

Abstract
Since the role of mechanical stretches in vascular tone regulation is poorly understood, we studied how stretch can influence endothelial tone.
Isometric contractions of isolated rat aortic helical strips were recorded. The resting tension was set at 0.7 g, 1.2 g or 2.5 g. Endothelium-preserved strips were precontracted with either phenylephrine or prostaglandin F2α (PGF2α).
In control conditions, acetylcholine (ACh) dose-dependently relaxed phenylephrine-precontracted strips independently of resting tension.
At 0.7 g resting tension, nitric oxide synthase (NOS) inhibitors did not reduce ACh-induced relaxation, while either a guanylyl cyclase inhibitor or a NO trapping agent prevented it. At 1.2 g and 2.5 g resting tensions, NOS inhibitors shifted the ACh dose-response curve to the right.
After preincubation with indomethacin (5 μM) or ibuprofen (10 and 100 μM), at 0.7 g and 1.2 g resting tensions, ACh induced an endothelium-dependent, dose-dependent contraction. ACh (10−6 M) increased the contraction up to two times greater the phenylephrine-induced one. Lipoxygenase inhibitors prevented it. At high stretch, the ACh vasorelaxant effect was marginally influenced by cyclooxygenase (COX) inhibition. Similar results were obtained when aortic strips were precontracted with PGF2α.
Our data indicate that when resting tension is low, ACh mobilizes a stored NO pool that, synergistically with COX-derived metabolites, can relax precontracted strips. COX inhibition up-regulates the lipoxygenase metabolic pathway, accounting for the ACh contractile effect. At an intermediate resting tension, NO production is present, but COX inhibition reveals a lipoxygenase-dependent, ACh-induced contraction. At high resting tension, NO synthesis predominates and COX metabolites influence ACh-induced relaxation marginally.
Keywords: rat aortic strips, resting tension, stored nitric oxide, lipoxygenase metabolite-induced contraction, nitric oxide synthase inhibitors, guanylyl cyclase inhibitor, nitric oxide trapping agent
Introduction
At least three different endothelial-derived relaxant factors have been thus far identified: prostacyclin (PGI2), nitric oxide (NO), and the endothelial-derived hyperpolarizing factor (EDHF). PGI2 is the main metabolite of cyclooxygenase (COX) in endothelial cells and its role as an anti-aggregating agent is well defined (Moncada & Vane, 1979). It seems, however, to affect vascular relaxation less, since in the presence of indomethacin, endothelial cell activation can still relax precontracted vascular preparations (Furchgott & Zawadzki, 1980). NO is produced by endothelial constitutive NO synthase (ecNOS) from L-arginine (Palmer et al., 1988). Throughout guanylyl cyclase activation, guanosine 3′:5′-cyclic monophosphate (cyclic GMP) increase and cyclic GMP-dependent protein kinase stimulation, NO can induce smooth muscle cell relaxation (Cornwell et al., 1991). EDHF has been identified as a cytochrome P450 metabolite of arachidonic acid (Fisslthaler et al., 1999), which, by activation of potassium channels, can induce hyperpolarization of vascular smooth muscle cell membrane and artery relaxation (Campbell & Harder, 1999).
Depending on the experimental and pathophysiological conditions, all these compounds can contribute to endothelial-dependent vasorelaxation.
Moreover, endothelial cells also produce contractile substances. Contracting factors include lipid-derived eicosanoids, such as arachidonic acid and lipoxygenase metabolites (Ezra et al., 1983; Rimele & Vanhoutte, 1983; Berkowitz et al., 1984; Marsault et al., 1997; Filipeanu et al., 1998). Leukotrienes are also produced by endothelial cells in culture (Kähler et al., 1993; Hollenberg et al., 1994). Therefore, from common precursor arachidonic acid, endothelial cells may produce either vasorelaxant factors or vasocontracting substances according to the prevalence of the different metabolic pathway.
Among the physiological regulators of endothelial-derived vasoactive substance production, shear stress and cell stretch may be of primary relevance. However, the influence of cell stretch on endothelial-derived vasoactive substance production and release is still partially unclear.
The effect of stretch has been investigated by Dainty et al. (1990) by changing either the resting length or the resting tension of rat aortic preparations in vitro. The authors show that the initial length of preparation (and hence stretch degrees) modifies the contractile response to phenylephrine. The decrease in tone induced by 1 μM acetylcholine (ACh), is indeed dependent on the initial stretch of endothelial-intact preparations. Although the authors focused their analysis on result variability from different laboratories, it emerged that variations in arterial tone and stretch may influence the response of smooth muscle cells to endothelial-derived vasoactive factors.
In coronary preparations from guinea-pigs and rats, stretch influences the relative contribution of endothelial-derived vasorelaxant substances to smooth muscle cell hyperpolarization (Parkington et al., 1993). Indeed, in unstretched guinea-pig coronary arteries, the hyperpolarization of smooth muscle cells induced by ACh is only slightly reduced by co-incubation with the ecNOS inhibitor ω-nitro-L-arginine methylester (L-NAME) and indomethacin. Indeed, exogenous NO and PGI2 are without effect. In contrast, in stretched preparations, exogenous NO and PGI2 induce hyperpolarization and co-incubation with L-NAME, and indomethacin abolishes the slow component of ACh-induced hyperpolarization. Delayed hyperpolarization is also induced by arachidonic acid, and indomethacin is able to antagonize its effect, suggesting that a metabolite of arachidonic acid may account for this effect. Similar results are also obtained in rat coronary arteries, although PGI2 is unable to induce hyperpolarization (Parkington et al., 1993). These data indicate that the contribution of different vasorelaxant products may vary according to the resting tension.
