X/IC50)n, where X is the drug concentration and n the Hill coefficient. Fitted values for n were 0.93±0.08. The IC50 for HERG1 blockade by mizolastine was 3.4 μM.
Figure 2.
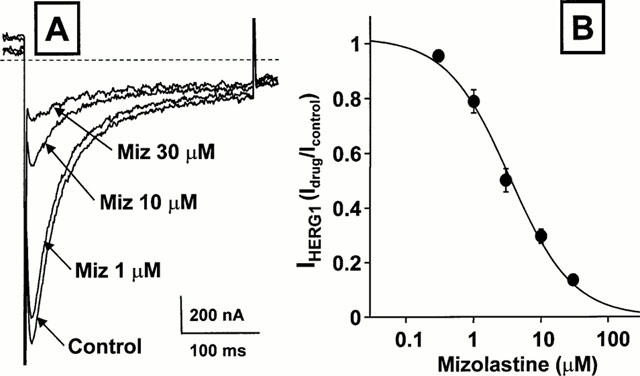
Dose-dependence of the inhibitory action of mizolastine on HERG1 K+ currents expressed in Xenopus oocytes. (A) Representative current traces recorded with the two-microelectrode voltage-clamp technique from a single HERG1-expressing oocyte exposed to control condition, and after perfusion with 1, 10 and 30 μM mizolastine (5 min for each concentration). Holding potential: −90 mV; test potential: 0 mV; return potential: −100 mV. Only the tail currents recorded upon return to −100 mV are shown. The dashed line in the panel indicate the zero current level. (B) Dose-dependence of HERG1 K+ currents inhibition by mizolastine. The inhibitory effect of the different concentration of the H1 receptor antagonist on the peak of the inward HERG1 K+ currents recorded at −100 mV was calculated for several cells and plotted against drug concentrations. The solid line represents the fit of the experimental data to the following binding isotherm: y=max/(1+X/IC50)n, where X is the drug concentration and n the Hill coefficient. The fitted value for n was between 0.72 and 0.96. Each point is the mean±s.e.mean of 3–6 determinations.
Since other second-generation antihistamines such as terfenadine and astemizole are known to potently inhibit HERG1 K+ channels, in the next series of experiments the kinetics of HERG1 K+ channels blockade by astemizole and mizolastine were compared. Figure 3 shows the time-course of the blockade of the inward HERG1 tail currents at −100 mV by increasing concentrations of mizolastine (0.3–3 μM), each perfused for 3 min followed by a washout period of variable duration (3–10 min). After achieving a steady-state block with each mizolastine concentration, the removal of the drug from the solution perfusing the oocytes caused a complete recovery of the K+ currents. When the time-course of such recovery was fit to a monoexponential function, the time constants of this monoexponential function did not depend on mizolastine concentration. In the experiment shown in Figure 3, the time constants were 46.6 s, 31.2 s, and 48.4 s for 0.3, 1 and 3 μM mizolastine, respectively. These rates likely reflect the kinetics of drug dissociation from the channel and could not be accounted for by the time-course of solution exchange in the recording bath (≈amp;15 s), as judged by the change in reversal potential of the HERG1 K+ current occurring upon switching solutions with different K+ concentrations (data not shown). On the other hand, the time-course of HERG1 K+ current recovery after 1 μM astemizole was much slower than that occurring with mizolastine, resulting in a value of 2340 s in the experiment shown in Figure 3. When similar experiments and analysis were performed on five different cells, the time constants for HERG1 K+ current recovery were 72.4±11.9 s (n=5) for 3 μM mizolastine, and 1361±306 s (n=5) for 1 μM astemizole. These results suggested that the lower potency for HERG1 K+ channels blockade exhibited by mizolastine when compared to astemizole (the IC50s were 3.4 μM vs 480 nM, respectively) (Taglialatela et al., 1998b) could be mainly ascribed to the faster dissociation rate of mizolastine from HERG1 K+ channels.
Figure 3.
