

Abstract
The metabotropic glutamate receptors (mGluRs) are a family of G-protein linked receptors that can be divided into three groups (group I, II and III). A number of studies have implicated group I mGluR activation in acute neuronal injury, but until recently it was not possible to pharmacologically differentiate the roles of the two individual subunits (mGluR1 and mGluR5) in this group.
We investigated the role of mGluR5 in acute NMDA and glutamate mediated neurodegeneration in cultured rat cortical cells using the mGluR5 antagonists MPEP and SIB-1893, and found that they provide significant protection at concentrations of 20 or 200 μM.
These compounds act as effective mGluR5 antagonists in our cell culture system, as indicated by the ability of SIB-1893 to prevent phosphoinositol hydrolysis induced by the specific mGluR5 agonist, (RS)-2-chloro-5-hydroxyphenylglycine (CHPG).
However, they also significantly reduce NMDA evoked current recorded from whole cells voltage clamped at −60 mV, and significantly decrease the duration of opening of NMDA channels recorded in the outside out patch configuration.
This suggests that although MPEP and SIB-1893 are effective mGluR5 antagonists, they also act as noncompetitive NMDA receptor antagonists. Therefore, the neuroprotective effects of these compounds are most likely mediated through their NMDA receptor antagonist action, and caution should be exercised when drawing conclusions about the roles of mGluR5 based on their use.
Keywords: Group I mGluRs, MPEP, SIB-1893, NMDA receptors, NMDA-induced toxicity, neuroprotection, patch clamp recording
Introduction
The metabotropic glutamate receptors (mGluRs) are a family of G-protein linked glutamate receptors, of which eight different subtypes have been described to date (Knöpfel et al., 1995; Pin & Duvoisin, 1995; Riedel et al., 1996; Saugstad et al., 1997). These eight subtypes can be divided into three groups (group I, II and III) based on sequence homology, signal transduction mechanisms and pharmacological profile. Group I mGluR, which comprises subtype mGluR1 and mGluR5, are coupled via phospholipase C to the inositol triphosphate/Ca2+ pathway and show sensitivity to (RS)-3,5-dihydroxyphenylglycine [(RS)-DHPG]: (Schoepp et al., 1994). Group II and III receptors are negatively coupled to the adenylyl cyclase second messenger system, and are sensitive to agonists such as (+)-1S,2S,5R,6S-2-aminobicyclo {3.1.0]hexane-2,6-dicarboxylic acid (LY354740) and L-2-amino-4-phosphonobutyric acid (L-AP4) respectively (Knöpfel et al., 1995; Pin & Duvoisin, 1995; Riedel et al., 1996; Saugstad et al., 1997; Schoepp et al., 1999).
A wide range of physiological roles has been ascribed to the mGluRs, including interactions with Ca2+ and K+ channels, and regulation of ionotropic glutamate receptors (for review see Anwyl, 1999). As a result, the mGluRs have been implicated in a number of pathophysiological and neurodegenerative disorders, including neuronal injury. A number of studies have shown that group I mGluR activation can exacerbate neuronal injury both in vitro (Bruno et al., 1995; 1996; 1999; Mukhin et al., 1996; 1997a,1997b; Agrawal et al., 1998; Strasser et al., 1998) and in vivo (Gong et al., 1995; Mukhin et al., 1996; Bruno et al., 1999). It has been suggested that mGluR1 is involved in this exacerbation of injury, as antisense-oligodeoxynucleotide (AS-ODN) directed to mGluR1 reduced injury, whereas AS-ODN directed to mGluR5 did not (Mukhin et al., 1996). However, the elucidation of the role of the individual subtypes has been hampered by the lack of availability of subtype-specific compounds. Recently, a number of mGluR5 specific antagonists, such as (E)-2-methyl-6-styryl-pyridine (SIB-1893) and its structural derivative 2-methyl-6- (phenylethynyl)-pyridine (MPEP) have become available. These compounds have been reported to be potent, selective and systemically active antagonists of the mGluR5 subtype, with no appreciable agonist or antagonist activity at recombinant mGluR1b, group II or III mGluRs or ionotropic glutamate receptors (Gasparini et al., 1999). It has been previously reported that MPEP may cause a small reduction (22%) in glutamate evoked responses in Xenopus oocytes expressing recombinant hNMDA1A/2B receptors, but this reduction was not considered significant (Gasparini et al., 1999).
Here we report that both MPEP and SIB-1893 provide neuroprotection against glutamate- or NMDA-induced toxicity in cultured rat cortical neurones in vitro. However, although these compounds act as effective mGluR5 antagonists in our system, they also appear to significantly modulate NMDA receptor activity. These observations raise questions about the receptor specificity of these purported mGluR5 selective receptor antagonists and suggest caution in drawing conclusions about the role of mGluRs based on the use of such compounds.
Methods
Cell culture
Primary cortical neuronal cultures were prepared as previously described (Mukhin et al., 1998), with some modifications. In brief, cortical hemispheres were isolated from 17–18 day embryonic Sprague-Dawley rats (Taconic, Germantown, NY, U.S.A.), and minced in Krebs-Ringer bicarbonate buffer containing 0.3% bovine serum albumin (BSA; Life Technologies, Gaithersburg, MD, U.S.A.). The cells were dissociated in 1800 u ml−1 trypsin (Sigma, St. Louis, MO, U.S.A.) at 37°C for 20 min; trypsinization was halted by the addition of 200 u ml−1 DNase I and 3600 u ml−1 soybean trypsin inhibitor (Sigma). Individual cells were obtained by trituration, and were then centrifuged through a 4% BSA layer. The cell pellet was resuspended in Neurobasal medium (Gibco BRL, Grand Island, NY, U.S.A.) supplemented with 25 μM glutamate (Sigma), 0.5 mM glutamine (Biofluids, Rockville, MD, U.S.A.), 1% antibiotic-antimycotic (Biofluids) and 2% B27 supplement (Gibco). Cells were seeded at a density of 5×105 cells per ml onto 96-well microplates precoated with poly-D-lysine (Sigma), or at a density of 2.5×105 cells per ml on 12-mm glass coverslips (Fisher Scientific, Pittsburgh, PA, U.S.A.; catalogue number: 12-545-82) in 24-well plates precoated with poly-D-lysine (mol. wt. 70,000–150,000, Sigma). Cultures were incubated at 37°C in 5% CO2, and were fed on day 4 in vitro (4 DIV) by addition of 50% volume of Neurobasal medium, 0.5 mM glutamine and 1% antibiotic-antimycotic to each well. Cultures were used for experiments on 7–10 DIV.
