

Abstract
The effects of inhibition of acetylcholine (ACh)-induced hyperpolarization on dilatation of submucosal arterioles were investigated in the guinea-pig ileum.
In smooth muscles of the arterioles depolarized by Ba2+ (0.5 mM) to about −40 mV, ACh (3 μM) repolarized the membrane to about −65 mV (hyperpolarization), irrespective of the absence or presence of L-Nω-nitroarginine (L-NOARG, 0.1 mM) and diclofenac (1 μM), and increased the diameter (dilatation).
Combined application of charybdotoxin (CTX, 50 nM) and apamin (0.1 μM), inhibitors of some types of K+-channels, abolished the ACh-induced hyperpolarization and dilatation.
18β-Glycerrhetinic acid (18β-GA, 30 μM), a known inhibitor of gap junctions, depolarized the membrane to about −36 mV, either in the absence or in the presence of Ba2+, with no associated contraction of the arterioles. In the presence of 18β-GA, ACh-induced hyperpolarization was abolished, however the dilatation was inhibited only partially, with associated inhibition of constriction produced by Ba2+ and NA.
18β-GA inhibited the dilatation produced by sodium nitroprusside, an NO donor.
The ACh-induced hyperpolarization and dilatation were abolished in the presence of 2-aminoethoxydiphenyl borate (30 μM), an inhibitory modulator of inositol trisphosphate receptor-mediated Ca2+ release from intracellular stores.
It is concluded that in submucosal arterioles, hyperpolarizations produced by ACh have causal relationship to the arteriolar dilatation. 18β-GA did not induce parallel relationship between hyperpolarization and dilatation produced by ACh. 18β-GA may have unidentified inhibitory effects on agonist-mediated actions, in addition to the inhibition of gap junctions.
Keywords: Arteriole, acetylcholine, hyperpolarization, vasodilatation, gap junction, K+ channels, InsP3 receptor, NO
Introduction
Endothelium-derived hyperpolarizing factor (EDHF) is one of the important factors involved in the endothelium-dependent relaxation of vascular smooth muscle (Chen et al., 1988; Chen & Suzuki, 1989; Suzuki & Chen, 1990; Garland et al., 1995). The chemical nature of EDHF remains unclear. Recent experiments suggest that metabolites of arachidonic acid such as 11,12-eicosatrienoic acids (Popp et al., 1996; Campbell et al., 1996) or endogenous cannabinoids (Randall & Kendall, 1997), or K+ ions released from endothelial cells (Edwards et al., 1998) may be involved as mediator of the endothelium-dependent hyperpolarization. In large arteries, the major endothelial vasodilator may be the endothelium-derived relaxing factor (EDRF; Furchgott & Zawadzski, 1980). The contribution of EDRF and EDHF in the endothelium-dependent relaxation differs between proximal and peripheral arteries, and EDHF may be more important than EDRF (or nitric oxide, NO) in peripheral arterioles (Garland et al., 1995: Shimokawa et al., 1996). It is therefore important to investigate the relationship between membrane hyperpolarization and relaxation in arterioles. However, parallel measurements of membrane responses and mechanical changes in arteriolar smooth muscles are not easy, mainly due to the small sizes of the vessels. The submucosal arteriole of the guinea-pig ileum is one such preparation in which arteriolar diameters and membrane potential of smooth muscle cells are measurable (Hirst, 1977; Hirst & Neild, 1980; Neild, 1989).
In smooth muscle of guinea-pig submucosal arterioles, acetylcholine (ACh) hyperpolarizes the membrane in an endothelium-dependent manner (Hashitani & Suzuki, 1997). The ACh-induced hyperpolarization is produced originally in the endothelial cells by activation of Ca2+-activated K+-channels, and it is conducted to smooth muscle cells through gap junctions (Yamamoto et al., 1999). ACh increases intracellular Ca2+ in endothelial cell due to release from the internal store through activation of inositol trisphosphate (InsP3) receptors (Schlling & Elliott, 1992). Attempts were made to inhibit the ACh-induced hyperpolarization by three different mechanisms, i.e. inhibition of K+-channels, inhibition of gap junctions and inhibition of the release of Ca2+ from the internal stores, and their effects on arteriolar dilatation were observed. K+ channels were inhibited by charybdotoxin (CTX) and apamin (Hashitani & Suzuki, 1997; Yamamoto et al., 1999). Myo-endothelial gap junctions were inhibited by 18β-glycyrrhetinic acid (18β-GA) (Yamamoto et al., 1998; 1999). The release of Ca2+ from internal stores was inhibited by 2-aminoethoxydiphenyl borate (2APB), an inhibitory modulator of InsP3 receptor-mediated Ca2+ release (Maruyama et al., 1997; Cui & Kanno, 1997). The electrical responses of arteriolar smooth muscle were recorded by conventional intracellular microelectrode methods and the isotonic mechanical responses were recorded by measuring the change in arteriolar diameter using DIAMTRAK system (Neild, 1989).
The results indicate that in submucosal arterioles of the guinea-pig ileum, the ACh-induced hyperpolarization of smooth muscle membrane is causally related to the dilatation. However, parallel inhibition of hyperpolarization and dilatation was not produced when 18β-GA was applied. Possible mechanisms related to this discrepancy are discussed.