Since the role of stretch in endothelial-derived vascular tone regulation has been only partially explored, we decided to investigate the ACh effect on contractile tone in rat aortic strips stretched at different resting tensions. Our data indicate that, according to stretch, the relative contribution of NO and arachidonic acid metabolites on ACh-induced relaxation varies. At low stretch, ACh can either relax or contract aortic preparations, considering that relaxation is obtained when COX products are present and contraction is obtained when COX is inhibited. Moreover, in this condition a stored, depletable NO pool is released independently of ecNOS inhibition. At high stretch, NO relaxation predominates and COX inhibition does not influence the ACh-induced relaxation.
Methods
This investigation conforms to European Community rules for the care and use of laboratory animals.
Microscopy
Helical preparations of rat thoracic aorta were fixed by immersion in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 6 h at room temperature, postfixed in 1% OsO4 in 0.1 M phosphate buffer (pH 7.4) dehydrated in acetone series, passed through propylene oxide, and embedded in epoxy resin (Epon 812, Fluka, Buchs, Switzerland). Semithin sections, 2 μm thick, were cut with an ultramicrotome, stained with toluidine blue-sodium tetraborate, viewed and photographed with a light microscope. Semithin sections were used because they allow for a clear-cut identification of the components of the arterial tunica intima, especially the endothelium.
Functional studies
Male Wistar rats (300–350 g) were anaesthetized and killed by cervical dislocation. Thoracic aortas between the aortic arch and diaphragm were isolated and placed in cold gassed (5% CO2 in O2) Krebs-Henseleit solution (K-H) of the following composition (mM): NaCl 110, NaHCO3 25, KCl 4.8, KH2PO4 1.2, MgSO4 1.2, D(+)glucose 11, CaCl2 2.5. The aorta was cleaned from adhering fat and connective tissue and cut into helical strips (3 mm width, 20 mm length). The strips were mounted in an organ bath (2 ml) with one end connected to a tissue holder and the other to a force transducer (Battaglia-Rangoni), then the strips were perfused with pre-warmed (37°) K-H, gassed with 5% CO2 in O2. The resting tension was set at 0.7, 1.2 or 2.5 g and the tissue was equilibrated for 60 min. These resting tensions were arbitrarily chosen in the range in which phenylephrine, prostaglandin F2α (PGF2α) and ACh induced, in control conditions, a similar dose-response curve (see below). Isometric contractions were recorded. After equilibration, dose-response curves to phenylephrine or PGF2α were performed and the maximal contraction obtained was expressed as 100% of contractile response. Using the maximal contracting dose of phenylephrine or PGF2α (100%), the cumulative dose-response curve of ACh was done. In several experiments, after extensive washing in the absence (control) or presence of ecNOS inhibitors, 10−7 M of phenylephrine was reapplied and the ACh dose-response curve repeated. The complete procedure was reproduced several times until a superimposable ACh dose-response curve was obtained. All dose-response curves were performed by administering drugs as a bolus injection. In each preparation, the presence of a functional endothelial lining was demonstrated in control conditions by a near-complete relaxation to ACh of precontracted strips. To further assess that the manipulation of aortic preparations had not caused a loss of the endothelial layer, light microscopic examination was carried out on aortic helical strips prepared as for the functional experiments. The relaxation or contraction induced by ACh was expressed as a decrease or an increase in the contractile force from the maximal contraction. Experiments were mostly performed on endothelium-intact preparations. In several cases, the endothelium was removed by rubbing the cells with cotton gauze, and the absence of endothelium was determined as described before, where ACh was unable to induce relaxation.
6-keto prostaglandin F1α (6-keto PGF1α) release
6-keto PGF1α was measured, using a commercial radioimmunoassay kit (Amersham Pharmacia Biotech, Buckinghamshire, U.K.), in the lyophilized perfusion medium obtained by closing the organ bath for 4 min. Standard concentrations of 6-keto PGF1α were also dissolved in K-H and lyophilized analogously to unknown samples. Determinations were carried out according to the instructions provided by the manufacturer.
Materials
Acetylcholine (ACh), phenylephrine, prostaglandin F2α (PGF2α), indomethacin, ibuprofen sodium salt, 5,8,11-triynoic acid (ETI), 2-phenyl-4,4,5-tetramethyl-imidazoline-1-oxyl-3-oxide (PTIO), ω-monomethyl-L-arginine monoacetate salt (L-NMMA) and L-N5-(1-iminoethyl)ornithine dihydrochloride (L-NIO) were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.), ω-nitro-L-arginine methylester (L-NAME), 6-anilino-5,8-quinolinequinone (LY83583) were purchased from Calbiochem (La Jolla, CA, U.S.A.). All other reagents were of analytical grade. 3-(1-(4-chlorobenzyl)-3t-butyl-thio-5-isopropylindol-2-yl)-2,2-dimethylpropanoic acid (MK-886) was the kind gift of Merck Sharp & Dohme, Italy (Rome).
Mathematical and statistical methods
Statistical comparisons between data groups were performed using paired Student's t-test. Median effective concentrations (EC50s) were calculated using the Implot (version4) program (GraphPad Software, San Diego, CA, U.S.A.). A P value <0.05 was considered significant.
Results
Morphology
Histological examination of semithin sections of aortic strips revealed the presence of large tracts of the aortic wall provided with a continuous layer of endothelial cells (not shown).
Functional studies
Phenylephrine caused a rapid onset, dose-dependent contraction of aortic strips independent of the resting tension; therefore, as shown in Figure 1, the dose-response curves were superimposable, obtaining maximal contraction at 10−7 M phenylephrine. Hence, in all experiments 10−7 M phenylephrine was used to precontract aortic strips.
Figure 1.