Cell viability assay
Rat cortical neuronal cells cultured in 96-well plates at 7–8 DIV were pretreated for 30 min with 0.2–200 μM of mGluR5 antagonists MPEP or SIB-1893, with or without MK801 following addition of 150 μM Na-glutamate (Sigma) or 50 μM NMDA (Tocris). After 24 h of incubation with drugs, cell viability was tested by measuring LDH release, using CytoTox 96 non-radioactive cytotoxicity assay kit (Promega), according to the manufacturer's protocol. Relative absorbance was measured at 490 nm using a Ceres 9000 microplate reader (Bio-Tek Instruments, Winooski, VT, U.S.A.). Background LDH release, determined in intact control cultures, was subtracted from all experimental values. We have previously shown that changes in LDH release accurately reflect neuronal cell death in this model, as shown using other markers such as trypan blue or ethidium homodimer (Mukhin et al., 1997b).
PI hydrolysis
PI hydrolysis was measured in cortical neuronal cultures at 7 DIV as described previously (Mukhin et al., 1996), with minor modifications. Cortical neuronal cells cultured in 96-well plates were incubated overnight with myo-[3H]-inositol (22.3 Ci mmol−1, NEN, Boston, MA, U.S.A.). One μCi well−1. Cells were washed twice with Locke's buffer and incubated at 37°C in the same buffer for 30 min, in the presence or absence of mGluR5 antagonist (0.2–200 μM SIB-1893). Subsequently, 1 mM of CHPG, an mGluR5 agonist, was added together with 20 mM LiCl, and incubation was continued for an additional 30 min. Thereafter, incubation buffer was aspirated, inositol phosphates were extracted by 0.1 M HCl, containing 2 mM CaCl2, and transferred to columns with AG 1-X8 anion-exchange resin (Bio-Rad, Hercules, CA, U.S.A.). After separation, according to the method described by Berridge et al. (1982), accumulated [3H]-inositol phosphates were measured using liquid scintillation counter LS 6500 (Beckman Instruments, Fullerton, CA, U.S.A.).
Electrophysiology
Cortical cultures on glass coverslips were transferred to a recording chamber that was continuously perfused with bath solution at room temperature (21°C), containing (in mM) NaCl 145, KCl 5, CaCl2 1, HEPES 5, Glucose 5, Sucrose 25, D-Serine 0.01, adjusted to pH 7.4 with NaOH. Electrodes were pulled from thin-walled borosilicate glass (Blu-Tip capillary tubes, Oxford Labware) in three stages on a horizontal pipette puller (Mecanex S.A., Switzerland). Patch pipette solution contained (in mM) K-gluconate 145, MgATP 5, GTP-Na 0.2, EGTA 5, HEPES 10, adjusted to pH 7.2 with KOH. Typical pipette resistance was 5–8 MΩ.
The patch clamp technique was used in the whole cell recording and outside-out patch recording configuration. Cells were voltage clamped at −60 mV and currents were monitored with an Axopatch 1D amplifier, filtered at 1 kHz and digitized at 10–20 kHz using an IBM-compatible computer equipped with a Digidata 1200 acquisition board (Axon Instruments, Foster City, CA, U.S.A.) and pClamp 8 software (Axon Instruments). Analysis and curve fitting for the single-channel current recordings were performed using Fetchan and pStat analysis programs (Axon Instruments). The threshold for measuring open and shut intervals was set at one-half the maximal amplitude of the main conductance level of the channel. Log-binned interval histograms for shut and open duration were plotted with a square root vertical axis and fitted using maximum likelihood fitting. Data are expressed as mean±s.e.mean; P values represent the results of individual t-tests.
All drugs were diluted to the required concentration in bath solution, and were applied by a gravity-fed Y-tubing delivery system (Murase et al., 1989) placed within 100 μM of the cell or outside-out patch. This delivery system allows rapid onset (<50 ms) and local distribution of the drugs.
Drugs
Na-glutamate was obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). N-methyl-D-aspartate (NMDA), (1S, 2R)-1-aminocyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD), (S)-dihydroxyphenylglycine [(S)-DHPG], (RS)-chloro-5-hydroxyphenylglycine (CHPG), (E)-2-methyl-6-styryl-pyridine (SIB-1893) and 2-methyl-6- (phenylethynyl)-pyridine (MPEP) were all purchased from Tocris (Ballwin, MO, U.S.A.).
Results
Effects of mGluR5 antagonists on glutamate- and NMDA-induced neuronal cell death in vitro
To evaluate the possible role of mGluR5s in neuronal cell death, the influence of the specific mGluR5 antagonists MPEP and SIB-1893 on cell survival in vitro was examined using rat cortical neuronal cultures subjected to glutamate- or NMDA-induced toxicity. LDH release based cell viability assay revealed significant neuroprotective effects of MPEP and SIB-1893 both in glutamate- (Figure 1A) and NMDA-treated cultures (Figure 1B). Neuroprotection was observed at concentrations of the antagonists of 20 μM and above (Figure 1). When the noncompetitive NMDA receptor antagonist MK801 (10 μM) was co-applied with MPEP or SIB-1893, no further significant neuroprotection was observed (data not shown).
Figure 1.

Treatment with MPEP and SIB-1893 attenuated glutamate- and LDH release in rat cortical neuronal cultures. At 7 DIV, indicated concentrations of MPEP or SIB-1893 were added to cultures 30 min prior to application of 150 μM of glutamate (A) or 50 μM of NMDA (B). LDH release was measured after 24 h of treatment. Background LDH release, determined in intact control cultures, was subtracted from all experimental values. Histograms represent LDH release as a percentage of control levels±s.d., n=8–16 cultures per condition. *P<0.05 versus controls, as compared by ANOVA followed by the Student-Newman-Keuls test.
