

Abstract
Mesangial cells play an active role in the inflammatory response to glomerular injury. We have studied in cultured human mesangial cells (CHMC) several effects of 9-cis retinoic acid (9-cRA), an activator of both retinoic acid receptors (RARs) and retinoid X receptors (RXRs).
9-cRA inhibited foetal calf serum-induced CHMC proliferation. It also prevented CHMC death induced by the inflammatory mediator H2O2. This preventive effect was not due to any increase in H2O2 catabolism and it persisted even when both catalase and glutathione synthesis were inhibited. Finally, 9-cRA diminished monocyte adhesion to FCS-stimulated CHMC. Interestingly, the retinoid also inhibited in FCS-stimulated cells the protein expression of two mesangial adhesion molecules, fibronectin and osteopontin, but it did not modify the protein expression of intercellular adhesion molecule-1 and vascular adhesion molecule-1.
All major RARs and RXRs isotypes were expressed in CHMC regardless of the presence or absence of 9-cRA. Transcripts to RAR-α, RAR-β and RXR-α increased after incubation with 9-cRA whereas RXR-γ was inhibited, suggesting a major role for RARs and RXRs in 9-cRA-anti-inflammatory effects.
9-cRA was toxic only at 50 μM (a concentration 50–5000 times higher than required for the effects above). Cell death occurred by apoptosis, whose onset was associated with a pronounced increase in catalase activity and reduced glutathione content, being more effectively induced by all-trans retinoic acid. Modulation of the oxidant/antioxidant balance failed to inhibit apoptosis.
We conclude that mesangial cells might be a target for the treatment of inflammatory glomerulopathies with 9-cRA.
Keywords: Mesangial cell, inflammation, 9-cis retinoic acid, all-trans retinoic acid, monocyte, osteopontin, vascular cell adhesion molecule-1, intercellular cell adhesion molecule-1, hydrogen peroxide, apoptosis
Introduction
Glomerular diseases remain the most common cause of progressive renal failure that may require dialysis or kidney transplantation. Remarkable recent advances in our understanding of the pathogenesis of various forms of glomerulonephritis have made it obvious that glomerular cells and especially mesangial cells play an active part in the inflammatory response to glomerular injury (Pfeilschifter, 1994). For instance, these cells contribute to phagocyte infiltration of the glomerulus during glomerular inflammation since they express cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (Denton et al., 1991; Springer, 1994, Khachigian et al., 1997) or secrete osteopontin (OP) (Nagasaki et al., 1997), a glycoprotein with cell adhesive properties which plays a role in macrophage infiltration (reviewed by Giachelli et al., 1995; Lan et al., 1998), and fibronectin (FN) (Kosmehl et al., 1996), a major glomerular matrix component which has been shown to mediate monocyte accumulation in the damaged kidney (Chana & Wheeler, 1999). Furthermore, the mesangial cells constitute a highly specialized pericapillary tissue and they have evolved three prominent proinflammatory features as a result of the cross-communication with invading immune cells (Kashgarian & Sterzel, 1992): (1) increased inflammatory mediator production; (2) increased matrix production by mesangial cells; and (3) increased mesangial cell proliferation. Interestingly, mesangial cells may also contribute to the repair process of experimental proliferative glomerulonephritis through a reduction of hypercellularity by means of the process of apoptosis (Shimizu et al, 1995).
We have recently shown (Moreno et al., 1999b) that all-trans-retinoic acid (tRA) inhibits OP mRNA expression in cultured human mesangial cells (CHMC) and prevents the cytotoxicity of H2O2–one of the mediators implicated in the pathophysiology of inflammatory renal diseases (Nath et al., 1994)–in CHMC, in parallel to an increase in catalase activity and reduced glutathione content (GSH). There is now strong evidence that two families of nuclear retinoic acid receptors, RAR-α, -β, -γ and RXR-α, -β, -γ mediate the effects of retinoids (reviewed by Giguere, 1994). tRA only activates RARs whereas 9-cis retinoic acid (9-cRA) is a pan-agonist and it activates both RARs and RXRs (Heyman et al., 1992; Levin et al., 1992). Thus 9-cRA may lead to a broader or a different spectrum of anti-inflammatory activity than tRA. Studies were therefore conducted to explore the ability of 9-cRA: (1) to modulate mesangial cell number through antiproliferative mechanisms; (2) to inhibit monocyte adhesion to CHMC; (3) to inhibit mesangial expression of molecules involved in monocyte adhesion to CHMC; and (4) to prevent the toxic effect of H2O2 on CHMC, which may be secreted by activated macrophages (Schena et al., 1997). To gain insight on the role of RARs and RXRs in the observed effects, we also studied the modulatory effect of 9-cRA on mRNA expression of these receptors. Finally, we studied retinoid toxicity on CHMC.