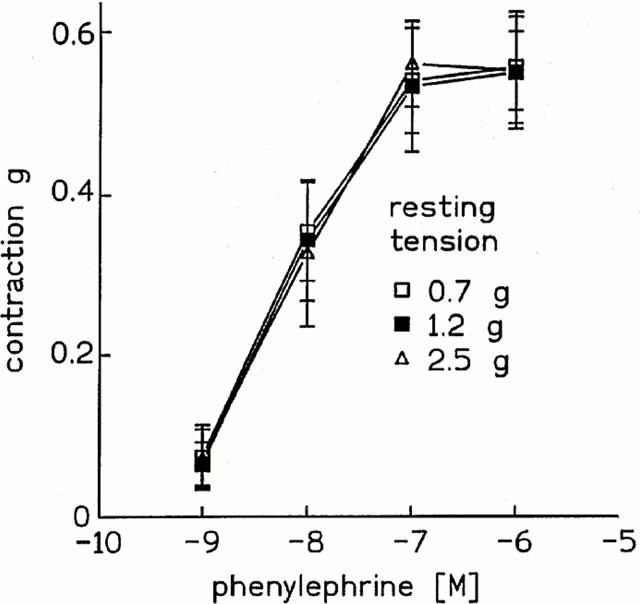
Effect of phenylephrine on isolated rat thoracic aortic strips. Cumulative dose-response curves of phenylephrine were performed at 0.7, 1.2 and 2.5 g resting tension. Values are the mean±s.e.mean of at least four experiments.
In phenylephrine precontracted aortic strips, administration of ACh induced a dose-dependent relaxation (Figure 2, panels A and B), 10−6 M ACh being sufficient to induce a complete relaxation of endothelium-bearing preparations. As predicted, in the endothelial-denuded preparations, ACh was ineffective independent of applied resting tension, while Na nitroprusside was still able to relax aortic strips (not shown). No differences in the dose-response curve of ACh were observed in relation to resting tension (Table 1). Moreover, in the presence of an intact endothelium, the Na nitroprusside relaxant effect was also not different in preparations stretched at 0.7 g and 2.5 g (EC50s: 44.1±4.01 and 41.8±3.80 nM respectively at 0.7 g and 2.5 g).
Figure 2.
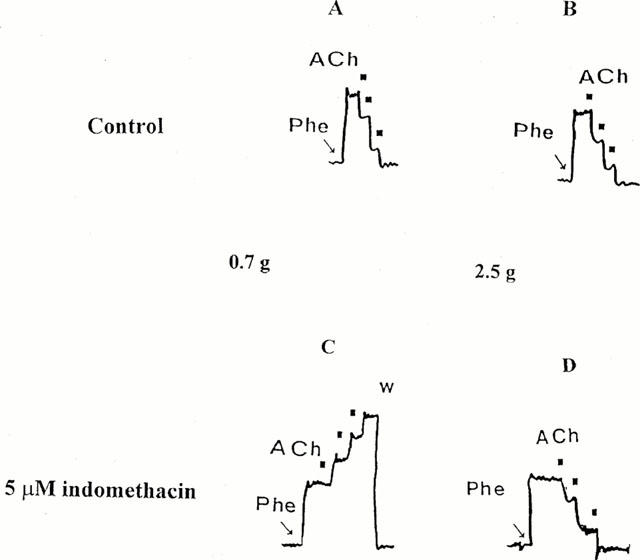
Effect of ACh on phenylephrine-precontracted rat aortic strips. Cumulative dose-response curves of ACh were performed in control conditions (A and B) and in the presence of 5 μM indomethacin (C and D). Aortic strips were stretched at a resting tension of 0.7 g (A and C panels) and 2.5 g (B and D) and precontracted with 10−7 M phenylephrine (Phe). Black squares indicate ACh (10−7–3×10−7–10−6 M) administration. Typical traces are shown. W=washing.
Table 1.
Acetylcholine median effective concentrations (EC50s) that induce relaxation in 10−7 M phenylephrine-precontracted rat aortic strips

The role of NO-mediated relaxation dependent on the activation of ecNOS at the different tensions was tested by inhibiting the enzyme with 100 μM L-NAME (Figure 3). At the lower resting tension, L-NAME (100 μM, 30 min preincubation) did not reduce the endothelium-dependent vasorelaxation induced by ACh (Figure 3A) and ACh EC50s were not significantly different (Table 1). Similar results were obtained by preincubating aortic strips with 200 μM L-NAME for 60 min.
Figure 3.
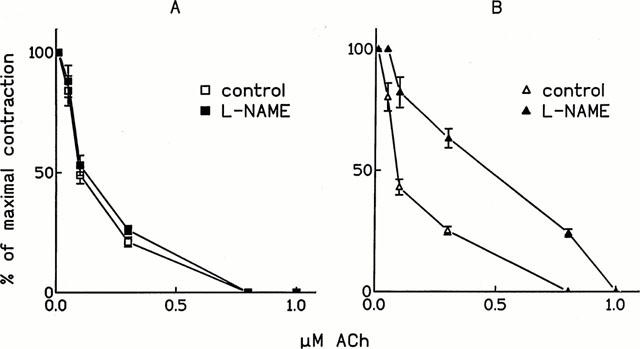
Effect of L-NAME on ACh-induced relaxation. Cumulative dose-response curves of ACh either in control conditions or after 30 min preincubation with 100 μM L-NAME were performed on 10−7 M phenylephrine-precontracted rat thoracic aortic strips stretched at different resting tensions (A: 0.7 g and B: 2.5 g). The contraction obtained after phenylephrine is set as 100%. Values are the mean±s.e.mean of at least four experiments.
We also tested the effect of two other ecNOS inhibitors, namely L-NMMA (100 μM, 60 min preincubation) and L-NIO (25 μM, 60 min preincubation). As reported in Table 2, at low resting tension ACh EC50s were not significantly different when they were calculated on the 1st curve performed in the presence of the indicated ecNOS inhibitor. However, by repeating ACh dose-response curves several times on the same preparation maintained in a continuous perfusion of ecNOS inhibitors, the ACh relaxant effect decreased and on the 4th curve the ACh EC50s were up to 4 fold higher than those calculated on the 1st curve (Table 2).