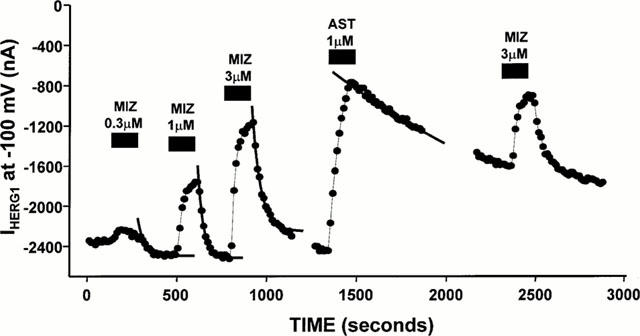
Kinetic difference between HERG1 K+ channel blockade by mizolastine and astemizole in Xenopus oocytes. The peak of the inward tail K+ current carried by HERG1 channels, measured upon return to −100 mV after depolarizing pulses of 1 s duration to 0 mV elicited every 15–20 s from an holding potential of −90 mV, is plotted versus time during the exposure to the different experimental conditions indicated by the black boxes. The solid lines are the fits of the experimental points recorded upon washout of each of the drug concentrations indicated to single exponential equations of the following form: y=A1*exp{−(t-K)/τ}+C, using the fitting routines of the Clampfit module of the pClamp suite (ver. 6.0.3), where A1 is the amplitude of the exponential function, (t−K) is the time range used fot the fitting (taking into consideration the start of the fit region, K), τ is the time constant, and C is an offset to account for the steady-state asymptote.
Effects of mizolastine on HERG1 K+ channels heterologously expressed upon stable transfection of HEK 293 cells with HERG1 cDNA or constitutively expressed in SH-SY5Y human neuroblastoma cells
In order to verify that the HERG1 inhibitory action exerted by mizolastine was not a characteristic of the Xenopus oocyte expression system, HEK 293 cells stably transfected with HERG1 cDNA (Zhou et al., 1998) were used. Figure 4 shows representative current traces from the same HERG1-transfected HEK 293 cell subsequently recorded in control conditions and after perfusion with increasing mizolastine concentrations (0.01–1 μM), each lasting about 4 min. From a holding potential of −80 mV, the K+ channels were activated by 1-s depolarizing pulses at potentials from −60 mV to +40 mV. At the end of the depolarizing pulses, the cell was repolarized to −100 mV, giving rise to a large inward tail current. In line with the results obtained in Xenopus oocytes, mizolastine displayed a dose-dependent inhibitory action on the HERG1 K+ currents heterologously expressed in HEK 293 cells, with an IC50 for such inhibitory action of 350±76 nM (n=5 cells). After perfusing the cells with the highest concentration of mizolastine (1 μM), 1 μM astemizole perfusion caused a further and almost complete blockade of HERG1 inward K+ currents (91.8±2.5%; n=8). The small residual outward and inward K+ currents recorded after astemizole perfusion are likely carried by endogenous outwardly-rectifying K+ channels constitutively expressed in HEK 293 cells, since it was insensitive to blockade by extracellular or intracellular application of astemizole and was identical to the K+ current recorded in untransfected HEK 293 cells (data not shown).
Figure 4.
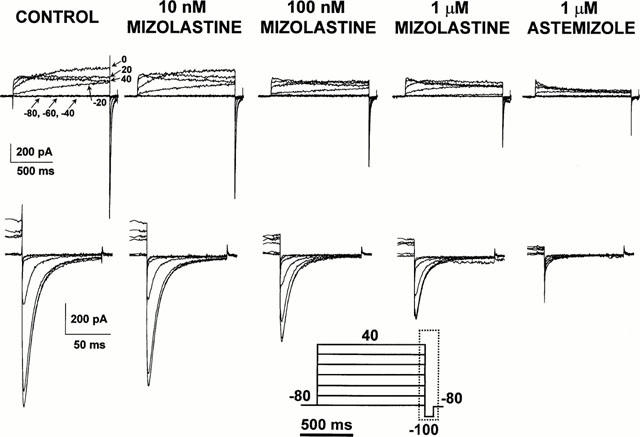
Effect of extracellular perfusion with mizolastine on IHERG1 heterologously expressed in HEK 293 cells stably transfected with HERG1 cDNA. Representative current traces recorded in the whole-cell configuration of the patch-clamp technique from a single HERG1-transfected HEK 293 cell. Records were obtained in control conditions, after 5 min perfusion with increasing concentrations of extracellular mizolastine (10 nM, 100 nM and 1 μM), and after 5 min exposure to 1 μM extracellular astemizole. The pulse protocol (shown at the bottom of the Figure) was the following: holding potential: −80 mV; test potentials from −80 mV to +40 mV in 20 mV steps (1 s duration); return potential: −100 mV. The bottom panels of the figure (corresponding to the time region indicated by the boxed area in the voltage pulse protocol) show the inward tail currents recorded upon repolarization to −100 mV on an expanded time and current scale.