Methods
Reagents
All-trans retinoic acid (tRA) and 9-cis retinoic acid (9-cRA) were a kind gift of Productos Roche S.A. (Spain). Unless otherwise stated, all of the biochemical reagents used in this study were purchased from the Sigma Chemical Co. Tissue-cultured materials, growth media and serum for cell cultures were obtained from Gibco Laboratories. All other chemicals used were of the purest grade commercially available.
cDNA probes and antibodies
VCAM-1 probe was a HindIII-XhoI fragment of the human cDNA consisting of nucleotides 132–1814 (Osborn et al., 1989), OP probe was a 1.4 kb BamHI-HindIII fragment of the rat OP cDNA (Giachelli et al., 1991), ICAM-1 probe was a 1.8 kb Sal1-Kpn1 fragment of the human cDNA (Stauton et al., 1989) and FN probe was a AvaI-HindIII fragment consisting of 649 nucleotides of the human FN (Meyers, 1993). The following probes, kindly provided by Dr Pierre Chambon (Institut de Genetique et de Biologie Moléculaire et Cellulaire, Strasbourg, France) were used for Northern blot hybridization of RARs and RXRs: RAR-α probe was a 1.7 kb EcoRI fragment of the human RAR-α cDNA (Petkovich et al., 1987); RAR-β probe was a 1.5 kb BamHI-SacI fragment of the human RAR-β cDNA (Brand et al., 1988); RAR-γ probe was a 1.5 kb EcoRI fragment of the human RAR-γ cDNA (Krust et al., 1989); RXR-α probe was a 1.4 kb XbaI-Xho fragment of the human RXR-α cDNA (Leid et al., 1992); RXR-β probe was a 1.8 kb fragment of the human RXR-β cDNA (Leid et al., 1992) and RXR-γ probe was a 2.0 kb EcoRI fragment of the human RXR-γ cDNA (Leid et al., 1992).
Four different primary antibodies were used in these studies: (1) Monoclonal mouse antibody anti-human VCAM-1 (catalogue no. BBA5, R&D Systems); (2) Monoclonal mouse antibody anti-human ICAM-1 (catalogue no. BBA3, R&D Systems); (3) Polyclonal rabbit antibody anti-human OP (catalogue no. AB1870, Chemicon); and (4) Monoclonal mouse antibody anti-human fibronectin cell binding domain (catalogue no. MAB1937, Chemicon). Secondary antibodies conjugated to horseradish peroxidase were either anti-mouse IgG–against primary monoclonal antibodies–(catalogue no. A4416, Sigma) or anti-rabbit IgG–against the primary polyclonal antibody – (catalogue no. AP132P, Chemicon).
Cell culture
Human mesangial cells were obtained from adult specimens, as we have previously described (Diez et al., 1995). The identity of the cells was confirmed by morphologic and functional criteria. Under phase-contrast microscopy all the cells appeared large and stellate and no cells with epithelial or endothelial morphologic characteristics were seen. Mesangial cells showed histochemical evidence of containing actomyosin fibres, and they did not stain for factor VIII, unlike endothelial cells. In addition all the cells examined contracted after incubation with platelet activating factor, angiotensin II and arginine-vasopressin. Cultured medium was made of RPMI 1640 supplemented with 10% foetal calf serum (FCS), 200 mM L-glutamine and antibiotics (penicillin 100 u ml−1, streptomycin 100 μg ml−1 and amphotericin B 0.25 μg ml−1). Confluent cells between 12th and 15th passages were used and they were made quiescent when appropriate by 48 h incubation with medium supplemented with 0.5% FCS.