Table 2.
Acetylcholine median effective concentrations (EC50s) that relax phenylephrine-precontracted rat aortic strips calculated on the 1st and 4th repeated curve

When resting tension was set at 2.5 g, the preincubation with 100 μM L-NAME for 30 min shifted the ACh dose-response curve to the right (Figure 3B), as demonstrated by the increase in ACh EC50s (Table 1). Similarly, the ACh dose-response curve was shifted to the right after pre-incubation with either 100 μM L-NMMA or 25 μM L-NIO (Table 2). By repeating ACh dose-response curves several times on the same preparation maintained in the constant presence of ecNOS inhibitors, ACh, EC50s were increased, although these increases were of a lesser extent than those observed at a lower resting tension (Table 2).
At an intermediate resting tension (1.2 g), the ACh EC50 calculated in the presence of 100 μM L-NAME was significantly increased from 0.25±0.015 (control) to 0.49±0.012 μM (P<0.001 vs control).
In order to better investigate the role of NO in the vasorelaxant effect of ACh, we performed experiments using either a guanylyl cyclase inhibitor (LY83583) or a NO trapping agent (PTIO). As shown in Figure 4, the inhibition of guanylyl cyclase or the trapping of NO prevented the vasorelaxant effect of ACh. LY83583 (10 μM) totally abolished the vasorelaxation induced by ACh, whereas 10 μM PTIO blocked it by 96%. At high resting tension, the ACh dose-response curve performed in the presence of L-NAME was not modified by co-incubation with either 10 μM PTIO or 10 μM LY83583.
Figure 4.
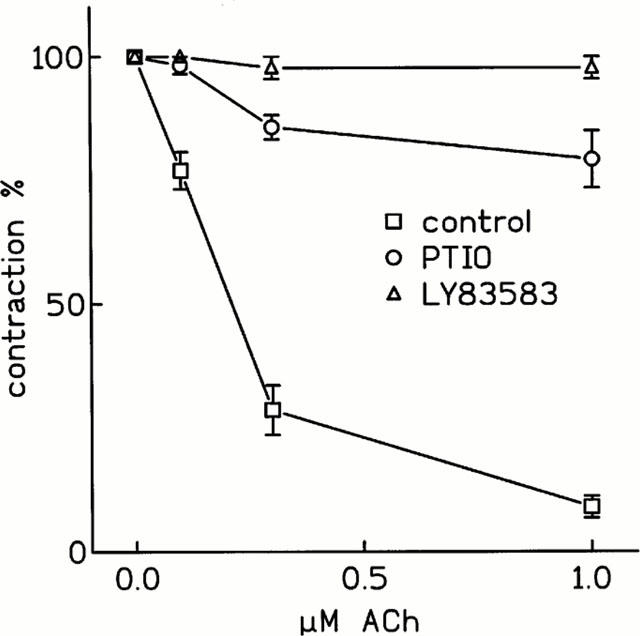
Effect of LY83583 and PTIO on ACh-induced relaxation. Cumulative dose-response curves of ACh in control conditions, after 30 min preincubation with either 10 μM LY83583 or 10 μM PTIO were performed on 10−7 M phenylephrine-precontracted rat thoracic aortic strips stretched at 0.7 g resting tension. The contraction obtained after phenylephrine is set as 100%. Values are the mean±s.e.mean of at least four experiments.
The role of COX metabolites in the vasorelaxation induced by ACh was then investigated by blocking COX isoenzymes with 5 μM indomethacin. In these conditions (pre-incubation with 5 μM indomethacin for 30 min), the contractile effect of phenylephrine was not modified at the assayed resting tensions. ACh dose-dependently relaxed phenylephrine-precontracted aortic strips only when resting tension was set at 2.5 g (Figure 2D). At the lower tension, ACh induced a strong, long-lasting dose-dependent contraction (Figure 2C). This increase in contraction was up to 2 fold the phenylephrine-induced one at 10−6 M ACh. Simultaneous incubation with 5 μM indomethacin and 100 μM L-NAME did not modify the contractile dose-response curve of ACh. When preparations were stretched at 0.7 g and incubated with ibuprofen (200 and 10 μM), another well known COX-inhibitor avoiding lipoxygenase cross-inhibition, 10−6 M ACh induced a dose-dependent vasoconstriction. In this condition, 10−6 M ACh induced an increase in contraction twice as much as phenylephrine alone (206±19.3% and 203±6.2% in the presence of 200 and 10 μM ibuprofen respectively). When aortic strips were stretched at an intermediate resting tension (1.2 g) and in the presence of 5 μM indomethacin, ACh also induced a strong contraction; 10−6 M ACh was able to increase up to twice as much as phenylephrine alone (197±22%). The ACh contractile effect observed in the presence of COX inhibitors was, however, absent when endothelium was mechanically removed. In the absence of endothelium, 10−4 M ACh was also unable to induce contraction or relaxation in either the presence or absence of indomethacin.
When aortic strips were precontracted with the maximal contracting dose of PGF2α (10−5 M), ACh induced a dose-dependent relaxation independent of resting tension (Figure 5A and B). When preparations were stretched at 0.7 g and preincubated with 5 μM indomethacin, ACh induced a dose-dependent contraction (Figure 5C), while when resting tension was at 2.5 g, ACh also induced relaxation in the presence of indomethacin.
Figure 5.