Methods
Male albino guinea-pigs, weighing 150–250 g, were anaesthetized with ether and then exsanguinated by bleeding from the femoral artery. Lengths of ileum were removed and cut into a small segment. The segment of ileum was slit open along the mesenteric border and pinned out in a dissecting chamber with the mucosa upside. The mucosal layer was removed gently by fine tweezers. A thin sheet of submucosal connective tissue containing arterioles was dissected from the underlying circular smooth muscle layer using methods reported by Hirst (1977) and pinned onto transparent silicone rubber in the base of a small recording chamber (volume, approximately 1 ml). The chamber was mounted on an inverted compound microscope (TMD-100, Nikon, Tokyo, Japan) and the preparation continuously superfused with warmed (35°C) Krebs solution at a constant flow rate of about 4 ml min−1. The ionic composition of the Krebs solution was (in mM); Na+ 137.4, K+ 5.9, Mg2+ 1.2, Ca2+ 2.5, H2PO4− 1.2, HCO3− 15.5, Cl− 134, glucose 11.5. The solution was aerated with 95% O2 containing 5% CO2, and the pH of the solution was maintained in the range 7.3–7.4. Since endothelial cells cannot be removed from these vessels without damaging the smooth muscle (Neild et al. 1990), all experiments were carried out in arterioles with intact endothelial cells.
Arteriolar smooth muscles were impaled with glass capillary microelectrode filled with 1 M KCl (tip resistance 100–250 MΩ). Membrane potential changes were recorded using a high input-impedance amplifier (Axoclamp-2B, Axon Instruments, CA, U.S.A.), and displayed on a cathode-ray oscilloscope (SS-9622, Iwatsu, Tokyo, Japan). After low-pass filtering (cut-off frequency, 40 Hz), the potential changes were digitized and stored with a Digidata 1200A data acquisition system (Axon Instruments, CA. U.S.A.). Segmented arterioles (about 100 μm long) were prepared by cutting with a fine knife, and the passive electrical properties of the arteriolar smooth muscle cells were determined by injecting a hyperpolarizing current, with a duration of 2 s, through the recording electrode. The injecting currents were controlled with pCLAMP software (Axon Instruments. CA, U.S.A.). The acceptable impalement of the electrode into arteriolar smooth muscle cell was confirmed using criteria reported previously (Hashitani & Suzuki, 1997).
The preparation was viewed with an inverted compound microscope equipped with a television camera (WV BP 310, Panasonic, Yokohama, Japan), and outside diameters of an arteriole were monitored using the DIAMTRAK system developed by Neild (1989). The results were displayed on a pen-writing recorder (VP-6524A, Matsushita Electric, Tokyo, Japan) and also stored with a Digidata 1200A data acquisition system.
Acetylcholine chloride (ACh), apamin, 18β-glycyrrhetinic acid (18β-GA), noradrenaline hydrochloride (NA), diclofenac sodium (all from Sigma, St. Louis, MO); 2-aminoethoxydiphenyl borate (2APB) (Ono, Osaka, Japan); barium chloride (Katayama, Osaka, Japan), charybdotoxin (CTX) and L-Nω-nitroarginine (L-NOARG) (Peptide institute, Osaka, Japan). These drugs were dissolved in distilled water (ACh, apamin, CTX, NA, barium chloride, diclofenac, L-NOARG) or dimethylsulphoxide (18β-GA, 2APB) at 1–0.1 mM. and diluted further in Krebs solution to obtain the desired concentration. The final concentrations of these solvents did not exceed 1 : 1000 in the Krebs solution, and these procedures did not alter the pH of the Krebs solutions.
Measured values were expressed as mean±standard deviation (s.d.). Statistical significance of the values was determined using paired and unpaired Student's t-test. Probabilities of less than 5% (P<0.05) were considered significant.
Results
Effects of K+-channel inhibitors on the ACh-induced hyperpolarization and relaxation
Smooth muscle cells of the submucosal arteriole showed properties similar to those reported previously (Hirst, 1977; Hirst & Neild, 1980; Hashitani & Suzuki, 1997). Briefly, the membrane was quiescent with occasional transient depolarization, and the resting membrane potential ranged from −70 and −82 mV (−75.4±3.2 mV, n=153). Application of ACh (3 μM) did not produce any detectable change in the membrane potential (control, −75.6±2.1 mV, n=5; in ACh, −75.6±1.7 mV, n=5, P>0.1). Inhibition of the inward rectifier K+ channels with 0.5 mM Ba2+ (Edwards & Hirst, 1988) depolarized the membrane with an initial generation of several action potentials to reach a stable level ranging between −38 and −45 mV (−41.8±0.4 mV, n=35, Figure 1A, a). In the presence of Ba2+, application of 0.1 mM L-Nω-nitroarginine (L-NOARG), an inhibitor of NO synthesis, and 1 μM diclofenac, an inhibitor of cyclooxygenase (Moncada et al., 1991), did not further alter the membrane potential of arteriolar smooth muscle cells (control, −43.3±3.2 mV, n=6; in L-NOARG and diclofenac, −43.0±1.5 mV, n=6, P>0.1). These results indicate that both NO and prostanoids do not contribute significantly to the Ba2+-induced depolarization. In the presence of Ba2+, L-NOARG and diclofenac, application of ACh (3 μM) produced a hyperpolarization with an initial transient and following sustained component (Figure 1A,a).
Figure 1.