Experimental design
9-cRA was dissolved in ethanol and appropriate controls consisting of either vehicle (final concentration 0.09%) or medium were always included (when necessary, retinoid effects were first tested in dose-response and time-response experiments (in some experiments tRA effects were also tested)). Studies performed were: (1) Modulation of cell number. We assessed the inhibitory effect of 9-cRA on FCS-stimulated mesangial cell proliferation by three different methods: direct cell count, [3H]-thymidine incorporation and reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). We also studied retinoid toxicity. These later studies were mainly performed with tRA (since it was a stronger inducer of apoptosis than 9-cRA) and they consisted of assessment of the loss of viable cells, identification of apoptotic death and changes in antioxidant defence systems such as catalase activity and reduced glutathione content; (2) Prevention of H2O2-induced cell death. Quiescent CHMC in 96-well plates (typically 20,000 cells per well) were preincubated either with 9-cRA or tRA for 24 h and then incubated with H2O2 for 24 h. H2O2-induced cytotoxicity was quantified, as previously described (Moreno et al., 1999b), by measuring LDH efflux from the cytosol of the damaged cells. Since tRA stimulates antioxidant defences (Moreno et al., 1999b), additional experiments were performed to compare the relative potency of tRA and 9-cRA in terms of maximal stimulation of the GSH system and to confirm if prevention was really due to increased H2O2 catabolism (by measuring the rate of comsuption of H2O2 exogenously added to CHMC preincubated for 24 h with the either 9-cRA or tRA); (3) Inhibition of monocyte adhesion to CHMC. Experiments to study the inhibition by 9-cRA of monocyte adhesion to CHMC stimulated with 20% FCS were performed as follows: confluent CHMC were incubated for 24 h in 20% FCS containing 9-cRA or vehicle. Cells were then washed and human monocytes, isolated from blood from healthy donors, were added to CHMC. After coincubation for 2 h, the adhesion of monocytes to CHMC was quantified as described below; (4) Inhibition of the expression of FN, OP, ICAM-1 and VCAM-1. Experiments similar to those described above were performed. At definite time intervals along the incubation period with 9-cRA, total RNA or protein was extracted and the expression of the mentioned molecules was assessed by Northern and Western blot techniques; (5) Expression of mRNA for RARs and RXRs in mesangial cells and its modulation by 9-cRA. Retinoid effects were tested in time-response experiments, using a concentration of 1 μM to ensure a maximal response. At definite time intervals along the incubation period, total RNA was extracted and the mRNA expression of the mentioned molecules was assessed by Northern blot techniques.
Analytic procedures
Measurement of cell proliferation
Cells at ≈80% confluence in 96-well plates (MTT assay) or in 24-well plates ([3H]-thymidine incorporation) or in 6-well plates (direct cell count) were incubated in RPMI 1640 medium +0.5% FCS for 48 h to induce quiescence. Cells were then preincubated for 2 h with the retinoids, and then with 20% FCS for 24 h. Monolayers were then tested for the ability to reduce exogenous 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan (McGahon et al., 1995) or to incorporate [3H]-thymidine. Each experimental condition was assayed in quadruplicate. The MTT assay was performed in cells incubated for 6 h with MTT (0.15 μg ml−1). The blue formazan product was solubilized with 0.04 M HCl in isopropanol and the plates were then read on a microELISA reader using a test wavelength of 595 nm (McGahon et al., 1995). Alternatively, treated cells were incubated for 6 h with [3H]-thymidine (1 μCi well−1, 10 Ci mmol−1, American Radiolabeled Chemicals, St Louis, MO, U.S.A.) and, after removing medium, cells were washed three times with ice-cold phosphate-buffered saline. Then ice-cold 10% trichloroacetic acid was added to remove acid-soluble materials and, 5 min later, it was removed. Cells were then solubilized overnight in 0.1 M NaOH and subjected to liquid scintillation counting. Finally, direct cell counting was performed on trypsinized cells from 6-well plates, non-viable cells (Trypan blue analysis) were excluded from the count.