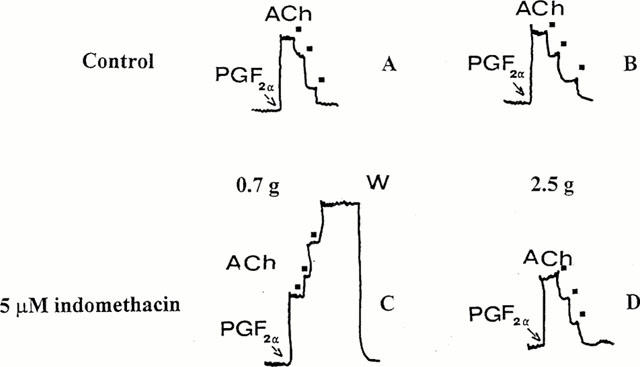
Effect of ACh on PGF2α-precontracted rat aortic strips. Cumulative dose-response curves of ACh were performed in control conditions (A and B) and in the presence of 5 μM indomethacin (C and D). Aortic strips were stretched at a resting tension of 0.7 g (A and C) and 2.5 g (B and D) and precontracted with 10−5 M PGF2α (PGF2α). Black squares indicate ACh (10−7–3×10−7–10−6 M) administration. Typical traces are shown. W=washing.
In order to explore the nature of contractile agents released by endothelium in the presence of 5 μM indomethacin at 0.7 g resting tension, the effects of two lipoxygenase inhibitors were tested. Both ETI (20 μM) and MK-886 (1 μM) antagonized the contractile effect of 10−6 M ACh (Table 3), but did not restore its relaxant effect. Similarly, at a very high dose, indomethacin (100 μM) prevented the contractile effect of 10−6 M ACh observed in its presence at a low dosage (5 μM indomethacin), and the administration of ACh did not modify the contracting tone induced by phenylephrine. Moreover, since the increase in contraction may be dependent on the calcium influx through the L-type calcium channel, we tested the effect of the L-type calcium channel blocker verapamil (1 μM) on the extra contraction induced by ACh in the presence of indomethacin. While the contractile effect of phenylephrine was not influenced by 1 μM verapamil, it prevented the contractile effect of ACh (Table 3). Lipoxygenase inhibitors also blocked the ACh-induced extra contraction when resting tension was 1.2 g.
Table 3.
Effect of 10−6 M acetylcholine on rat aortic strips preincubated with 5 μM indomethacin in the absence (none) or presence of lipoxygenase inhibitors or verapamil

In a PGF2α precontracted preparation, preincubation with 100 μM indomethacin also abolished the extra contraction induced by ACh at 0.7 g resting tension.
We then evaluated the effect of a simultaneous inhibition of COX and NO-dependent relaxation. When resting tension was set to 0.7 g, and 10 μM PTIO and 5 μM indomethacin were simultaneously perfused, the ACh contractile dose-response curve was only marginally modified, having ACh EC50s not significantly different (0.41±0.13 μM, indomethacin alone vs 0.35±0.14 μM, indomethacin plus PTIO). When resting tension was set to 1.2 g, the ACh dose-response curve was shifted to the left by pre-incubating with 5 μM indomethacin and 100 μM L-NAME; however, 3×10−7 M ACh induced the maximal contraction. When resting tension was set to 2.5 g, the simultaneous perfusion of 5 μM indomethacin and 100 μM L-NAME totally prevented the relaxant effect of ACh. Still, ACh did not induce a significant increase in contractions (not shown).
PGI2 release
We also measured the release of PGI2 in the organ bath by measuring its main metabolite 6-keto PGF1α at the lower resting tension. As shown in Figure 6, after the administration of phenylephrine, a significant increase in PGI2 was observed. The successive administration of ACh was unable to further increase the release of PGI2 in the organ bath. After washing ACh and phenylephrine, the same aortic strips were preincubated with 5 μM indomethacin. As predicted, indomethacin strongly reduced the production of PGI2. Nor the consecutive administration of phenylephrine and ACh, nor preincubation with ETI restored the release measured in control conditions.
Figure 6.
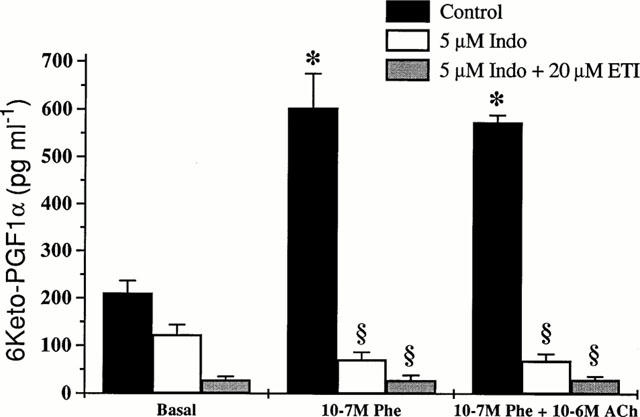
Release of 6-keto PGF1α from isolated rat aortic strips. 6-keto PGF1α was measured by RIA either under basal conditions, after administration of 10−7 M phenylephrine or after administration of 10−7 M phenylephrine and 10−6 M ACh on the supernatant obtained from the same isolated rat thoracic aortic strips preloaded at 0.7 g. Basal value indicates the level of 6-keto PGF1α after reaching a steady-state tension in each experimental condition. For more details, see Methods. Values are the mean±s.e.mean of four experiments. *P<0.001 vs basal conditions; §P<0.001 vs control.
Discussion
Our data show new aspects of the endothelial-mediated regulation of rat aorta tone primarily dependent on resting tension.