In addition to the fundamental role of the IKr current in regulating action potential repolarization in cardiac cells, recent evidence suggests that HERG1 K+ channels are also expressed in other excitable tissues such as the brain (Wymore et al., 1997), in several neuroblastoma cell lines (Arcangeli et al., 1995), as well as in most tumor cell lines of different histogenesis (IHERG1) (Bianchi et al., 1998). For this reason, the SH-SY5Y clone of human neuroblastoma cells was used to study the effect of mizolastine on mammalian cells constitutively expressing HERG1 K+ channels. In order to avoid contamination by other K+ currents also expressed in these cells, IHERG1 was studied by means of a voltage-clamp protocol in which the cell was depolarized for 10 s to 0 mV, a membrane potential which fully activated IHERG1 and completely inactivated the delayed rectifier K+ current (Arcangeli et al., 1995), and then repolarized to increasingly negative voltages (from 0 to −120 mV) for 365 ms. This voltage protocol allows to detect a K+-selective inward current displaying the biophysical properties of IHERG1 (Arcangeli et al., 1995; Bianchi et al., 1998). Figure 5 shows representative current responses from a single SH-SY5Y human neuroblastoma cell exposed to increasing mizolastine concentrations (0.1–1 μM). Similarly to the results obtained in HEK 293 HERG1-transfected cells, 0.1 μM mizolastine causes approximately a 50% blockade of the inward HERG1 K+ currents; and a further increase in IHERG1 blockade was observed by increasing the concentration of the second-generation H1 receptor blocker to 1 μM. Pooling together the results obtained in five different cells in which experiments similar to that shown in Figure 5 were performed, the per cent of inhibition of the peak inward IHERG1 of current recorded at −120 mV after 0.1 and 1 μM mizolastine, respectively, were 60.9±9.9% (n=5) and 78.5±2.6% (n=5) of control currents. It should be underlined that the intracellular solution for SH-SY5Y cells contained Cs+ and TEA to minimize the contribution of the delayed rectifier current during the long depolarizing pulse (10 s at 0 mV) required to activate IHERG1, as previously described (Taglialatela et al., 1998b). Due to the intrinsic voltage-dependence and to the fast kinetics of dissociation for both TEA and Cs+, the slow inward currents recorded at negative potentials should be minimally affected by the blocking ions. In fact, similar inhibition by mizolastine was obtained using an intracellular solution for SH-SY5Y recording identical to that for HEK 293 cells (data not shown).
Figure 5.
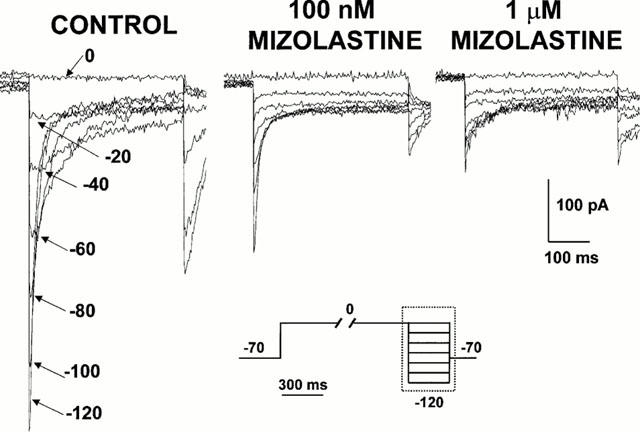
Effect of mizolastine on IHERG1 constitutively expressed in SH-SY5Y human neuroblastoma cells. Representative current traces recorded in the whole-cell configuration of the patch-clamp technique from an SH-SY5Y human neuroblastoma cell recorded in control conditions and after subsequent perfusion with 100 nM and 1 μM mizolastine (5 min each). The pulse protocol (shown at the bottom of the Figure) was the following: holding potential: −70 mV; depolarizing test potential: 0 mV for 10 s; hyperpolarizing return potentials: from 0 to −120 mV in −20 mV steps for 365 ms; return potential: −70 mV. Only approximately the last 450 ms of each pulses are shown, as indicated by the boxed area in the voltage pulse protocol.
Discussion
The present study aimed to investigate the possible interference of mizolastine, a novel second-generation H1 receptor antagonist recently available in Europe, with the K+ currents carried by the HERG1 gene product. These channels, which provide the molecular basis for the cardiac repolarizing current IKr that plays a crucial role in controlling the duration of the cardiac action potential (Sanguinetti & Jurkiewicz, 1990; Sanguinetti et al., 1995), have been focused on since several reports have appeared in the literature of the last decade warning about the potential pro-arrhythmic effects of drugs such as some first- and second-generation antihistamines (astemizole and terfenadine, in particular), prokinetics, antipsychotics, and antidepressants that derive from HERG1 K+ channels blockade (Taglialatela et al., 1998a).