Apoptosis assays
(1) Microscopic analyses: Morphologic examination was performed by light and fluorescent microscopy. Cells for light microscopy were fixed using 10% buffered formaline and stained for 60 min with Giemsa reagent. Cells were also examined under fluorescent microscopy, after staining with acridine orange (4 μg ml−1) for 10 min. Apoptosis was identified using morphological criteria including pycnotic nuclei, marked chromatin condensation, nuclear fragmentation into spherical structures, and apoptotic bodies. We used as a positive control, mesangial cells cultured without foetal calf serum for 6 days, and as a negative control mesangial cells cultured with 15% of foetal calf serum. (2) In situ detection of apoptosis: Mesangial cells were washed and fixed during 20 min at room temperature with 4% formaldehyde containing phosphate-buffered saline. Then, in situ end labelling (TUNEL) was performed by using the In situ cell death detection kit (Boehringer Mannheim) according to the manufacturer's instructions. (3) Trypan blue analysis: Cells were trypsinized, stained with trypan blue dye and total cell count was performed in a Neubauer chamber. Assays were repeated four times. (4) Measurement of antioxidant defences: (i) Catalase activity (Aebi, 1983): Sample was diluted in phosphate buffer (KH2PO4 50 mM, pH 7.0) containing 0.2% Triton-X-100 and 2 ml were added to 1 ml of 30 mM H2O2. Changes in absorbance at 240 nm were measured for 30 s. The rate constant of a first-order reaction (k) was used as a unit by the equation
where t2−t1 is the measured interval in seconds and A1 and A2 are the absorbances at initial and final measurement points, respectively. Results were corrected to cellular protein content (Lowry et al., 1951): (ii) Reduced glutathione content: Cells were lysed in a cold room and proteins were precipitated with 0.9 ml perchloric acid. After neutralization with 0.3 ml 1 M KOH/KHCO3 and centrifugation, 0.15 ml of supernatant were collected on plastic tubes, o-phtal-dialdehyde was added (0.15 ml of a 7.46 μM solution) and tubes were incubated at room temperature for 15 min. GSH was measured in a scanning fluorescence spectrophotometer (Perkin-Elmer model LS-5B) at 420 nm with excitation wavelength of 350 nm, using a standard curve (Hissin & Hilf, 1976). Results were corrected for cellular protein.
H2O2-induced cytotoxicity
The cytotoxicity detection kit (LDH) (supplied by Boehringer Mannheim) was used according to the manufacturer's instructions. Each experimental condition was assayed in quadruplicate.
H2O2 assay
Consumption of H2O2 was measured by the horseradish peroxidase-mediated oxidation of phenol red (Pick & Keisari, 1980), as described for renal tissue (Ricardo et al., 1994): H2O2 (100 μM final concentration) was added to CHMC and, at the appropriate times, 40 μl of medium were removed and added to 1.12 ml of the phenol red solution (0.1 g phenol red l−1), 140 mM NaCl, 10 mM potassium phosphate buffer and 5.5 mM dextrose) with 40 μl of horseradish peroxidase (1.5 g/l). The cuvettes were incubated for 5 min at room temperature and brought to pH 12.5 by the addition of 72 μl of 1 N NaOH. Absorbance was read at 610 nm against an H2O2-standard curve (Ricardo et al., 1994).
Human monocyte isolation and adhesion assays
Human monocytes were isolated from 25 ml heparinized blood from healthy donors (Heidenreich et al., 1996). In brief, mononuclear cells were prepared by Ficoll-Hypaque density gradient centrifugation (400×g, 30 min), washed and seeded on plastic culture plates (Falcon, U.S.A.) containing RPMI1640 medium and 20% FCS in 5% CO2/95% air atmosphere in order to enrich the sample in monocytes by 1 h adherence to culture plates (Heidenreich et al., 1996). This procedure gave a purity >90% as determined by flow cytometry using CD14 antibodies as an specific monocyte-macrophage marker (Passlick et al., 1989), and a viability >95% as detected by trypan blue exclusion.