PI hydrolysis
To determine whether the antagonists can interact with PLC-coupled mGluR5s in our in vitro system, we examined effect of the SIB-1893 on agonist-induced inositol phosphates (IP) accumulation in cultured rat cortical neuronal cells. As shown in Figure 2, treatment of cortical neuronal cultures with 0.2–200 μM of SIB-1893 completely abolished IP accumulation induced by CHPG, a highly specific mGluR5 agonist (Doherty et al., 1997). This finding was further confirmed by PI hydrolysis measurement using similar concentrations of MPEP (Movsesyan et al., unpublished observation).
Figure 2.
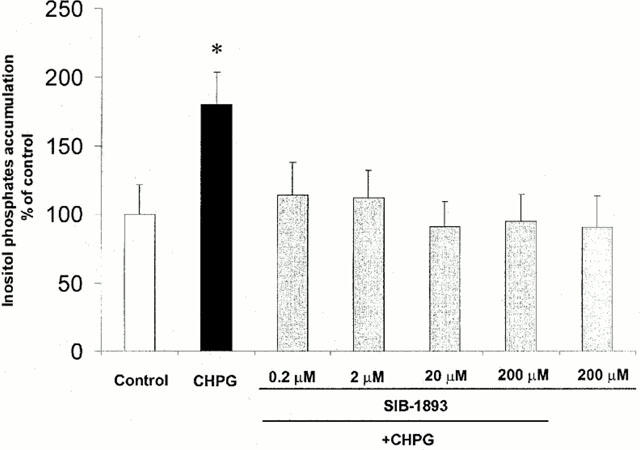
Treatment with MPEP or SIB-1893 abolished CHPG-induced PI hydrolysis. At 7 DIV, indicated concentrations of SIB-1893 were added to rat cortical neuronal cultures 30 min prior to stimulation with 1 mM CHPG. PI hydrolysis was measured by inositol phosphates accumulation within 30 min after addition of CHPG. Histograms represent inositol phosphates levels as a percentage of controls±s.d., n=6–12 cultures per condition. *P<0.05 versus controls, as compared by ANOVA followed by the Student-Newman-Keuls test.
Electrophysiology
Whole cell current recording
Currents evoked by the rapid application of 50 μM NMDA were recorded in the absence and presence of the mGluR5 antagonists. The steady state response to the initial application of NMDA was expressed as 100% (control) and subsequent responses to drug application were expressed as a percentage of this control value. The mean current evoked by application of 50 μM NMDA was 554±67.6 pA (n=15). In each cell, the initial control NMDA application was followed, at 2–3 min intervals, by co-application of 50 μM NMDA with 20 and 200 μM of an mGluR5 antagonist (with or without pre-perfusion of the antagonist) and a second application of 50 μM NMDA, to test for recovery. Cells in which this second NMDA response was significantly different from the control NMDA evoked response were discarded.
Representative traces, showing the effect of pre-perfusion of 20 and 200 μM MPEP on the NMDA evoked whole cell response, are illustrated in Figure 3A. The initial peak response and the steady state response to NMDA application were measured, and the data are summarized in Figure 3B.
Figure 3.
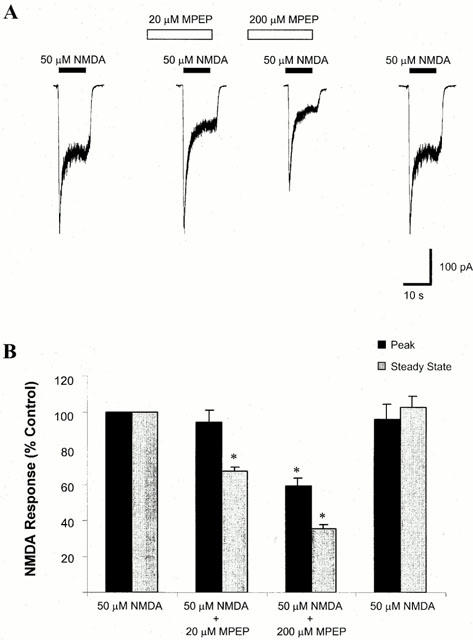
Effect of MPEP on NMDA-evoked whole cell current. (A) Representative traces of NMDA-evoked whole cell currents in the absence or presence of MPEP. Whole cell currents were recorded from cultured cortical neurones at 7–10 DIV, with voltage clamped at −60 mV in Mg2+ free solution. MPEP (20 or 200 μM) was delivered to the cell 30 s prior to delivery of 50 μM NMDA. (B) A summary of the effect of MPEP on NMDA-evoked current. Both peak and steady state responses were recorded. At a concentration of 20 μM, MPEP did not significantly alter peak NMDA current, but reduced the steady state current to 67.4±2.3% of control values (n=5, *P<0.05). At a concentration of 200 μM, MPEP significantly reduced both peak and steady state NMDA-evoked current to 59.4±4.2 and 35.1±2.5% of control values respectively (n=5, *P<0.05).
Pre-perfusion of 20 and 200 μM MPEP for 30 s before, and during, the application of 50 μM NMDA resulted in a steady state NMDA current response that was significantly different from that evoked by 50 μM NMDA alone. In the presence of 20 μM NMDA, the steady state current was 67.4±2.3% compared to control values (n=5, P<0.05, Figure 3B, grey bars), whereas in the presence of 200 μM MPEP, the NMDA evoked whole cell current was 35.1±2.5% compared to control (n=5, P<0.05, Figure 3B, grey bars). The peak NMDA evoked response was not significantly altered by pre-perfusion of 20 μM MPEP (94.4±6.8% compared to control, n=5, Figure 3B, black bars), whereas the peak NMDA evoked current was significantly altered by pre-perfusion of 200 μM MPEP (59.4±4.2% compared to control, n=5, P<0.05, Figure 3B, black bars).