The results of this study suggest that mizolastine is able to block in some degree both constitutively and heterologously expressed HERG1 K+ channels. In fact, the benzimidazole blocked HERG1 K+ channels either heterologously expressed in amphibian (Xenopus oocytes) or in mammalian (HEK 293) cells, as well as constitutively present in SH-SY5Y human neuroblastoma cells. The existence of a 10 fold difference in the IC50 values obtained for mizolastine blockade of HERG1 K+ channels expressed in Xenopus oocytes and in HEK 293 cells could be explained by the different properties of the amphybian membrane when compared to that of mammalian cells. In fact, the follicular cell layer and the vitelline membrane surrounding the oocytic membrane might to a certain degree impede the access of compounds at the level of the ion channels expressed in the amphybian membrane. Furthermore, it should be reminded that this difference is known to exist also for other HERG1 K+ channel blockers; in fact, the antiarrhythmic methanesulphonanilide dofetilide displayed an IC50 value for HERG1 in whole Xenopus oocytes of 320 nM (Ficker et al., 1998), whereas in HEK-transfected mammalian cells this value is approximately 30 times lower, being 12 nM (Snyders et al., 1996). In addition, the potency of mizolastine in inhibiting HERG1 K+ channels appears to be at least 10 times lower than that of the well-known cardiotoxic molecule astemizole. In fact, the IC50s for HERG1 inhibition in Xenopus oocytes were 3 μM for mizolastine and 300 nM for astemizole; furthermore, in HERG1-transfected HEK 293 cells, the IC50s for the two second-generation antihistamines were 300 and 1 nM, respectively (Zhou et al., 1998). The lower potency of HERG1 inhibition by mizolastine could be at least in part ascribed to its faster dissociation rate from its receptor site on the channel when compared to astemizole.
The potential clinical relevance of a certain inhibitory action by mizolastine on HERG1 K+ channels is worth considering, since the drug reached a CMAX of 0.3–1 μM during standard therapy (10–20 mg die−1) (Chosidow et al., 1996; Chaufour et al., 1999), a concentration range which clearly showed significant HERG1 inhibitory action, particularly in eukaryotic cells. However, it should be noted that the steady-state levels achieved by the drug in therapeutic settings are much lower than the CMAX, and that the potency for HERG1 K+ channels blockade by mizolastine is much lower than that exerted by the same drug at the level of H1 receptors in vitro (the KD for mizolastine antagonism at H1 receptors is 1 nM) (Benavides et al., 1995).
In addition, the present study also shows that mizolastine is able to interfere with the K+ channels encoded by the Ether-à-go-go (EAG) and by the Kv3.1 genes; the potency of mizolastine in inhibiting EAG channels seems to be comparable to that displayed by the same molecule on the K+ channels encoded by HERG1, whereas Kv3.1 was only inhibited by 15% at the maximal drug concentration tested (10 μM). However, it should be underlined that, in several species including humans (Pardo et al., 1999), EAG K+ channels seem not to be expressed in cardiac tissue (Ludwig et al., 1994), but limited to the brain. It is therefore possible to speculate that EAG blockade is unlikely to cause an increased risk of arrhythmic events. On the other hand, the K+ channels encoded by the Kv3.1 gene also do not seem to play a major role in the control of cardiac excitability, even though one study has suggested the participation of Kv3.1-like channels in the ultrarapid delayed rectifier K+ current found in canine atrial myocytes (Yue et al., 1996). More importantly, mizolastine (up to 10 μM) was unable to affect the potassium channels encoded by the Kv2.1 gene, which is expressed in the heart and participates in the control of cardiac repolarization (Xu et al., 1999).