When resting tension is low (0.7 g), COX-derived metabolites, synergistically with NO, seem to play an important role in the relaxation induced by ACh. In these experimental conditions, a stored pool of NO is released, while NO neo-synthesis is practically absent. Indeed, although ecNOS inhibitors, namely L-NAME, L-NMMA and L-NIO, do not modify the dose-response curve of ACh, LY83583 abolishes ACh-induced relaxation of phenylephrine-precontracted aortic strips, indicating a relevant role for guanylyl cyclase. Moreover, the finding that PTIO strongly reduces ACh-induced relaxation suggests that ACh can mobilize a stored NO pool, which, synergistically with PGI2, can relax precontracted strips. This stored NO pool is, however, limited. Indeed, by repeating the ACh dose-response curve several times in the continuous presence of ecNOS inhibitors, the relaxing effect of ACh is strongly reduced as demonstrated by the powerful increase in ACh EC50s. A similar stored nitric oxide pool has been described in rabbit aortic smooth muscle (Venturini et al., 1993), rat aortic non-endothelial cells when the aorta is treated for 18 h with LPS and L-Arginine (Muller et al., 1996), in vivo in L-NAME treated rats (Davisson et al., 1996) and, more recently, in human platelets (Hirayama et al., 1999). The exact biological meaning of this pool is still unclear, but according to our data, it can be hypothesized that it plays a role when stretch is low. In vivo this condition may be mimicked when blood pressure is low and cells of resistance vessels are marginally stretched. In this condition, NO can be supplied without a constant ecNOS activation. Moreover, as judged by the data obtained in the presence of LY83583, it seems that guanylyl cyclase activation is the main relaxant pathway in rat aortic preparations stretched at low resting tension.
The dosage of PGI2 through its metabolite 6-keto PGF1α shows that a consistent PGI2 amount is released immediately after phenylephrine administration. The subsequent administration of ACh is unable to achieve a further increase in PGI2 production in the perfusion medium. As expected, in the presence of 5 μM indomethacin, the production of PGI2 is strongly inhibited and the small trend to a further decrease in PGI2 can probably be attributed to the washing of small quantities of either PGI2 or its metabolites trapped inside cells and tissue.
On the other hand, as shown in Figures 1 and 2, phenylephrine induces a consistent contraction, which is only marginally affected by COX inhibition. Therefore, the self-produced PGI2 is incapable of inducing a relevant relaxation. However, this self-produced PGI2 seems to be essential, since, at low resting tension, relaxation disappears in the presence of COX inhibitors and it is not restored also when the ACh-induced extra contraction is prevented (see below).
Thus at low stretch, ACh can induce a release of stored NO, which synergistically with self-produced PGI2 induces relaxation. A synergistic antiaggregatory effect of PGI2 and NO has been originally described in human platelets: low doses of both compounds inhibit aggregation and induce disaggregation of human platelets, while either PGI2 or NO alone at the same dose are ineffective (Radomski et al., 1987).
Our data also show that at a lower resting tensions (0.7 g and 1.2 g), when COX metabolism is inhibited, ACh strongly contracts phenylephrine-precontracted vessels. In this condition, the arachidonic acid metabolic pathway through lipoxygenase can predominate and lipoxygenase-derived contracting factors account for this paradoxical effect of ACh. Although we are unable to measure by radioimmunoassay an in-the-range leukotriene amount (D4, C4 and E4 leukotrienes, that, according to Kähler et al. (1993), can be synthesized by bovine aortic endothelial cells), the finding that lipoxygenase inhibitors block the ACh-induced extra contraction strongly suggests this hypothesis. Also, the suppression of ACh-induced contraction by 100 μM indomethacin is in line with this interpretation: the high dose of indomethacin can also inhibit lipoxygenase (Siegel et al., 1979). Moreover, since MK-886 is a specific inhibitor of 5-lipoxygenase (Gillard et al., 1989), this isoform plays a major role in our preparations. A similar diversion of arachidonic acid to the lipoxygenase pathway has been observed in canine saphenous veins: the maximal contraction evoked by ACh and high extracellular potassium concentration (Rimele & Vanhoutte, 1983) and bradykinin (Marsault et al., 1997) is enhanced in the presence of indomethacin in comparison to that obtained in control conditions. Since in all cases the control contraction is restored in the presence of indomethacin and nordihydroguaiaretic acid, an inhibitor of lipoxygenase, it has been argued that the inhibition of COX, turning arachidonic acid metabolism to lipoxygenase, produces lipoxygenase vasoconstrictor metabolites (Rimele & Vanhoutte, 1983; Marsault et al., 1997). Lipoxygenase metabolites could increase intracellular calcium by directly activating voltage-sensitive calcium channels (Vanhoutte et al., 1985). Indeed, in canine saphenous veins, diltiazem inhibits the increase in tension due to indomethacin preincubation during ACh- and potassium-induced contractions (Rimile & Vanhoutte, 1983).
In our experiments, verapamil prevents the ACh-induced contraction, suggesting that the paradoxical effect of ACh (observed at low resting tension in the presence of 5 μM indomethacin and blocked by lipoxygenase inhibitors) is mediated by an influx of extracellular calcium through L-type, voltage-dependent calcium channels. Therefore, a relevant target for lipoxygenase products can be L-type calcium channels. Since the ACh contractile effect is abolished in the absence of endothelium, the contractile lipoxygenase metabolite(s) seem(s) to be produced by endothelial cells.
At high resting tension, the NO pathway predominates and COX metabolites seem to influence ACh-induced relaxation only marginally. Indeed, indomethacin does not modify the relaxant effect of ACh, while L-NAME, L-NMMA and L-NIO do. These data suggest that the ‘de novo' synthesis of NO is mainly responsible for ACh-induced relaxation when a high resting tension is applied. Indeed, the inhibition of guanylyl cyclase by LY83583 or NO trapping by PTIO does not potentiate the L-NAME inhibitory effect. Moreover, by repeating the ACh dose-response curve on the same preparation in the continuous presence of ecNOS inhibitors, the ACh EC50s are increased, but these increases are quite limited.
The residual relaxation obtained at high ACh doses (10−6 M) in the presence of L-NAME is prevented by 5 μM indomethacin, but ACh never induces contractions. This result suggests that the lipoxygenase products responsible for ACh-induced contractions are either not produced or the mechanism controlling the contractions is inactivated.