Similar results were observed when MPEP was co-applied with 50 μM NMDA, without pre-perfusion. Co-application of 20 or 200 μM MPEP with 50 μM NMDA significantly reduced the NMDA evoked whole cell response (71.8±3.7 and 41.3±3.5% compared to control respectively, n=5, P<0.05). In the presence of 20 μM MPEP, the peak NMDA evoked current was not significantly altered (81.3±10.8% compared to control values, n=5), whereas in the presence of 200 μM MPEP, the NMDA evoked response was significantly reduced (75.7±5.8% compared to control values, n=5, P<0.05).
Representative traces, showing the effect of pre-perfusion of 20 and 200 μM SIB-1893 on the NMDA evoked whole cell response, are illustrated in Figure 4A. The initial peak response and the steady state response to NMDA application were measured, and the data are summarized in Figure 4B.
Figure 4.
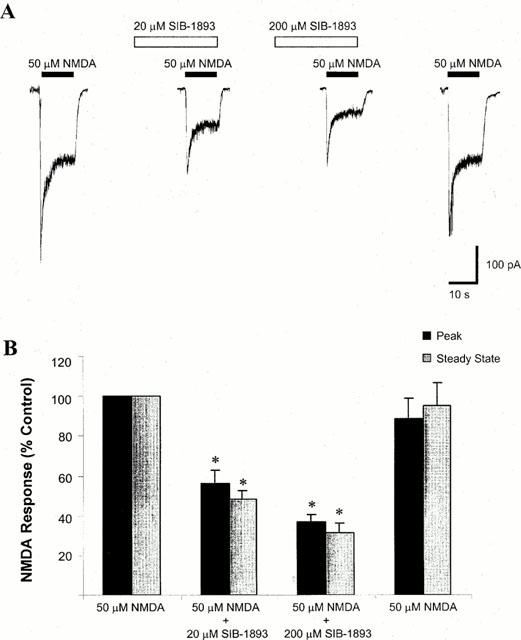
Effect of SIB-1893 on NMDA-evoked whole cell current. (A). Representative traces of NMDA-evoked whole cell currents in the absence and presence of SIB-1893. Whole cell currents were recorded from cultured cortical neurones at 7–10 DIV, with voltage clamped at −60 mV in Mg2+ free solution. SIB-1893 (20 or 200 μM) was delivered to the cell 30 s prior to delivery of 50 μM NMDA. (B) A summary of the effect of SIB-1893 on NMDA-evoked current. Both peak and steady state responses were recorded. At a concentration of 20 μM, SIB-1893 significantly reduced peak NMDA current to 55.9±6.5% and reduced the steady state current to 48.4±4.3% of control values (n=5, *P<0.05). At a concentration of 200 μM, SIB-1893 significantly reduced both peak and steady state NMDA-evoked current to 36.6±3.7 and 31.3±5.0% of control values respectively (n=5, *P<0.05).
Pre-perfusion of 20 and 200 μM SIB-1893 for 30 s before, and during, the application of 50 μM NMDA resulted in a steady state NMDA current response that was significantly different from that evoked by 50 μM NMDA alone. In the presence of 20 μM SIB-1893, the steady state current was 48.4±4.3% compared to control (n=5, P<0.05, Figure 4B, grey bars), and in the presence of 200 μM SIB-1893, the NMDA evoked whole cell current was 31.3±5.0% compared to control (n=5, P<0.05, Figure 4B, grey bars). The peak NMDA evoked response was also significantly altered by pre-perfusion of both 20 μM SIB-1893 (55.9±6.5% compared to control, n=5, Figure 4B, black bars), and 200 μM SIB-1893 (36.6±3.7% compared to control, n=5, P<0.05, Figure 4B, black bars).
Similar results were observed when SIB-1893 was co-applied with 50 μM NMDA, without pre-perfusion. Co-application of 20 or 200 μM SIB-1893 with 50 μM NMDA significantly reduced the steady state NMDA evoked whole cell response (71.8±3.7 and 41.3±3.5% compared to control respectively, n=5, P<0.05). In the presence of 20 μM SIB-1893 (compared to control, n=5) and 200 μM NMDA, the peak NMDA evoked response was significantly reduced (81.3±10.8 and 75.7±5.8% compared to control respectively, n=5, P<0.05).
Single channel current recordings
To examine the effect of MPEP and SIB-1893 on NMDA receptor function in greater detail, the effect of MPEP and SIB-1893 on NMDA channel activity in outside-out patches, excised from cortical neurones, was determined.
The effect of MPEP on NMDA channel open time is illustrated in Figure 4. A representative recording in response to application of 2 μM NMDA to an outside-out patch pulled from cultured cortical cells at 7–10 DIV is shown in Figure 4A, with the indicated segments of channel currents shown at expanded time scales. Channel currents recorded in response to application of 2 μM NMDA and 200 μM MPEP are illustrated in Figure 4B, showing a marked reduction of the duration of openings of the channels. Distributions of the channel open time were determined and are illustrated in Figure 4C,D. Two main open states were observed. To facilitate comparison, drug effects were expressed as a percentage of the duration of opening of the channels in response to 2 μM NMDA alone. The per cent changes of the mean open time of the NMDA channel in the absence and presence of 20 and 200 μM MPEP are shown in Figure 4E. At lower concentration (20 μM), MPEP significantly reduced channel open time, to 79.36±5.13% of control (n=6, P<0.05. Mean control channel open time for 11 patches was 3.24±0.37 ms). Application of 200 μM MPEP also caused a significant reduction in channel open time to 49.40±4.12% of control (n=6, P<0.05). MPEP also significantly reduced the open probability of the NMDA channel at concentrations of both 20 and 200 μM, from 0.09±0.01 to 0.06±0.01 and to 0.03±0.01 respectively (n=6, P<0.05).