In consideration of the rare, yet serious adverse cardiac effects occurring with terfenadine and astemizole, European and US Health Regulating Authorities have recommended a careful examination of the cardiac safety of newly introduced drugs in preclinical and clinical studies, and in particular the study of their possible interference with ion channels controlling the cardiac action potential (EMEA, 1997). In vitro studies performed on guinea-pig ventricular myocytes have suggested that mizolastine is unable to lengthen cardiac action potential duration, even at concentrations 10–30 times above the standard therapeutic range (Biton et al., 1998). Furthermore, clinical studies in both healthy volunteers (Chaufour et al., 1999) and in allergic patients (Delauche-Cavallier et al., 1999) have shown that mizolastine showed no significant effect on cardiac repolarization. Therefore, the results of both animal and clinical studies argue against the possible occurrence of cardiotoxic events during standard therapy with the novel H1 receptor blocker. However, it should be underlined that the recognition of the potential for cardiotoxicity exerted by terfenadine and astemizole required more than 10 years of extensive world-wide clinical use of these drugs. Interestingly, the initial warnings issued by the Regulatory Agencies against the potential risks associated to terfenadine and astemizole assumption were based on the reviews of only a couple of dozen clinical cases (among the millions of daily drug doses sold per year) in which the correlation between drug assumption and adverse cardiotoxic effects had been clearly demonstrated (Honig & Baraniuk, 1996).
A direct comparison of HERG1-blocking properties by different H1 receptor blockers also allows to provide further insight into the structure-activity relationships for these molecules. In fact, it has been suggested that the HERG1 K+ channel-blocking properties of terfenadine and its structural analogue ebastine are at least in part related to the tertiary amine substituent of the molecule rather than to the presence of the piperidine ring, or to the aromatic ring structures conferring H1 receptor blocking activity (Zhang, 1997); no correlation has been recently found between the ability to prolong the cardiac action potential duration, an effect possibly related to HERG1 K+ channels blockade, and the H1 antagonistic activity by several antihistamines (Zhang, 1997; Taglialatela et al., 2000). Lipophilicity and bulkiness appear to be the two crucial parameters in the tertiary amine group substituent conferring HERG1 K+ channel-blocking capacity to the antihistaminic molecule. In fact, the second-generation antihistamine cetirizine, which appears to lack of significant HERG1-inhibitory properties (Taglialatela et al., 1998b), has polar and smaller substitutions at the nitrogen atom (amido and carboxyl groups, respectively); on the other hand, terfenadine, astemizole, and ebastine, the H1 receptor antagonists most effective in inhibiting HERG1 K+ channels, have less polar and bulkier phenyl rings in the substituting side chains. Mizolastine, on the other hand, is characterized by a lower lipophylicity and thus by a low tissular penetration and distribution volume (Delauche-Cavallier et al., 1999). The lower lipophilicity of mizolastine could explain the lower potency of this novel congener in inhibiting HERG1 K+ channels when compared to astemizole or terfenadine. Furthermore, HERG1 K+ channels blockade by terfenadine (Roy et al., 1996) and astemizole (Taglialatela et al., 1998b), as well as by the antiarrhythmic dofetilide (Kiehn et al., 1996), seems to occur at a site located on the cytoplasmic side of the channel. However, the more hydrophilic profile, together with the relatively fast dissociation from the receptor site on the HERG1 K+ channels when compared to astemizole, seems to suggest the possible extracellular location of the blocking site for mizolastine on the channel protein.
In conclusion, the results of the present study suggest that the new second-generation H1 receptor antagonist mizolastine displays a certain degree of inhibitory action on HERG1 K+ channels, which underlie the cardiac repolarizing current IKr and represent a major target for the arrhythmogenic action exerted by other antihistamines such as terfenadine and astemizole. Although caution should be exercised when extrapolating the possible in vivo consequences of the results achieved from in vitro studies, and in consideration of the relatively high concentrations of mizolastine required to inhibit HERG1 K+ channels, which appear to be above the steady-state plasma levels required to exert its therapeutical action, the present results might be of some clinical significance for those patients at risk of developing cardiac arrhythmias undergoing therapy with mizolastine.
Acknowledgments
The authors are indebted to Dr M.T. Keating (Salt Lake City, UT, U.S.A.) for kindly providing HERG1 cDNA; Dr P. Cilli (Janssen-Cilag, Rome, Italy) for the generous supply of astemizole; and Drs M. Olivotto and A. Arcangeli (Firenze, Italy) for providing the SH-SY5Y human neuroblastoma cells and helpful discussions. The study was supported by the following grants: Telethon 1058, National Research Council (CNR) 97.04512.CT04, 97.01233.PF49, 98.03149.CT04, 99.02614.CT04, 99.00495.PF49, and Ministero dell'Università e della Ricerca Scientifica e Tecnologica (MURST) COFIN 99 to MT; and by CNR 97.04559.CT04, 98.01048.CT04, 98.00062.PF31, 99.02371, MURST COFIN 98 and Regione Campania (L. 41 and POP) to LA.