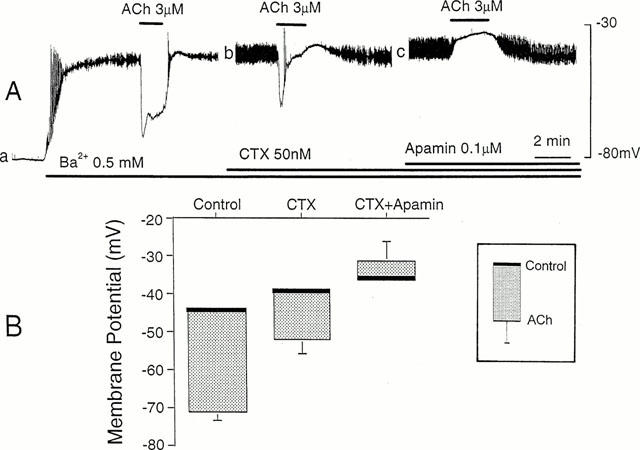
Effects of CTX and apamin on ACh-induced hyperpolarization. (A) ACh (3 μM) was applied for 2 min in the cumulative presence of 0.5 mM Ba2+ (a), 50 nM CTX (b) and 0.1 μM apamin (c). The diameter of arteriole, 50 μm. All the responses were recorded from the same cell. The resting membrane potential, −78 mV. (B) Membrane potentials measured before and during application of 3 μM ACh, in the cumulative presence of 0.5 mM Ba2+ (Control), 50 nM CTX (CTX) and 0.1 μM apamin (CTX+apamin). Each column indicates the mean value of the membrane potential (thick bar) with +/−s.d. (n=5–6). L-NOARG (0.1 mM) and diclofenac sodium (1 μM) were present throughout.
The effects of charybdotoxin (CTX), an inhibitor of Ca2+-activated K+-channels, and apamin, an inhibitor of small conductance Ca2+-activated K+ channels (Nelson & Quayle, 1995), on the ACh-induced hyperpolarization were observed in arterioles exposed to L-NOARG (0.1 mM) and diclofenac (1 μM). In the presence of Ba2+ (0.5 mM), CTX (50 nM) depolarized the membrane (control, −43.2±5.1 mV, n=5; in CTX, −39.0±5.8 mV, n=5, P<0.05). Combined application of CTX and apamin (0.1 μM) also depolarized the membrane (control, −44.8±5.1 mV, n=5; in both apamin and CTX, −36.8±7.0 mV, n=5, P<0.05). In the presence of CTX, the ACh-induced hyperpolarization was altered to a transient form with reduced amplitude (amplitude of the initial component, control 27.0±2.3 mV, n=6; in CTX, 12.8±3.9 mV, n=6; P<0.05) (Figure 1A, b). In the presence of both CTX and apamin, ACh produced no alteration in the membrane potential (n=5) or depolarized the membrane (4.6±5.4 mV, n=5) (Figure 1A, c). The ACh-induced changes in the membrane potential in the presence and absence of CTX and apamin are summarized in Figure 1B.
At rest, submucosal arterioles did not produce any spontaneous change in diameter. The arteriolar diameters were not altered by exposure to solutions containing 0.1 mML-NOARG and 1 μM diclofenac (control, 48.1±4.8 μm; in L-NOARG and diclofenac, 48.2±5.1 μm, n=9, P>0.1). These results suggested that both NO and prostanoids did not contribute significantly to maintain the diameter of arterioles in resting conditions. ACh (3 μM) did not alter the diameter of arterioles (control, −51.3±12.3 μm, n=3; in ACh, −51.0±12.3 μm, n=3, P<0.01), and this remained unchanged in the presence of L-NOARG and diclofenac (n=3, data not shown). In the presence of L-NOARG and diclofenac, application of Ba2+ (0.5 mM) containing noradrenaline (NA, 1 μM) reduced the diameter of arterioles (constriction), which effect reached a stable value within 2–3 min (control, 49.9±5.8 μm; in B2+ plus NA. 25.3±3.2 μm, n=10, P<0.05). In the constricted arterioles, ACh (3 μM) increased the diameter (i.e., dilatation), which reached a stable value within 2 min (control, 25.3±3.2 μm; in the presence of ACh, 42.6±5.1 μm, n=10, P<0.05) (Figure 2A, a). These results indicate that ACh induces dilatation of submucosal arterioles in the absence of any production of NO and prostanoids, and therefore the possible involvement of EDHF in the ACh-induced dilatation should be considered.
Figure 2.
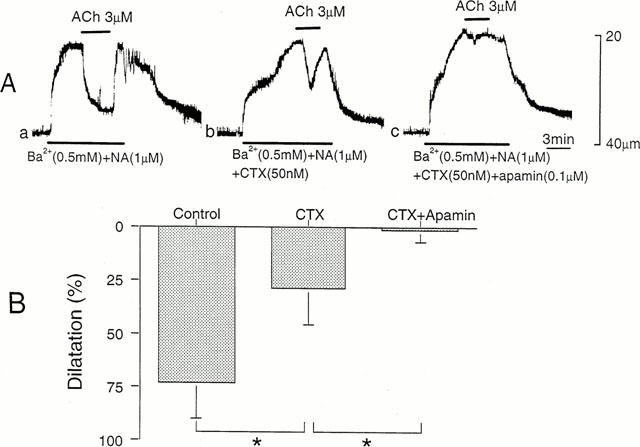
Effects of CTX and apamin on ACh-induced dilatation. (A) In an arteriole constricted with solution containing NA (1 μM) and Ba2+ (0.5 mM), dilatation produced by ACh (3 μM) was observed before (a) and after application of 50 nM CTX (b) and 50 nM CTX with 0.1 μM apamin (c). ACh was applied for 2 to 3 min. The resting diameter of arteriole, 40 μm. (B) Dilatation produced by ACh before (Control) and after application of CTX and apamin was measured as per cent of constriction produced before application of ACh. Mean±s.d. (n=4–10). *Significant reduction (P<0.05).