As illustrated in Figure 5, SIB-1893 also reduced NMDA channel opening duration. Representative recordings in response to application of 2 μM NMDA and to application of 2 μM NMDA and 200 μM SIB-1893 are illustrated in Figure 5A,B. A marked reduction of the open time of the channel currents can be seen in the presence of SIB-1893. Average per cent changes for the open time of the NMDA channel in the absence and presence of 20 and 200 μM SIB-1893 are shown in Figure 5E. At lower concentration (20 μM) SIB-1893 significantly reduced channel open time, to 73.02±10.20% of control (n=5, P<0.05). Application of 200 μM SIB-1893 also caused a significant reduction in channel open time to 47.43±10.03% of control (n=5, P<0.05). SIB-1893 also significantly reduced the open probability of the NMDA channel at a concentration of 200 μM, from 0.045±0.01 to 0.014±0.01 (n=5, P<0.05). In the presence of 20 μM SIB-1893, a slight, but not significant, reduction in open probability was recorded (from 0.045±0.01 to 0.042±0.01, n=5).
Figure 5.
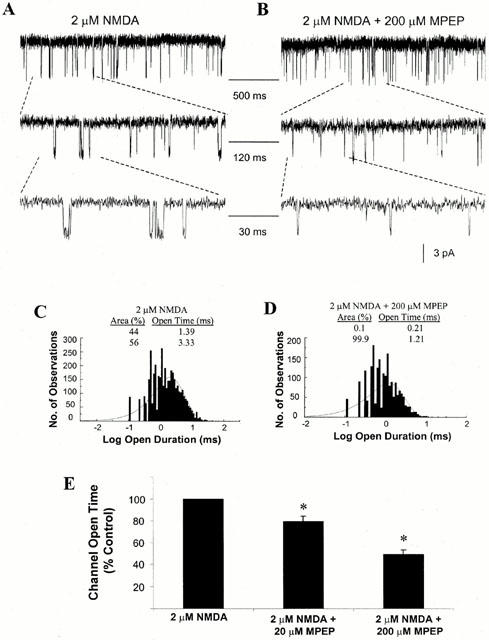
Effect of MPEP on NMDA single channel open time. (A) Representative current traces evoked by 2 μM NMDA, obtained from an outside-out patch recorded from cultured cortical neurones at 7–10 DIV, with voltage clamped at −60 mV in Mg2+ free solution. Indicated channel current segments are shown on different time scales. (B) Representative current traces evoked by 2 μM NMDA and 200 μM MPEP, obtained from an outside-out patch. (C) Single channel open time was recorded from the patches in the presence of 2 μM NMDA and the distribution of the open intervals was fit with two exponentials with time constants and relative areas of 1.39 ms (44%) and 3.33 ms (56%). (D) Distribution of open intervals from the same patches in the presence of 2 μM NMDA and 200 μM MPEP. Distribution was fit with two exponentials with time constants and relative areas of 0.21 ms (0%) and 1.21 ms (100%). (E) Summary of the effect of 20 and 200 μM MPEP on the NMDA-evoked single channel open time. In the presence of 20 μM MPEP, the channel open time was reduced to 79.36±5.13% of control values (n=6, *P<0.05). In the presence of 200 μM MPEP, the channel open time was reduced to 49.40±4.12% of control values (n=6, *P<0.05).
Discussion
There is compelling evidence implicating glutamate release and activation of glutamate receptors in the pathophysiology of CNS injury. Trauma to the brain or spinal cord causes marked elevations in the extracellular levels of glutamate in both animal models and humans (Faden et al., 1988; 1989; Katayama et al., 1990; Panter & Faden, 1992; Bullock et al., 1998). As a result, a large number of studies have investigated the role of the various glutamate receptors in traumatic neuronal injury, with the ultimate goal of developing potential drug therapies to treat CNS injury. MGluRs provide an attractive target for potential drug therapies, as they are primarily localized in the CNS, thereby reducing the likelihood of peripheral side effects. Interference with mGluRs seems to have only a modest impact on fast excitatory transmission, which would also be a potential advantage for chronic therapies (Knöpfel et al., 1995; Pin & Duvoisin, 1995; Nicoletti et al., 1996; Toms et al., 1996; Conn & Pin, 1997; Anwyl, 1999; Hölscher et al., 1999).
A number of studies have examined the role of group I mGluRs in neuronal injury. In mouse cortical cultures, mGluR group I agonists amplify neuronal cell death induced by a pulse of NMDA (Bruno et al., 1995; 1999; Strasser et al., 1998), whereas LY367385, a potent antagonist of mGluR1a, provided neuroprotection (Bruno et al., 1999). In rat mixed cortical/glial cultures, group I mGluR activation exacerbated injury induced by an in vitro punch injury model (Mukhin et al., 1996; 1997a,1997b). A small number of in vivo studies also support a role for group I mGluR activation in neuronal injury. MCPG, a weak group I/II antagonist exhibiting greater antagonistic effects at mGluR1 than at mGluR5 (Brabet et al., 1995; Joly et al., 1995; Kingston et al., 1995), improves neurological recovery and post-injury behavioural deficits, and decreases hippocampal cell loss in vivo after lateral fluid percussion injury (Gong et al., 1995; Mukhin et al., 1996). The mGluR1a antagonist LY367385, also provides significant neuroprotection when administered intrastriatally in rats infused with NMDA or in gerbils subjected to global ischaemia (Bruno et al., 1999).
Although considerable evidence implicates mGluR1 activation in acute neurodegeneration, until the recent development of agents such as MPEP and SIB-1893 it was not possible to determine the exact role of mGluR5 in neuronal injury. These drugs have been described as highly selective and potent antagonists, with no appreciable agonist or antagonist activity at mGluR1 receptors (Gasparini et al., 1999; Varney et al., 1999). MPEP and SIB-1893 do not share the α-amino acid moiety and the distal carboxylic acid function common to all known competitive mGluR antagonists, and Schild analysis indicates that the structurally related compound, SIB-1757, inhibits mGluR5 in a noncompetitive manner (Varney et al., 1999). Varney et al. (1999) suggest that because these compounds are noncompetitive, they may have the advantage of antagonizing the receptor even in the presence of high levels of glutamate likely to be present in the injured state. MPEP has also been shown to be centrally active following intravenous administration in rats (Gasparini et al., 1999). Given the noncompetitive mechanism of action and ability to cross the blood brain barrier, these compounds are extremely attractive potential therapeutic agents.