Abbreviations
- DMEM
Dulbecco's Modified Eagle Medium
- DMSO
dimethylsulphoxide
- EMEA
European Agency for the Evaluation of Medicinal Products
- FCS
foetal calf serum
- HEK293 cells
human embryonic kidney 293 cells
- HERG1
Human Ether-à-go-go-Related Gene 1
- IKr
rapid component of the cardiac repolarizing potassium current
- MEM
modified eagle medium
References
- ARCANGELI A., BIANCHI L., BECCHETTI A., FARAVELLI L., CORONNELLO M., MINI E., OLIVOTTO M., WANKE E. A novel inward-rectifying K+ current with a cell cycle-dependence governs the resting potential of neuroblastoma cells. J. Physiol. (London) 1995;489:455–471. doi: 10.1113/jphysiol.1995.sp021065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENAVIDES J., SCHOEMAKER H., DANA C., CLAUSTRE Y., DELAHAYE M., PROTEAU M., MANOURY P., ALLEN J., SCATTON B., LANGER S.Z. In vivo and in vitro interaction of the novel selective histamine H1 receptor antagonist mizolastine with H1 receptors in the rodent. Arzneimittelforschung. 1995;45:551–558. [PubMed] [Google Scholar]
- BERUL C.I., MORAD M. Regulation of potassium channels by nonsedating antihistamines. Circulation. 1995;91:2220–2225. doi: 10.1161/01.cir.91.8.2220. [DOI] [PubMed] [Google Scholar]
- BIANCHI L., WIBLE B., ARCANGELI A., TAGLIALATELA M., MORRA F., CASTALDO P., CROCIANI O., ROSATI B., FARAVELLI L., OLIVOTTO M., WANKE E. Herg encodes a K+ current highly conserved in tumors of different histogenesis: a selective advantage for cancer cells. Cancer Res. 1998;58:815–822. [PubMed] [Google Scholar]
- BITON B., MAITRE S., GODET D., DEPOORTERE H., ARBILLA S., O'CONNOR S.E., AVENET P., SCATTON B. Effect of the novel non-sedative histamine H1 receptor antagonist mizolastine on cardiac potassium and sodium currents. Allergy. 1998;43 Suppl.:160. [Google Scholar]
- BROSTOFF J., FITZHARRIS P., DUNMORE C., THERON M., BLONDIN P. Efficacy of mizolastine, a new antihistamine, compared with placebo in the treatment of chronic idiopathic urticaria. Allergy. 1996;51:320–325. [PubMed] [Google Scholar]
- CHAUFOUR S., CAPLAIN H., LILIENTHAL N., L'HERITIER C., DESCHAMPS C., DUBRUC C., ROSENZWEIG P. Study of cardiac repolarization in healthy volunteers performed with mizolastine, a new H1-receptor antagonist. Br. J. Clin. Pharmacol. 1999;47:515–520. doi: 10.1046/j.1365-2125.1999.00927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOSIDOW O., DUBRUC C., DANJOU P., FUSEAU E., ESPAGNE E., BIANCHETTI G., THENOT J.P., HERSON S., ROSENZWEIG P., REVUZ J. Plasma and skin suction-blister-fluid pharmacokinetics and time course of the effects of oral mizolastine. Eur. J. Clin. Pharmacol. 1996;50:327–333. doi: 10.1007/s002280050117. [DOI] [PubMed] [Google Scholar]
- CRAFT T.M. Torsade de pointes after astemizole overdose. Br. Med. J. 1986;292:660. doi: 10.1136/bmj.292.6521.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANJOU P., MOLINIER P., BERLIN I., PATAT A., ROSENZWEIG P., MORSELLI P.L. Assessment of the anticholinergic effect of the new antihistamine mizolastine in healthy subjects. Br. J. Clin. Pharmacol. 1992;34:328–331. doi: 10.1111/j.1365-2125.1992.tb05638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIES A.J., HARINDA V., MCHEWAN A., GHOSE R.R. Cardiotoxic effect with convulsions in terfenadine overdose. Br. Med. J. 1989;302:1469. doi: 10.1136/bmj.298.6669.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DELAUCHE-CAVALLIER M.C., CHAUFOUR S., GUERALT E., LACROUX A., MURRIETA M., WAJMAN A. QT interval monitoring during clinical studies with mizolastine, a new H1 antihistamine. Clin. Exp. Allergy. 1999;29:206–211. doi: 10.1046/j.1365-2222.1999.0290s3206.x. [DOI] [PubMed] [Google Scholar]
- EUROPEAN AGENCY FOR THE EVALUATION OF MEDICINAL PRODUCTS: COMMITTEE FOR PROPRIETARY MEDICINAL PRODUCTS Points to consider: the assessment of the potential for QT interval prolongation by non-cardiovascular medicinal products 1997. CPMP/986/96