The effects of CTX (50 nM) and apamin (0.1 μM) on the ACh-induced dilatation were observed in arterioles constricted with Ba2+ and NA, in the presence of L-NOARG and diclofenac. In the presence of CTX, the ACh-induced dilatation was changed to a transient form with reduced amplitude (Figure 2A, b). In the presence of both CTX and apamin, the ACh-induced dilatation was abolished (Figure 2A, c). These effects of CTX and apamin were reversible, requiring 20–30 min for the recovery (data not shown). The ACh-induced dilatation was quantified by expressing the amplitude as relative to the constriction produced before application of ACh. As shown in Figure 2B, the ACh-induced dilatation reached nearly 75% of constriction, and the value was reduced to about 25% in the presence of CTX and abolished by additional application of apamin.
Effects of 18β-GA on the ACh-induced hyperpolarization and relaxation
In smooth muscle cells of the submucosal arterioles, 18β-GA (30 μM) depolarized the membrane, either in the absence (control, −72.8±2.7 mV. n=6; in 18 β-GA, −32.0±6.3 mV. n=6, P<0.05) or presence of 0.5 mM Ba2+ (in Ba2+, −42.6±5.7, n=16; Ba2+ plus 18β-GA, −27.0±5.0, n=5, P<0.05). The depolarizing actions of 18 β-GA remained unaltered in the absence or presence of L-NOARG and diclofenac (n=3, data not shown). In either case, depolarization of the membrane induced by 18 β-GA was accompanied by increased noise levels. In the segmented arterioles, stimulation of arteriolar muscles with a rectangular inward current pulse (2 nA), which was supplied through the recording electrode, produced electrotonic potentials (amplitude, 5.3±2.7 mV, n=4). The decay of the electrotonic potential after cessation of the current pulse was not exponential (n=4, data not shown). The amplitude of electrotonic potentials was significantly increased during depolarization with 18β-GA (40.0±12.7 mV. n=4, P<0.05). The results indicate that the depolarization produced by 18-β GA accompanies an increase in the input resistance of smooth muscle membrane, possibly due to the inhibition of gap junctional connections with surrounding cells (Yamamoto et al., 1998).
The ACh-induced hyperpolarization was abolished (Figure 3A, b) or sometimes reduced greatly to 1–5 mV by 30 μM 18β-GA. The inhibitory effect of 18β-GA was reversible, with complete recovery requiring 15–20 min (Figure 3A, c). These effects of 18β-GA on ACh-induced hyperpolarization appeared in a concentration dependent manner between 10 and 30 μM, and the highest concentration was required to induce complete inhibition of the hyperpolarization response (Figure 3B).
Figure 3.
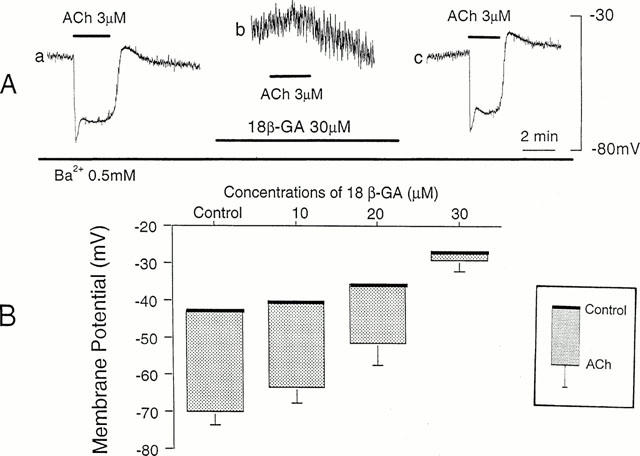
Effects of 18β-GA on ACh-induced hyperpolarization. (A) In the presence of Ba2+ (0.5 mM), ACh (3 μM) was applied for 2 min before (a), during application of 18β-GA (30 μM) for 20 min (b), and after removal of 18β-GA for 40 min (c). All responses were recorded from the same cell. The diameter of arteriole, 40 μm. (B) Membrane potentials measured before and during application of ACh (3 μM), in the presence of 0.5 mM Ba2+ alone (Control) and additional presence of 18β-GA (10–30 μM). Each column indicates the peak value of the ACh-induced hyperpolarization expressed by means+s.d. (n=5–6).
In the presence of L-NOARG and diclofenac, application of 18β-GA (30 μM) for over 30 min did not produce any detectable change in the diameter of arterioles (control, 45.3±3.8 μm; in 18 β-GA, 45.0±2.9 μm, n=7, P>0.1). However, the constriction of arterioles produced by Ba2+ and NA was inhibited by 18β-GA in a concentration-dependent manner. Amplitude of the constriction was reduced to 88±25 % (n=3) of control by 10 μM 18β-GA, to 50±15 % (n=4) by 20 μM 18β-GA, and to 65±23 % (n=8) by 30 μM 18β-GA (P<0.05 for each). The ACh-induced dilatation of arterioles was also reduced significantly by 18β-GA in a reversible manner (Figure 4A). The inhibitory effects of 18β-GA on the ACh-induced dilatation increased in a concentration dependent manner between 10 and 30 μM, and the maximum inhibition of about half of control was elicited by 30 μM 18β-GA (Figure 4B).