With this in mind, we determined the effect of MPEP and SIB-1893 on glutamate and NMDA induced toxicity in cultured rat cortical neurones in vitro. Both compounds provided significant protection against either glutamate or NMDA induced injury. Given that previous work in our laboratory indicated that mGluR5 was not likely to be involved in mediating neuronal injury (Mukhin et al., 1996), we were surprised by this marked neuroprotective effect. Therefore, we examined the specificity of the antagonists in our system. Since SIB-1893 and MPEP share close structural homology (Gasparini et al., 1999) and act similarly in our culture system, in this work we utilized only SIB-1893 for experiments on modulation of the mGluR5 agonist induced PI hydrolysis.
In agreement with previous reports (Gasparini et al., 1999; Varney et al., 1999), we found that SIB-1893 act as effective mGluR5 antagonist, as indicated by the ability of this compound to inhibit CHPG-induced phosphoinositide hydrolysis. It should be pointed out that SIB-1893 abolished agonist-induced PI hydrolysis at concentrations much lower than that required for neuroprotection. These results correspond to those observed for MPEP in the similar system (Movsesyan et al. unpublished observation). CHPG has been reported to selectively activate mGluR5a, while being devoid of activity at mGluR1a expressed in CHO cells (Doherty et al., 1997). We did not determine the efficacy or potency of MPEP or SIB-1893 at other mGluRs, although it has been reported that SIB-1893 may have agonist activity at mGluR4: at a concentration of 100 μM, SIB-1893 significantly reduced forskolin-elevated cyclic AMP levels in HEK-293 cells expressing human mGluR4a (Varney et al., 1999). This indicated that SIB-1893 acts as a mGluR4 agonist with an efficacy of 50% of that seen with L-AP4. In addition, it has been reported that activation of group III mGluRs can provide significant neuroprotection in vitro (Faden et al., 1997). However, given that MPEP and SIB-1893 provide similar neuroprotection in our system, and that MPEP does not affect forskolin-stimulated cyclic AMP levels (Gasparini et al., 1999), it appears unlikely that the main neuroprotective effect of these compounds is mediated through an mGluR4 agonist action.
It is more likely that MPEP and SIB-1893 mediate their neuroprotective effects through an NMDA antagonist action. Both compounds significantly reduced the steady state NMDA-evoked whole cell current at concentrations that provide significant neuroprotection. The compounds produced a greater effect on steady state NMDA current than on peak NMDA current, and a greater effect on steady state current was maintained whether the mGluR antagonists were pre-perfused or not. MPEP and SIB-1893, at a concentration of 200 μM, also reduced the open time of the NMDA channel by approximately 50%, and markedly reduced the open probability of the channel. This further supports the theory that both agents are acting as noncompetitive NMDA receptor antagonists. Whether the modulatory site on the NMDA receptor at which these compounds act is structurally related to the modulatory site on the mGluR5 receptor at which these compounds act, cannot be determined. A known, noncompetitive NMDA receptor antagonist, MK801, has been shown to markedly reduce glutamate or NMDA mediated neuronal cell death in vitro (Regan & Choi, 1994; Mukhin et al., 1997a; 1998). No further additive neuroprotective effect was observed when MPEP and SIB-1893 were applied to injured cells in the presence of MK801 in our system (glutamate-, NMDA-induced toxicity) or after mechanical injury (Movsesyan et al. unpublished observation).
It has previously been reported that MPEP and SIB-1893 have no appreciable agonist or antagonist activity at ionotropic receptors (Gasparini et al., 1999). However, it should be noted that the authors report a 22% reduction of glutamate evoked whole cell current recorded from Xenopus oocytes expressing the human NMDA1A/2B receptor complex, a reduction that failed to reach significant levels. It appears that this MPEP antagonism of the ionotropic glutamate response is magnified in the rat cortical cells. Why such a magnification would occur is unclear. It is possible that differences in homology, stoichiometry or post-translational modifications between the human recombinant NMDA receptors and the native rat NMDA receptors could confer differences in their affinity for MPEP. Although this factor has been ruled out for differences between the affinity of human recombinant mGluR5 and rat mGluR5 for SIB-1893 (Varney et al., 1999), it has not been addressed in the case of NMDA receptors. It also appears that SIB-1893, which has been shown to be less potent at mGluR5 (Varney et al., 1999), may be a more effective neuroprotective agent than MPEP which suggests that these compounds may provide neuroprotection via NMDA receptor, but not mGluR5 modulation.
In conclusion, we have shown that while MPEP and SIB-1893 act as effective mGluR5 antagonists in cultured rat cortical neurones in vitro, they also act as noncompetitive NMDA receptor antagonists. This brings into question the potential usefulness of these compounds for determining the functions of the mGluR5 receptor in vitro and in vivo and causes concerns regarding conclusions based on their use.
Figure 6.
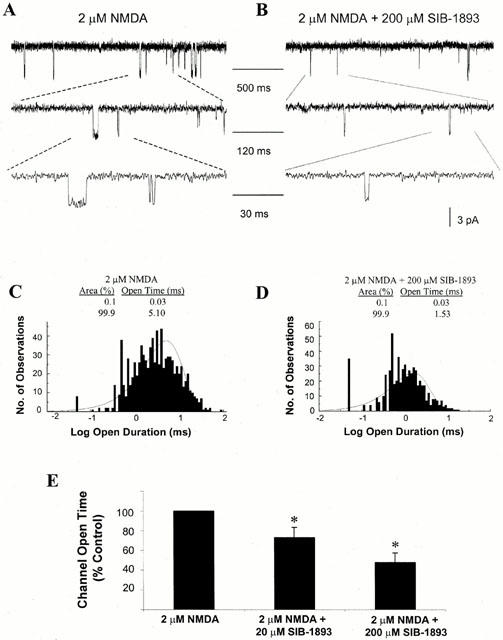
Effect of SIB-1893 on NMDA single channel open time. (A) Representative current traces evoked by 2 μM NMDA, obtained from an outside-out patch recorded from cultured cortical neurones at 7–10 DIV, with voltage clamped at −60 mV in Mg2+ free solution. Indicated channel current segments are shown on different time scale. (B) Representative current traces evoked by 2 μM NMDA and 200 μM SIB-1893 obtained from an outside-out patch. (C) Single channel open time was recorded from the patches in the presence of 2 μM NMDA and the distribution of the open intervals was fit with two exponentials with time constants and relative areas of 0.03 ms (0%) and 5.10 ms (100%). (D) Distribution of open intervals from the same patches in the presence of 2 μM NMDA and 200 μM SIB-1893. Distribution was fit with two exponentials with time constants and relative areas of 0.03 ms (0%) and 1.53 ms (100%). (E) Summary of the effect of 20 and 200 μM SIB-1893 on the NMDA-evoked single channel open time. In the presence of 20 μM SIB-1893, the channel open time was reduced to 73.02±10.02% of control values (n=5, *P<0.05). In the presence of 200 μM SIB-1893, the channel open time was reduced to 47.43±10.03% of control values (n=5, *P<0.05).