- FICKER E., JAROLIMEK W., KIEHN J., BAUMANN A., BROWN A.M. Molecular determinants of dofetilide block of HERG K+ channels. Circ. Res. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- FRECH G.C., VANDONGEN A.M.J., SCHUSTER G., BROWN A.M., JOHO R.H. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 1989;340:642–645. doi: 10.1038/340642a0. [DOI] [PubMed] [Google Scholar]
- FRINGS S., BRULL N., DZEJA C., ANGELE A., HAGEN V., KAUPP U.B., BAUMANN A. Characterization of ether-a-go-go channels present in photoreceptors reveals similarity to IKx, a K+ current in rod inner segments. J. Gen. Physiol. 1998;111:583–599. doi: 10.1085/jgp.111.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY E., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membranae patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HONIG P., BARANIUK J.N. Histamine and H1-receptor antagonists in allergic diseases 1996New York: Marcel Dekker Inc; 383–412.ed. Estelle, F., Simons, R. pp [Google Scholar]
- KIEHN J., LACERDA A., WIBLE B., BROWN A.M. Molecular physiology and pharmacology of HERG. Single channel currents and block by dofetilide. Circulation. 1996;94:2575–2579. doi: 10.1161/01.cir.94.10.2572. [DOI] [PubMed] [Google Scholar]
- LEYNADIER F., BOUSQUET J., MURRIETA M., ATTALI P. Efficacy and safety of mizolastine in seasonal allergic rhinitis. The Rhinase Study Group. Ann. Allergy Asthma Immunol. 1996;76:163–168. doi: 10.1016/s1081-1206(10)63417-5. [DOI] [PubMed] [Google Scholar]
- LUDWIG J., TERLAU H., WUNDER F., BRÜGGEMANN A., PARDO L.A., MARQUARDT A., STUHMER W., PONGS O. Functional expression of a rat homologue of the voltage gated ether a go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart. EMBO J. 1994;13:4451–4458. doi: 10.1002/j.1460-2075.1994.tb06767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONAHAN B.P., FERGUSON C.L., KILLEAVY E.S., LLOYD B.K., TROY J., CANTILENA L.R. Torsade de pointes occurring in association with terfenadine use. J.A.M.A. 1990;264:2788–2790. [PubMed] [Google Scholar]
- PARDO L.A., DEL CAMINO D., SANCHEZ A., ALVES F., BRUGGEMANN A., BECKH S., STUHMER W. Oncogenic potential of EAG K(+) channels. EMBO J. 1999;18:5540–5547. doi: 10.1093/emboj/18.20.5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATAT A., PERAULT M.C., VANDEL B., ULLIAC N., ZIELENIUK I., ROSENZWEIG P. Lack of interaction between a new antihistamine, mizolastine, and lorazepam on psychomotor performance and memory in healthy volunteers. Br. J. Clin. Pharmacol. 1995;39:31–38. doi: 10.1111/j.1365-2125.1995.tb04406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROY M-L. , DUMANIE R., BROWN A.M. HERG, a primary human ventricular target of the non sedating antihistamine terfenadine. Circulation. 1996;94:817–823. doi: 10.1161/01.cir.94.4.817. [DOI] [PubMed] [Google Scholar]
- SALATA J.J., JURKIEWICZ N.K., WALLACE A.A., STUPIENSKI R.F., III, GUINOSSO P.J., LYNCH J.J., JR Cardiac electrophysiological actions of the histamine H1-receptor antagonists astemizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ. Res. 1995;76:110–119. doi: 10.1161/01.res.76.1.110. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JIANG C., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JURKIEWICZ N.K. Two components of cardiac delayed rectifier K+ current: differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 1990;96:835–863. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLATER J.W., ZECHNICH A.D., HAXBY D.G. Second-generation antihistamines: a comparative review. Drugs. 1999;57:31–47. doi: 10.2165/00003495-199957010-00004. [DOI] [PubMed] [Google Scholar]
- SNYDERS D.J., CHAUDHARY A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol. Pharmacol. 1996;49:949–955. [PubMed] [Google Scholar]