Figure 4.
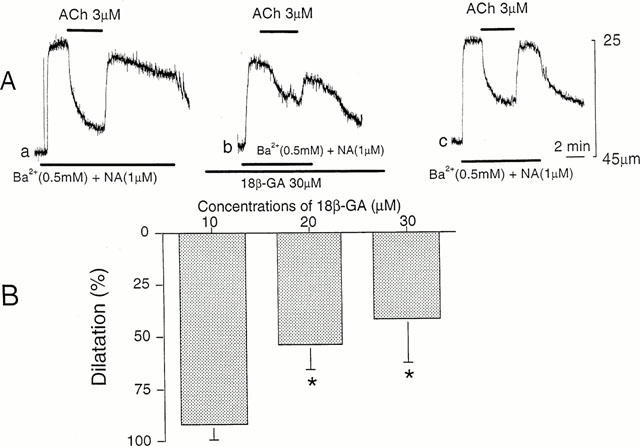
Effects of 18β-GA on ACh-induced dilatation. (A) In constricted arteriole with solution containing Ba2+ (0.5 mM) and NA (1 μM), ACh (3 μM) was applied for 2–3 min before (a), during presence of 18β-GA (30 μM) for 60 min (b) and after removal of 18β-GA for 60 min (c). All responses were recorded from the same arteriole. The resting diameter of arteriole, 43 μm. (B) Summary of the effects of 18β-GA on dilatation produced by ACh in submucosal arterioles. Each column indicates the per cent change of dilatation compared to contraction produced by solution containing Ba2+ (0.5 mM) and NA (1 μM). Mean±s.d. (n=4–10). *Significant reduction vs control (P<0.05).
Effects of 18β-GA on relaxation produced by NO donor
The results indicated that the inhibitory actions of 18β-GA appeared strongly on the ACh-induced hyperpolarization but they were weak on mechanical responses of submucosal arterioles. Attempts were made to observe the effects of 18β-GA on dilatation produced by sodium nitroprusside (SNP), an NO donor, on these arterioles. In the presence of Ba2+ and NA, the arteriolar dilatation produced by SNP (1 μM) was 48±5 % (n=7), and the value was reduced to 21±8 % (n=3) by 10 μM 18β-GA, to 23±7 % (n=4) by 20 μM 18β-GA, and to 18±15 % (n=7) by 30 μM 18β-GA. Although the inhibitory effects of 18β-GA on the SNP-induced dilatation were significant (P<0.05), there was no significant difference in the dilatation between different concentrations of 18β-GA (P>0.05).
Effects of 2APB on the ACh-induced hyperpolarization and relaxation
In the presence of Ba2+ (0.5 mM), membrane potentials ranged between −35 mV and −45 mV (mean, −40.8±4.1 mV, n=6), and 2-aminoethoxydiphenyl borate (2APB, 30 μM) depolarized the membrane to the range between −7 mV and −25 mV (mean, −17.7±7.6 mV, n=6, P<0.05). In the presence of 2APB, the ACh-induced hyperpolarization was changed to a transient form with reduced amplitude (control, 22.6±3.1 mV, n=5; in 2APB, 8.8±3.0 mV, n=5, P<0.05) (Figure 5A, b). The inhibitory effects of 2APB were reversible, with a complete recovery requiring 15–20 min (Figure 5A, c).
Figure 5.
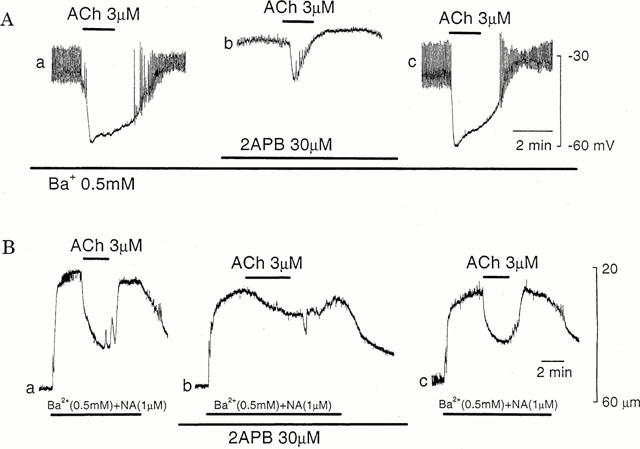
Effects of 2APB on ACh-induced hyperpolarization and relaxation. (A) In the presence of Ba2+ (0.5 mM), ACh (3 μM) was applied for 2 min before (a), during application of 2APB (30 μM) for 15 min (b), and after removal of 2APB for 40 min (c). All responses were recorded from the same cell. The diameter of arteriole, 50 μm. (B) In arterioles constricted with solution containing Ba2+ (0.5 mM) and NA (1 μM), ACh (3 μM), was applied for 2 min before (a), during presence of 2APB (30 μM) for 15 min (b) and after removal of 2APB for 60 min (c). All responses were recorded from the same arteriole. The resting diameter of arteriole, 56 μm.
At rest, 2APB (30 μM) did not produce any change in the arteriolar diameters (control, 45.5±11.7 μm, n=4; in 2APB, 44.8±11.0 μm, n=4, P>0.1). In the presence of 2APB, the amplitude of contraction produced by Ba2+ containing NA was reduced (control, 25.3±5.7 μm, n=4; in 2APB, 16.3±8.5 μm, n=4, P<0.05). 2APB also inhibited the amplitude of the ACh-induced dilatation (control, 76.3±18.5%, n=4; in 2APB, 35.0±10.8 %, n=4, P<0.05). The remarkable effects of 2APB also appeared on the shape of dilatation elicited by ACh, with the reduction in the rate of onset of the response (Figure 5B, b). The inhibitory effect of 2APB was reversible, with a complete recovery requiring 15–20 min (Figure 5B, c).