Acknowledgments
This study was supported by grants from the Department of Defense (DAMD-17-93-V-3018) and the NIH (RO1NS37313) to A.I. Faden.
Abbreviations
- AS-ODN
antisense-oligodeoxynucleotide
- CHPG
(RS)-2-chloro-5-hydroxyphenylglycine
- CNS
central nervous system
- DIV
days in vitro
- IP
inositol phosphates
- LDH
lactate dehydrogenase
- mGluR
metabotropic glutamate receptors
- MK801
(5R,10S)-(+)5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine
- MPEP
2-methyl-6-(phenylethynyl)-pyridine
- NMDA
N-methyl-D-aspartate
- PI
phosphoinositides
- (S)-DHPG
(S)-dihydroxyphenylglycine
- SIB-1893
(E)-2-methyl-6-styryl-pyridine
- 1S,3R-ACPD
(1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid
References
- AGRAWAL S.K., THERIAULT E., FEHLINGS M.G. Role of group I metabotropic glutamate receptors in traumatic spinal cord white matter injury. J. Neurotrauma. 1998;15:929–941. doi: 10.1089/neu.1998.15.929. [DOI] [PubMed] [Google Scholar]
- ANWYL R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res. Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- BERRIDGE M.J., DOWNES C.P., HANLEY M.R. Lithium amplifies agonist-dependent phosphatidylinositol responses in brain and salivary glands. Biochem. J. 1982;206:587–595. doi: 10.1042/bj2060587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRABET I., MARY S., BOCKAERT J., PIN J.-P. Phenylglycine derivatives discriminate between mGluR1- and mGluR5-mediated responses. Neuropharmacology. 1995;34:895–903. doi: 10.1016/0028-3908(95)00079-l. [DOI] [PubMed] [Google Scholar]
- BRUNO V., BATTAGLIA G., KINGSTON A., O'NEILL M.J., CATANIA M.V., DI GREZIA R., NICOLETTI F. Neuroprotective activity of the potent and selective mGlu1a metabotropic receptor antagonist, (+)-2-methyl-4 carboxyphenylglycine ( LY367385): comparison with LY357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology. 1999;38:199–207. doi: 10.1016/s0028-3908(98)00159-2. [DOI] [PubMed] [Google Scholar]
- BRUNO V., COPANI A., BONANNO L., KNÖPFEL T., HUHN R., ROBERTS P.J., NICOLETTI F. Activation of group III metabotropic glutamate receptors is neuroprotective in cortical cultures. Eur. J. Pharmacol. 1996;310:61–66. doi: 10.1016/0014-2999(96)00358-5. [DOI] [PubMed] [Google Scholar]
- BRUNO V., COPANI A., KNÖPFEL T., KUHN R., CASABONA G., DELL'ALBANI P., CONDORELLI D.F., NICOLETTI F. Activation of metabotropic glutamate receptors coupled to inositol phospholipid hydrolysis amplifies NMDA-induced neuronal degeneration in cultured cortical cells. Neuropharmacology. 1995;34:1089–1098. doi: 10.1016/0028-3908(95)00077-j. [DOI] [PubMed] [Google Scholar]
- BULLOCK R., ZAUNER A., WOODWARD J.J., MYSEROS J., CHOI S.C., WARD J.D., MARMAROU A., YOUNG H.F. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 1998;89:507–518. doi: 10.3171/jns.1998.89.4.0507. [DOI] [PubMed] [Google Scholar]
- CONN P.J., PIN J.-P. Pharmacology and functions of metabotropic glutamate receptors. Ann. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- DOHERTY A.J., PALMER M.J., HENLEY J.M., COLLINGRIDGE G.L., JANE D.E. (RS)-2-Chloro-5-Hydroxyphenylglycine (CHPG) activates mGlu5, but not mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology. 1997;36:265–267. doi: 10.1016/s0028-3908(97)00001-4. [DOI] [PubMed] [Google Scholar]
- FADEN A.I., DEMEIUK P., PANTER S.S., VINK R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- FADEN A.I., IVANOVA S.A., YAKOVLEV A.G., MUKHIN A.G. Neuroprotective effects of group III mGluR in traumatic neuronal injury. J. Neurotrauma. 1997;14:885–895. doi: 10.1089/neu.1997.14.885. [DOI] [PubMed] [Google Scholar]
- FADEN A.I., LEMKE M., SIMON R.P., NOBLE L.J. N-methyl-D-aspartate antagonist MK801 improves outcome following traumatic spinal cord injury in rats: behavioral, anatomic, and neurochemical studies. J. Neurotrauma. 1988;5:33–45. doi: 10.1089/neu.1988.5.33. [DOI] [PubMed] [Google Scholar]
- GASPARINI F., LINGENHÖL K., STOEHR N., FLOR P.J., HEINRICH M., VRANESIC I., BIOLLAZ M., ALLGEIER H., HECKENDORN R., URWYLER S., VARNEY M.A., JOHNSON E.C., HESS S.D., RAO S.P., SACAAN A.I., SANTORI E.M., VELIÇELEBI G., KUHN R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGluR5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- GONG Q.Z., DELAHUNTY T.M., HAMM R.J., LYETH B.G. Metabotropic glutamate antagonist, MCPG, treatment of traumatic brain injury in rats. Brain Res. 1995;700:299–302. doi: 10.1016/0006-8993(95)01081-6. [DOI] [PubMed] [Google Scholar]
- HÖLSCHER C., GIGG J., O'MARA S.M. Metabotropic glutamate receptor activation and blockade: their role in long-term potentiation, learning and neurotoxicity. Neurosci. Biobehav. Rev. 1999;23:399–410. doi: 10.1016/s0149-7634(98)00045-1. [DOI] [PubMed] [Google Scholar]