Monocyte adhesion to CHMC was tested by a previously described method (Shih et al., 1999) to study monocyte/endothelial interactions, with several modifications. Enriched monocytes (800,000 ml−1) in complete RPMI 1640 medium were layered onto confluent monolayers of CHMC in 35 mm Petri plates (total volume: 400 μl per plate) and cells were co-incubated at 37°C for 2 h (this time was chosen on the basis of preliminary time-course experiments). The suspension was then aspirated, and non-adherent monocytes were dislodged by gently rinsing the monolayers three times with sterile phosphate-buffered saline (pH 7.3). Cells were fixed with methanol/ethanol 1 : 1 (v v−1) for 30 min on ice and then stained with Giemsa during 3 h. These experiments were performed in triplicate plates for every experimental condition. Under light microscopy (×100), CHMC grew in monolayers and showed a characteristic stellate appearance with smooth peripheral borders, whereas monocytes exhibited a round profile, with little presence of cytoplasm and a pycnotic nucleus, and a cell size around 3–5 times smaller than CHMC. Identification of these cells as monocytes was confirmed by staining for nonspecific esterase. Counts of adherent monocytes was performed by three independent observers in a double-blind fashion (every observer selected in a random way 10 microscopic fields in every plate).
Northern blots
mRNA levels were measured in total RNA extracts isolated from CHMC (Chomczynsky & Sacchi, 1987). Total RNA was electrophoresed in a denaturing 1% agarose gel, transferred to a nylon membrane and probed with [32P]-labeled-probes. The filters were washed at room temperature twice with 2× standard saline citrate (1× standard saline citrate: 0.15 M NaCl, 0.015 M Na3-citrate, pH 7.0), at 65°C twice with 1× standard saline citrate containing 1% sodium dodecylsulfate and at 65°C twice with 0.1× standard saline citrate containing 0.1% sodium dodecyl sulphate. As a loading control, blots were also probed with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. The blots were exposed to film (Kodak, XOMAT) or visualized in a Phosphorimager (Molecular Dynamics, Sunnyvale, CA, U.S.A.).
Western blots
Mesangial cells were solubilized with a combination of detergents and protease inhibitors, as described previously (Shih et al., 1999), and boiled for 5 min before being separated on a 5–10% SDS–PAGE; then the proteins were transferred onto nitrocellulose membranes. Membranes were incubated for 1 h with the antibodies at a concentration of 1–2 μg ml−1. Blots were developed using secondary antibodies conjugated to horseradish peroxidase for viewing with enhanced chemiluminescence (Amersham).
Statistics
Data were compared using Student's t-test, and P<0.05 was considered statistically significant. When multiple mean values were involved, an ANOVA was first performed followed by post hoc comparisons performed with a Bonferroni/Dunn test.
Results
Inhibition of cell proliferation and retinoid toxicity
Inhibition of cell proliferation
We asked if 9-cRA would inhibit CHMC proliferation. We therefore added 20% FCS to quiescent CHMC preincubated or not with 9-cRA. FCS added alone stimulated a 15–18% increase in MTT reduction and cell count increased from 335,209±18,204 cells per well to 381,204±23,326 cells per well (n=3). 9-cRA caused dose-dependent inhibition of MTT reduction (Figure 1) within a narrow dose range: threshold inhibition was found at 10 nM 9-cRA and maximal inhibition of FCS-stimulated MTT reduction occurred at 100 nM 9-cRA (cell count was 346,207±21,409 cells per well). In a similar way, maximal inhibition of FCS-stimulated [3H]-thymidine incorporation occurred at 100 nM 9-cRA (the per cent of inhibition was 61±5, n=3) Maximal inhibition by tRA (the per cent of inhibition was 69±8, n=3) was of similar magnitude to that found with 9-cRA but the inhibitory threshold was found to be 5 μM, that is to say 500 fold higher than the threshold for 9-cRA antiproliferative effect. 9-cRA at 1 nM to 25 μM neither reduced mesangial cell viability as judged by trypan blue dye exclusion nor induced any apoptosis in the range within the range of concentrations tested for the proliferation studies (results are not shown).
Figure 1.