Discussion
In small arterioles, application of conventional microelectrode techniques often penetrate electrode to endothelial cells instead of smooth muscle cells (Emerson & Segal, 2000). This may be also the case in the present experiment, since submucosal arterioles of the guinea-pig possess only a single layer of smooth muscle cells (Hirst, 1977). Thus, although attempts were made to record membrane potentials from smooth muscle cells, it may be reasonable to understand that the responses are obtained from either endothelial or smooth muscle cells. However in these arterioles, both endothelial and smooth muscle cells may behave synchronously due to tight electrical connections with gap junctions (Yamamoto et al., 1998). Therefore, the data obtained may be applicable for the assessment of the relationship between membrane potential and dilatation in smooth muscles, in the absence of any functional alteration of myo-endothelial gap junctions. However, it is argued that the evaluation of the effects of the modulators of gap junctions such as 18β-GA requires additional histological confirmations.
In the submucosal arterioles, ACh did not produce any change in the membrane potential of smooth muscle, probably because of the large potential due to activation of inward rectifying K+-channels (Edwards & Hirst, 1988). As reported previously (Hashitani & Suzuki, 1997), the ACh-induced hyperpolarization was detected only after inhibiting the inward rectifying K+-channels with Ba2+. The hyperpolarization was inhibited by CTX and apamin, the results again confirming our previous observations (Hashitani & Suzuki, 1997). The K+-channels inhibited by CTX and apamin are activated by an elevation of intracellular Ca2+ (Nelson & Quayle, 1995). In the submucosal arterioles, ACh increases endothelial Ca2+ with no elevation of smooth muscle Ca2+ (Fukuta et al., 1999a). Thus, it is reasonable to speculate that the K+-channels activated by ACh are located in endothelial cells. However, it is also worth to note that the ACh-induced Ca2+-transient of endothelial cells does not change even after the hyperpolarization has been inhibited with CTX (Fukuta et al., 1999a). Similar suggestions could also be inferred from the effects of 2APB on the ACh-induced responses. 2APB inhibits InsP3-receptor mediated Ca2+ release (Maruyama et al., 1997; Cui et al., 1997). ACh increases endothelial Ca2+ by accelerated production of InsP3 (Schilling & Elliot, 1992). Thus, it seems likely that, in the submucosal arterioles, ACh induces detectable responses only in endothelial cells and these are conducted to smooth muscle cells electrotonically through gap junctions.
The present experiments show that in submucosal arterioles, inhibition by CTX or apamin of the ACh-induced hyperpolarization is associated with the reduction of dilatation. The experiments are carried out in the presence of L-NOARG and diclofenac sodium, suggesting that the possible mediator for the ACh-induced dilatation is EDHF. Thus, arteriolar dilatation has a causal relationship with hyperpolarization of the membrane, as is the case with large arteries (Chen & Suzuki, 1989; Suzuki & Chen, 1990; Garland et al., 1995). The ACh-induced hyperpolarization was also abolished by 18β-GA, probably by inhibiting gap junctions between endothelial and smooth muscle cells (Yamamoto et al., 1998). However, 18β-GA inhibited the ACh-induced dilatation of arterioles partially. Thus, there was a discrepancy in the relationship between membrane potential and dilatation of arterioles when the ACh-induced hyperpolarization was inhibited by 18β-GA.
The endothelial membrane possesses no voltage-activated Ca2+ channels, and influx of Ca2+ may be facilitated when potential differences across the membrane is high (Laskey et al., 1990; Schlling & Elliott, 1992). CTX inhibits hyperpolarization of endothelial cells (Yamamoto et al., 1998), and this will weaken the driving force for Ca2+ influx in the cell. On the other hand, inhibition of intercellular gap junctions by 18β-GA does not prevent the ACh-induced hyperpolarization in endothelial cells (Yamamoto et al., 1999), suggesting that the driving force for influx of Ca2+ remains unaltered in the presence of 18β-GA. Thus, the inhibition of the ACh-induced hyperpolarization of arterial smooth muscle membrane is produced by different mechanisms between K+ channel inhibitors and 18β-GA. In the present study, 2APB inhibited the ACh-induced hyperpolarization and dilatation to a similar extent. This suggests that the endothelial Ca2+ supplied from the internal store directly relate to the dilatation of arterioles. Many endothelial vasodilators require elevated endothelial Ca2+ for the release (Suzuki & Chen, 1990), and this is confirmed by the inhibitory effect of BAPTA on the ACh-induced hyperpolarization in the submucosal arterioles (Hashitani & Suzuki, 1997). If this is the case, the ACh-induced dilatation of submucosal arterioles in the presence of 18β-GA is produced by unidentified substance (or mechanisms) independent of membrane hyperpolarization. Alternatively, hyperpolarization propagated from endothelial cells through gap junctions may only be partially responsible for the ACh-induced dilatation.