- JOLY C., GOMEZA J., BRABET I., CURRY K., BOCKAERT J., PIN J.-P. Molecular, functional, and pharmacological characterisation of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J. Neurosci. 1995;15:3970–3981. doi: 10.1523/JNEUROSCI.15-05-03970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATAYAMA Y., BECKER D.P., TAMURA T., HOVDA D.A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 1990;73:889–900. doi: 10.3171/jns.1990.73.6.0889. [DOI] [PubMed] [Google Scholar]
- KINGSTON A.E., BURNETT J.P., MAYNE N.G., LODGE D. Pharmacological analysis of 4-carboxyphenylglycine derivatives: comparison of effects on mGluR1a and mGluR5a subtypes. Neuropharmacology. 1995;34:887–894. doi: 10.1016/0028-3908(95)00069-i. [DOI] [PubMed] [Google Scholar]
- KNÖPFEL T.S., KUHN R., ALLEGIER H. Metabotropic glutamate receptors: Novel targets for drug development. J. Med. Chem. 1995;38:1417–1426. doi: 10.1021/jm00009a001. [DOI] [PubMed] [Google Scholar]
- MUKHIN A., FAN L., FADEN A.I. Activation of metabotropic glutamate receptor subtype mGluR1 contributes to post-traumatic neuronal injury. J. Neurosci. 1996;16:6012–6020. doi: 10.1523/JNEUROSCI.16-19-06012.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUKHIN A.G., IVANOVA S.A., ALLEN J.W., FADEN A.I. Mechanical injury to neuronal/glial cultures in microplates: role of NMDA receptors in secondary neuronal cell death. J. Neurosci. Res. 1998;51:748–758. doi: 10.1002/(SICI)1097-4547(19980315)51:6<748::AID-JNR8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- MUKHIN A.G., IVANOVA S.A., FADEN A.I. mGluR modulation of post-traumatic neuronal death: role of NMDA receptors. NeuroReport. 1997b;8:2561–2566. doi: 10.1097/00001756-199707280-00028. [DOI] [PubMed] [Google Scholar]
- MUKHIN A.G., IVANOVA S.A., KNOBLACH S.M., FADEN A.I. New in vitro model of traumatic neuronal injury: Evaluation of secondary injury and glutamate receptor-mediated neurotoxicity. J. Neurotrauma. 1997a;14:651–663. doi: 10.1089/neu.1997.14.651. [DOI] [PubMed] [Google Scholar]
- MURASE K., RYU P.D., RANDIC M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely isolated rat spinal dorsal horn neurons. Neurosci. Lett. 1989;103:56–63. doi: 10.1016/0304-3940(89)90485-0. [DOI] [PubMed] [Google Scholar]
- NICOLETTI F., BRUNO V., COPANI A., CASABONA G., KNÖPFEL T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders. Trends Neurosci. 1996;19:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- PANTER S.S., FADEN A.I. Pretreatment with NMDA antagonists limits release of excitatory amino acids following traumatic brain injury. Neurosci. Lett. 1992;136:165–168. doi: 10.1016/0304-3940(92)90040-e. [DOI] [PubMed] [Google Scholar]
- PIN J.-P., DUVOISIN R. The metabotropic glutamate receptors: Structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- REGAN R.F., CHOI D.W. The effect of NMDA, AMPA/kainate and calcium channel antagonists on traumatic cortical neuronal injury in culture. Brain Res. 1994;633:236–242. doi: 10.1016/0006-8993(94)91544-x. [DOI] [PubMed] [Google Scholar]
- RIEDEL G., WETZEL W., REYMANN K.G. Comparing the role of metabotropic glutamate receptors in long-term potentiation and in learning and memory. Prog. Neuro-Psychopharmacol. Psychiat. 1996;20:761–789. doi: 10.1016/0278-5846(96)00058-9. [DOI] [PubMed] [Google Scholar]
- SAUGSTAD J.A., KINZIE J.M., SHINOHARA M.M., SEGERSON T.P., WESTBROOK G.L. Cloning and expression of rat mGluR8 reveals a distinct pharmacological profile. Mol. Pharmacol. 1997;51:119–125. doi: 10.1124/mol.51.1.119. [DOI] [PubMed] [Google Scholar]
- SCHOEPP D.D., GOLDSWORTHY J., JOHNSON B.G., SALHOFF C.R., BAKER S.R. 3,5-dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J. Neurochem. 1994;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- SCHOEPP D.D., JANE D.E., MONN J.A. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1473. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- STRASSER U., LOBNER D., BEHRENS M.M., CANZONIERO L.M., CHOI D.W. Antagonists for group I mGluRs attenuate excitotoxic neuronal death in cortical cultures. Eur. J. Neurosci. 1998;10:2848–2855. doi: 10.1111/j.1460-9568.1998.00291.x. [DOI] [PubMed] [Google Scholar]
- TOMS N.J., ROBERTS P.J., SALT T.E., STATON P.C. Latest eruptions in metabotropic glutamate receptors. TIPS. 1996;17:429–435. doi: 10.1016/s0165-6147(96)01006-1. [DOI] [PubMed] [Google Scholar]
- VARNEY M.A., COSFORD N.P.D., JACHEC J., RAO S.P., SACAAN A.I., LIN F.-F., BLEICHER L., SANTORI E.M., FLOR P.J., ALLGEIER H., GASPARINI F., KUHN R., HESS S.D., VELIÇELEBI G., JOHNSON E.C. SIB-1757 and SIB-1893: Selective, noncompetetive antagonists of metabotropic glutamate receptor type 5. J. Pharmacol. Exp. Ther. 1999;290:170–181. [PubMed] [Google Scholar]