At an intermediate tension (1.2 g), the ACh dose-response curve performed in the presence of the ecNOS inhibitor L-NAME is shifted to the right, as shown when preparations are stretched at high resting tension. However, in the presence of 5 μM indomethacin, ACh strongly contracted aortic strips and the preparations behave as those stretched at low resting tension. Therefore, although we do not explore a linear range of resting tensions, it seems that the stretch-dependent behaviour of aortic preparations modifies gradually.
These stretch-dependent behaviours are not dependent on a different sensitivity to NO, since Na nitroprusside relaxation is not influenced by resting tension.
Mechanical stretch and/or swelling can activate ionic channel activity in endothelial and smooth muscle cells (Davis et al., 1992; Sackin, 1995). In cultured pig aortic endothelial cells, stretch can activate calcium channels (Lansman, 1987) and large-conductance, calcium-activated K+ channels in freshly isolated smooth muscle cells from rabbit mesenteric artery (Dopico et al., 1994). These alterations in ion channel activity could modify resting membrane potential and, therefore, modify the L-type, voltage-dependent calcium channel properties, that, as shown, seem to play a role in the lipoxygenase-dependent contraction. Further research is needed to clarify this point.
In airway epithelium cells, cyclic stretch inhibits COX metabolite synthesis (mainly PGE2, 6-keto PGF1α and TxB2) (Savla et al., 1997), and mechanical stretch due to osmotic swelling increases intracellular calcium concentration in isolated human umbilical cord vein endothelial cells (Oike et al., 1994). This increase in intracellular calcium concentration is blocked by 4-bromophenacylbromide, a PLA2 inhibitor, suggesting that the increase in intracellular calcium is mediated by arachidonic acid (Oike et al., 1994). These data, obtained in very different experiment conditions, imply that stretch can modulate the arachidonic acid metabolism.
In conclusion, our data demonstrate that the synthesis, release and effect of different endothelial-derived vasoactive factors (namely NO, PGI2 and lipoxygenase metabolites) are strictly linked to the stretch status of the aorta. All these components may be relevant in the pathophysiological regulation of blood pressure.
Acknowledgments
This work was supported by a grant from the University of Florence. We would like to thank Prof Fabrizio Ledda and Prof Roberto Levi for reading the manuscript and Ms Sharon Blumenstock for editing.
Abbreviations
- ACh
acetylcholine
- COX
cyclooxygenase
- cyclic GMP
guanosine 3′:5′-cyclic monophosphate
- ecNOS
endothelial constitutive NO synthase
- EC50s
median effective concentrations
- EDHF
endothelial-derived hyperpolarizing factor
- EDRFs
endothelial-derived relaxant factors
- ETI
5,8,11-triynoic acid
- 6-keto PGF1α
6-keto prostaglandin F1α
- L-NAME
ω-nitro-L-arginine methylester
- L-NMMA
ω-monomethyl-L-arginine
- L-NIO
L-N5-iminoethylornithine
- LY83583
6-anilino-5,8-quinolinequinone
- MK-886
3-(1-(4-chlorobenzyl)-3t-butyl-thio-5-isopropylindol-2-yl)-2,2-dimethylpropanoic acid
- PGF2α
prostaglandin F2α
- PGI2
prostacyclin
- PTIO
2-phenyl-4,4,5-tetramethyl-imidazoline-1-oxyl-3-oxide
References
- BERKOWITZ B.A., ZABKO-POTAPOVICH B., VALOCIK R., GLEASON J.G. Effects of leukotrienes on the vasculature and blood pressure of different species. J. Pharmacol. Exp. Ther. 1984;229:105–112. [PubMed] [Google Scholar]
- CAMPBELL W.B., HARDER D.R. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circulation. 1999;84:484–488. doi: 10.1161/01.res.84.4.484. [DOI] [PubMed] [Google Scholar]
- CORNWELL T.L., PRYZWANSKY K.B., WYATT T.A., LINCOLN T.M. Regulation of sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Mol. Pharmacol. 1991;40:923–931. [PubMed] [Google Scholar]
- DAINTY I.A., MCGRATH J.C., SPEDDING M., TEMPLETON A.G.B. The influence of the initial stretch and the agonist-induced tone on the effect of basal and stimulated release of EDRF. Br. J. Pharmacol. 1990;100:767–773. doi: 10.1111/j.1476-5381.1990.tb14090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS M.J., DONOVITZ J.A., HOOD J.D. Stretch-activated single-channel and whole cell currents in vascular smooth muscle cells. Am. J. Physiol. 1992;262:C1083–C1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- DAVISSON R.T., BATES J.N., JOHNSON A.K., LEWIS S.J. Use-dependent loss of acetylcholine- and bradykinin-mediated vasodilatation after nitric oxide synthase inhibition. Evidence for performed stores of nitric oxide-containing factors in vascular endothelial cells. Hypertension. 1996;28:354–360. doi: 10.1161/01.hyp.28.3.354. [DOI] [PubMed] [Google Scholar]
- DOPICO A.M., KIRBER M.T., SINGER J.J., WALSH J.V., Jr Membrane stretch directly activates large conductance Ca2+-activated K+ channels in mesenteric artery smooth muscle cells. Am. J. Hyperents. 1994;7:82–89. doi: 10.1093/ajh/7.1.82. [DOI] [PubMed] [Google Scholar]