In the submucosal arterioles, depolarization of the membrane and resulting constriction with Ba2+ was required to observe the ACh-induced hyperpolarization and dilatation. Similar or greater amplitude of depolarization was also produced by 18β-GA, irrespective of the presence or absence of Ba2+. However, 18β-GA alone could not induce arteriolar constriction. Thus, there is a discrepancy between membrane depolarization and constriction in submucosal arterioles. The Ba2+-induced depolarization may be produced by inhibition of inward rectifying K+-channels (Edwards & Hirst, 1988; Nelson & Quayle, 1995). It remains unclear why the membrane is depolarized by the inhibition of gap junctions with 18β-GA. This was accompanied by an increase in electrotonic potentials produced by inward current pulse, indicating that the input resistance of smooth muscle membrane was increased, presumably due to isolation of the recording cell from surrounding cells. In the rabbit conduit arteries, Gap 27, a specific inhibitory gap junction peptide, does not attenuate the contraction produced by phenylephrine (Chaytor et al., 1998). Thus, an alternative proposal is that the depolarization produced by 18β-GA is not directly related to inhibition of gap junctions.
Inhibitory actions of 18β-GA also appeared on the mechanical responses of submucosal arterioles, such as an inhibition of constriction produced by Ba2+ and NA or of dilatation produced by SNP. The ACh-induced dilatation of submucosal arterioles may not involve endothelial NO, since the dilatation is not altered in the presence of L-NOARG. However, SNP was a potent dilator of these arterioles, suggesting that smooth muscles of these arterioles posses an ability to produce dilatation through NO-cyclic GMP pathways. Inhibition by 18β-GA of the SNP-induced dilatation suggests that this saponin derivative has multiple inhibitory actions on arteriolar tissues, in addition to the inhibition of gap junctions. Similar multiple actions of 18β-GA including inhibition of the relaxation produced by K+-channel opener has also been reported in aorta from the guinea-pig (Fukuta et al., 1999b). However, water-soluble derivative of 18β-GA, carbenoxolone, does not prevent hyperpolarization and dilatation produced by K-channel opener in the hepatic and mesenteric arteries of rat (Edwards et al., 1999).
Although hyperpolarization of the membrane is considered one of the important factors to cause dilatation in vascular smooth muscles (Kuriyama et al., 1995), the underlying cellular mechanisms are controversial. Experiments using K+-channel openers suggest that there are at least three mechanisms involved in the hyperpolarization-induced relaxation of vascular smooth muscle. Hyperpolarization of the membrane inhibits Ca2+ influx through voltage-dependent Ca2+-channels in vascular smooth muscles (Nelson et al., 1990). The production of second messengers such as InsP3 is reduced during hyperpolarization, thus causing the reduction of Ca2+ release from the internal stores in the rabbit mesenteric artery (Itoh et al., 1992). Reduction by K+-channel opener of the sensitivity of contractile protein to Ca2+ is also suggested in the canine coronary artery (Okada et al., 1993). However, it remains unclear which of any of these mechanisms are involved in the EDHF-induced vasodilatation.
It is concluded that, in guinea-pig submucosal arterioles, inhibition of the ACh-induced hyperpolarization by K+-channel blockade or Ca2+-release blockade is causally related to the inhibition of vasodilatation. The inhibition of intercellular gap junctions by 18β-GA also attenuates hyperpolarization and dilatation responses produced by ACh, but the effects are much weaker on mechanical responses than on electrical responses. The effects of 18β-GA seem to involve several unidentified factors, in addition to the inhibition of gap junctions.
Acknowledgments
The authors were grateful to Professor G.D.S. Hirst, The University of Melbourne, and Dr T.O. Neild, The Flinders University, for DIAMTRAK facility. The experiments were supported by the grants from the Ministry of Education, Sports and Culture of Japan to H. Suzuki (09470011) and the Uehara Memorial Foundation. 2APB was kindly supplied from Dr Takayuki Maruyama (Minase Research Institute, Ono Pharmacutical Comp., Osaka). Dr T. Kanno (Yanaihara Institute, Shizuoka) kindly helped for facilities of 2APB.
Abbreviations
- ACh
acetylcholine
- 2APB
2-aminoethoxydiphenyl borate
- CTX
charybdotoxin
- EDHF
endothelium-derived hyperpolarizing factor
- EDRF
endothelium-derived relaxing factor
- 18β-GA
18β-glycyrrhetinic acid
- InsP3
inositol trisphosphate
- L-NOARG
L-Nω-nitroarginine
- NA
noradrenaline
- NO
nitric oxide
- SNP
sodium nitroprusside
- TTX
tetrodotoxin
References
- CAMBELL W.G., GEBREMEDHIN D., PRATT P.F, HARDER D.R. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- CHAYTOR A.Y., EVANS W.H, GRIFFITH T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998;508:561–573. doi: 10.1111/j.1469-7793.1998.561bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G, SUZUKI H. Some electrical properties of the endothelium-dependent hyperpolarization recorded from rat arterial smooth muscle cells. J. Physiol. 1989;410:91–106. doi: 10.1113/jphysiol.1989.sp017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H, WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUI Z.J, KANNO T. Photodynamic triggering of calcium oscillation in the isolated rat pancreatic acini. J. Physiol. 1997;504:47–55. doi: 10.1111/j.1469-7793.1997.047bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J, WESTON A.H. K+ is an endothelium-derived hyperporarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., FELETOU M., GARLAND M.J., THOLLON C., VANHOUTTE P.M, WESTON A.H. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS F.R, HIRST G.D.S. Inward rectification in submucosal arterioles of guinea-pig ileum. J. Physiol. 1988;404:437–454. doi: 10.1113/jphysiol.1988.sp017298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMERSON G.G, SEGAL S.S. Endothelial cell pathway for conduction of hyperpolarization and vasodilatation along hamster feed artery. Circ. Res. 2000;86:94–100. doi: 10.1161/01.res.86.1.94. [DOI] [PubMed] [Google Scholar]