- SORKIN E.M., HEEL R.C. Terfenadine: a review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1985;29:34–56. doi: 10.2165/00003495-198529010-00002. [DOI] [PubMed] [Google Scholar]
- SUESSBRICH H., WALDEGGER S., LANG F., BUSCH A.E. Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett. 1996;385:77–80. doi: 10.1016/0014-5793(96)00355-9. [DOI] [PubMed] [Google Scholar]
- TAGLIALATELA M., CASTALDO P., PANNACCIONE A., GIORGIO G., ANNUNZIATO L. HERG K+ channels as pharmacological targets: present and future implications. Biochem. Pharmacol. 1998a;55:1741–1746. doi: 10.1016/s0006-2952(98)00002-1. [DOI] [PubMed] [Google Scholar]
- TAGLIALATELA M., PANNACCIONE A., CASTALDO P., GIORGIO G., ZHOU Z., JANUARY C.T., GENOVESE A., MARONE G., ANNUNZIATO L. Molecular basis for the lack of HERG K+ channel block-related cardiotoxicity by the H1 receptor blocker cetirizine compared with other second-generation antihistamines. Mol. Pharmacol. 1998b;54:113–121. doi: 10.1124/mol.54.1.113. [DOI] [PubMed] [Google Scholar]
- TAGLIALATELA M., TIMMERMAN H., ANNUNZIATO L. Cardiotoxic potential and CNS effects of first-generation antihistamines. Trends Pharmacol. Sci. 2000;21:52–56. doi: 10.1016/s0165-6147(99)01437-6. [DOI] [PubMed] [Google Scholar]
- TAGLIALATELA M., VANDONGEN A.M.J., DREWE J.A., JOHO R.H., BROWN A.M., KIRSCH G.E. Patterns of internal and external tetraethylammonium block in four homologous K+ channels. Mol. Pharmacol. 1991;40:229–307. [PubMed] [Google Scholar]
- TASMAN A.J., WEBER P. A double blind placebo controlled multicentered study on mizolastine, a new non- sedative H1-receptor antagonist, in patients suffering from perennial allergic rhinoconjunctivitis. Allergy. 1995;50:79. [Google Scholar]
- TRUDEAU M.C., WARMKE J.W., GANETZKI B., ROBERTSON G.A. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- VUURMAN E.F., UITERWIJK M.M., ROSENZWEIG P., O'HANLON J.F. Effects of mizolastine and clemastine on actual driving and psychomotor performance in healthy volunteers. Eur. J. Clin. Pharmacol. 1994;47:253–259. doi: 10.1007/BF02570505. [DOI] [PubMed] [Google Scholar]
- WARMKE J.W., GANETZKY B. A family of potassium channel genes related to eag in Drosofila and mammals. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOOSLEY R.L., CHEN Y., FREIMAN J.P., GILLIS R.A. Mechanism of the cardiotoxic action of terfenadine. J.A.M.A. 1993;269:1532–1536. [PubMed] [Google Scholar]
- WOOSLEY R.L. Cardiac actions of antihistamines. Annu. Rev. Pharmacol. Toxicol. 1996;36:233–252. doi: 10.1146/annurev.pa.36.040196.001313. [DOI] [PubMed] [Google Scholar]
- WYMORE R.S., GINTANT G.A., WYMORE R.T., DIXON J.E., MCKINNON D., COHEN I.S. Tissue and species distribution of mRNA for the IKr-like K+ channel, erg. Circ. Res. 1997;80:261–268. doi: 10.1161/01.res.80.2.261. [DOI] [PubMed] [Google Scholar]
- XU H., BARRY D.M., LI H., BRUNET S., GUO W., NERBONNE J.M. Attenuation of the slow component of delayed rectification, action potential prolongation, and triggered activity in mice expressing a dominant-negative Kv2 alpha subunit. Circ. Res. 1999;85:623–633. doi: 10.1161/01.res.85.7.623. [DOI] [PubMed] [Google Scholar]
- YUE L., FENG J., LI G.R., NATTEL S. Characterization of an ultrarapid delayed rectifier potassium channel involved in canine atrial repolarization. J. Physiol. (London). 1996;496:647–662. doi: 10.1113/jphysiol.1996.sp021716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG M-Q. Chemistry underlying the cardiotoxicity of antihistamines. Curr. Med. Chem. 1997;4:187–200. [Google Scholar]
- ZHOU Z., GONG Q., YE B., FAN Z., MAKIELSKI J.C., ROBERTSON G.A., JANUARY C.T. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 1998;74:230–241. doi: 10.1016/S0006-3495(98)77782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]