- EZRA D., BOYD L.M., FEUERSTEIN G., GOLDSTEIN R.E. Coronary constriction by leukotriene C4, D4 and E4 in the intact pig heart. Am. J. Cardiol. 1983;51:1451–1454. doi: 10.1016/0002-9149(83)90328-4. [DOI] [PubMed] [Google Scholar]
- FILIPEANU C.M., BRAILOIU E., PETRESCU G., NELEMANS S.A. Extracellular and intracellular arachidonic acid-induced contractions in rat aorta. Eur. J. Pharmacol. 1998;349:67–73. doi: 10.1016/s0014-2999(98)00180-0. [DOI] [PubMed] [Google Scholar]
- FISSLTHALER B., POPP R., KISS L., POTENTE M., HARDER D.R., FLEMING I., BUSSE R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GILLARD J., FORD-HUTCHINSON W., CHAN C., CHARLESON S., DENIS D., FOSTER A., FORTIN R., LEGER S., MCFARLANE C.S., MORTON H., PIECHUTA H., ROUZER A., ROUZER C.A., ROKACH J., YOUNG R., MACINTYRE D.E., PETERSON L., BACH G., EIERMANN G., HOPPLE S., HUME J., HUPE L., LUELL R., METZGER J., MEURER R., MILLER D.K., OPAS E., PACHOLOK S. L-663, 536 (MK-886) (3-(1-(4-chlorobenzyl)-3t-butyl-thio-5-isopropylindol-2-yl)-2,2-dimethylpropanoic acid), a novel, orally active leukotriene biosynthesis inhibitor. Can. J. Physiol. Pharmacol. 1989;67:456–464. doi: 10.1139/y89-073. [DOI] [PubMed] [Google Scholar]
- HIRAYAMA A., NORANHA-DUTRA A.A., GORDGE M.P., NEILD G.H., HOTHERSALL J.S. S-Nitrosothiols are stored by platelets and released during platelet-neutrophil interactions. Nitric-oxide. 1999;3:95–104. doi: 10.1006/niox.1999.0208. [DOI] [PubMed] [Google Scholar]
- HOLLENBERG S.M., TONG W., SHELHAMER J.H., LAWRENCE M., CUNNION R.E. Eicosanoid production by human aortic endothelial cells in response to endothelin $$$- Am. J. Physiol. 1994;267:H2290–H2296. doi: 10.1152/ajpheart.1994.267.6.H2290. [DOI] [PubMed] [Google Scholar]
- KÄHLER J., CONFORTO A., DUDEK R., TERMIN A., BING R.J. Synthesis of leukotrienes by freshly harvested endothelial cells. J. Mol. Cell. Cardiol. 1993;25:647–653. doi: 10.1006/jmcc.1993.1077. [DOI] [PubMed] [Google Scholar]
- LANSMAN J.B. Single stretch-activated ion channels in vascular endothelial cells as mechanotransducers. Nature. 1987;325:811–813. doi: 10.1038/325811a0. [DOI] [PubMed] [Google Scholar]
- MARSAULT R., ILLIANO S., VANHOUTTE P.M. Bradykinin-induced contractions of canine saphenous veins: mediation by B2 receptors and involvement of eicosanoids. Br. J. Pharmacol. 1997;120:215–220. doi: 10.1038/sj.bjp.0700898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., VANE J.R. Pharmacology and endogenous roles of prostaglandin peroxides, thromboxane A2 and prostacyclin. Pharmacol. Rev. 1979;30:293–331. [PubMed] [Google Scholar]
- MULLER B., KLESCHYOV A.L., STOCLET J.C. Evidence for N-acetylcysteine-sensitive nitric oxide storage as dinitrosyl-iron complexes in lipopolysaccharide-treated rat aorta. Br. J. Pharmacol. 1996;119:1281–1285. doi: 10.1111/j.1476-5381.1996.tb16034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OIKE M., DROOGMANS G., NILIUS B. Mechanosensitive Ca2+ transients in endothelial cells from human umbilical vein. Proc. Natl. Acad. Sci., U.S.A. 1994;91:2940–2944. doi: 10.1073/pnas.91.8.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALMER R.M., ASHTON D.S., MONCADA S. Vascular endothelial cells synthetize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- PARKINGTON H.C., TARE M., TONTA M.A., COLEMAN H.A. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J. Physiol. 1993;465:459–476. doi: 10.1113/jphysiol.1993.sp019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RADOMSKI M.W., PALMER R.M., MONCADA S. The anti-aggregating properties of vascular endothelium: interaction between prostacyclin and nitric oxide. Br. J. Pharmacol. 1987;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIMELE T.J., VANHOUTTE P.M. Effect of inhibitors of arachidonic acid metabolism and calcium entry of responses to acetylcholine, potassium and norepinephrine in the isolated canine saphenous vein. J. Pharmacol. Exp. Ther. 1983;225:720–728. [PubMed] [Google Scholar]
- SACKIN H. Mechanosensitive channels. Ann. Rev. Physiol. 1995;57:333–353. doi: 10.1146/annurev.ph.57.030195.002001. [DOI] [PubMed] [Google Scholar]
- SIEGEL M.I., MCCONNELL R.T., PORTER N.A., CUATRECASAS P. Arachidonate metabolism via lipoxygenase and 12L-hydroperoxydy-5,8,10,14-icosatetraenoic acid peroxide sensitive to anti-inflammatory drugs. Proc. Natl. Acad. Sci., U.S.A. 1979;76:3774–3778. doi: 10.1073/pnas.77.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAVIA U., SPORN P.H., WATERS C.M. Cyclic stretch of airway epithelium inhibits prostanoid synthesis. Am. J. Physiol. 1997;273:L1013–L1019. doi: 10.1152/ajplung.1997.273.5.L1013. [DOI] [PubMed] [Google Scholar]
- VANHOUTTE P.M., RIMELE T.J., FLAVAHAN J. Lipoxygenase and calcium entry in vascular smooth muscle. J. Cardiovasc. Pharmacol. 1985;7:S47–S52. doi: 10.1097/00005344-198500076-00009. [DOI] [PubMed] [Google Scholar]
- VENTURINI C.M., PALMER R.M.J., MONCADA S. Vascular smooth muscle contains a depletable store of a vasodilator which is light-activated and restored by donors of nitric oxide. J. Pharmacol. Exp. Ther. 1993;266:1497–1500. [PubMed] [Google Scholar]