- FUKUTA H., HASHITANI H., YAMAMOTO Y, SUZUKI H. Calcium responses induced by acetylcholine in submucosal arterioles of the guinea-pig small intestine. J. Physiol. 1999a;515:489–499. doi: 10.1111/j.1469-7793.1999.489ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FUKUTA H., KOSHITA M., YAMAMOTO Y, SUZUKI H. Inhibition of the endothelium-dependent relaxation by 18β-glycyrrhetinic acid in the guinea-pig aorta. Jpn. J. Physiol. 1999b;49:267–274. doi: 10.2170/jjphysiol.49.267. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F, ZAWADZSKI J.V. The obligatory role of the endothelial cells in the relaxation of arterial smooth muscle cells by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K, COCKS T.M. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trend. Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- HASHITANI H, SUZUKI H. K+ channels which contribute to the acetylcholine-induced hyperpolarization in smooth muscle of the guinea-pig submucosal arteriole. J. Physiol. 1997;501:319–329. doi: 10.1111/j.1469-7793.1997.319bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRST G.D.S. Neuromuscular transmission in arterioles of guinea-pig submucosa. J. Physiol. 1977;273:263–275. doi: 10.1113/jphysiol.1977.sp012093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRST G.D.S, NEILD T.O. Some properties of spontaneous excitatory junctional potentials recorded from arterioles guinea-pig. J. Physiol. 1980;303:43–60. doi: 10.1113/jphysiol.1980.sp013269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITOH T., SEKI N., SUZUKI S., ITO S., KAJIKURI J, KURIYAMA H. Membrane hyperpolarization inhibits agonist-induced synthesis of inositol 1,4,5-trisphosphate in rabbit mesenteric artery. J. Physiol. 1992;451:307–328. doi: 10.1113/jphysiol.1992.sp019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KURIYAMA H., KITAMURA K, NABATA H. Pharmacological and physiological significance of ion channels and factors that modulate them in vascular tissues. Pharmacol. Rev. 1995;47:387–573. [PubMed] [Google Scholar]
- LASKEY R.E., ADAMS D.J., JOHNS A., RUBANYI G.M, VAN BREEMEN C.Regulation of [Ca2+]i in endocardial cells by membrane potential Endothelium-Derived Relaxing Factors 1990Basel: Karger; 128–135.ed. Rubanyi, G.M. & Vanhoutte, P. M. pp [Google Scholar]
- MARUYAMA T., KANAJI T., NAKADE S., KANNO T, MIKOSHIBA K. 2APB, 2-aminoethoxdiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J, HIGGS E.A. Nitric oxide: physiology pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- NEILD T.O. Measurement of arteriole diameter changes by analysis of television images. Blood Vessels. 1989;26:48–52. [PubMed] [Google Scholar]
- NEILD T.O., SHEN K.-Z, SURPRENANT A.M. Vasodilatation of arterioles by acetylcholine released from single neuron in the guinea-pig submucosal plexus. J. Physiol. 1990;420:247–265. doi: 10.1113/jphysiol.1990.sp017910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON M.T., PATLAK J.B., WORLEY J.F, STANDEN N.B. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am. J. Physiol. 1990;259:C3–C18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- NELSON M.T, QUAYLE J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- OKADA Y., YANAGISAWA T, TAIRA N. BRL 38227 (levcromakalim)-induced hyperpolarization reduces the sensitivity to Ca2+ of contractile elements in canine coronary artery. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;347:438–444. doi: 10.1007/BF00165396. [DOI] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., HECKER M., FLEMING I, BUSSE R. A transferable β -naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J. Physiol. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANDALL M.D, KENDALL D.A. Involvement of a cannabinoid in endothelium-derived hyperpolarizing factor-mediated coronary vasorelaxation. Eur. J. Pharmacol. 1997;335:205–209. doi: 10.1016/s0014-2999(97)01237-5. [DOI] [PubMed] [Google Scholar]
- SCHILLING W.P, ELLIOTT S.J. Ca2+ signaling mechanisms of vascular endothelial cells and their role in oxidant-induced endothelial dysfunction. Am. J. Physiol. 1992;262:H1617–H1630. doi: 10.1152/ajpheart.1992.262.6.H1617. [DOI] [PubMed] [Google Scholar]
- SHIMOKAWA H., YASUTAKE H., FUJII K., OWADA M.K., NAKAIKE R., FUKUMOTO Y., TAKAYANAGI T., NAGAO T., EGASHIRA K., FUJISHIMA M, TAKESHITA A. The importance of the hyperpolarization mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J. Cardiovasc. Pharmacol. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- SUZUKI H, CHEN G. Endothelium-derived hyperpolarizing factor (EDHF): an endogenous potassium-channel activator. News Physiol. Sci. 1990;5:212–215. [Google Scholar]
- YAMAMOTO Y., FUKUTA H., NAKAHIRA Y, SUZUKI H. Blockade by 18β-glycyrrhetinic acid of intercellular electrical coupling in guinea-pig arterioles. J. Physiol. 1998;511:501–508. doi: 10.1111/j.1469-7793.1998.501bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAMOTO Y., IMAEDA K, SUZUKI H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999;514:505–513. doi: 10.1111/j.1469-7793.1999.505ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]