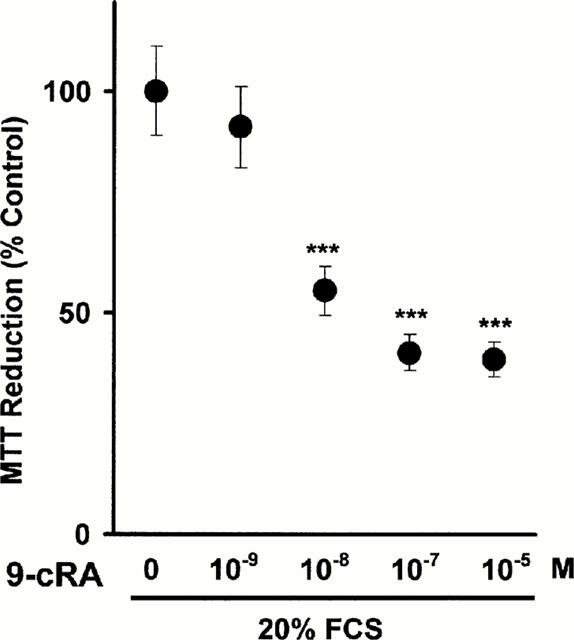
Inhibition by 9-cRA of FCS-stimulated mesangial cell proliferation (measured as MTT reduction). Quiescent mesangial cells were incubated for 2 h with 9-cRA. FCS (20%) or an equivalent amount of medium (to obtain the basal values of every experimental condition) was then added and cells were incubated for 24 h. Cell proliferation was quantified by measuring the ability of monolayers to reduce exogenous MTT to formazan after 6 h incubation (as described in Methods). In control cells, the difference between the absorbances corresponding to 20% FCS-stimulated cells versus 0.5% FCS-quiescent cells was considered as 100%. In 9-cRA treated cells, the difference between absorbances found in 20% FCS-stimulated cells versus 0.5% FCS cells was obtained and then results were expressed as per cent of change with respect the 100% established for control cells. There were no statistically significant differences between the absorbances found in 0.5% FCS cells in the various experimental conditions tested. Results are the mean±s.d of three separate experiments in quadruplicate. ANOVA, ***P<0.001 vs 0.
Effects of high concentrations of retinoids on the morphology and viability of mesangial cells
We first performed microscopic examination of whether occurrence of apoptosis after treatment with retinoids occurred in CHMC or not. Because of the absence of definitive molecular markers, morphologic examination is the only way to be sure that cells are undergoing apoptosis (McGahon et al., 1995). Before treatment, the mesangial cells grew in monolayers until confluence was reached. After exposure to 25 μM tRA or 50 μM 9-cRA for 24 to 72 h, however, significant numbers of cells showed the typical morphology of apoptosis including pycnotic nuclei, marked chromatin condensation, nuclear fragmentation into spherical structures, and apoptotic bodies (Figure 2A, left side, upper panels). Such cells also exhibited condensed chromatin acridine orange dye in contrast to the control (Figure 2A, left side, lower panels). We also assessed retinoid-induced apoptotic cell death in mesangial cells by TUNEL determination of DNA fragmentation (Figure 2B). The number of TUNEL-positive cells – this indicating the occurrence of DNA fragmentation – was significantly increased by incubation of cells for 24 to 72 h either in the presence of 50 μM 9-cRA or in the absence of FCS. Again 50 μM 9-cRA produced similar effects than 25 μM tRA (results not shown). Few positive cells were observed when cells were cultured in the presence of 20% FCS, and this was a consistent background level of apoptosis. Given that tRA was a stronger apoptosis inductor than 9-cRA, most of the remaining studies were performed with tRA. To quantify the effect of tRA on mesangial cell death, we incubated cells for various intervals of time with 25 μM tRA and then measured both total cell count and cell viability: the incubation of mesangial cells with tRA resulted in loss of viable cells (Figure 2A, right side).
Figure 2.
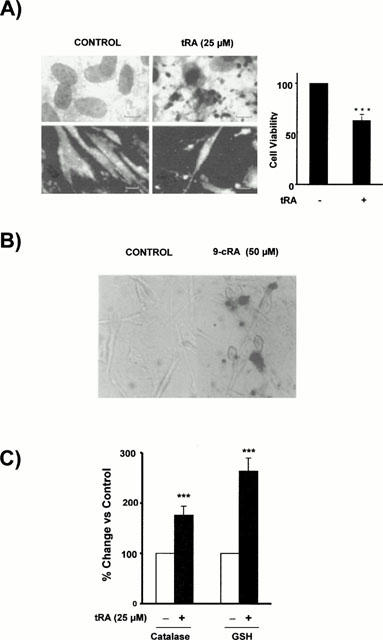
Retinoid-induced apoptosis in CHMC. (A) Effect of high concentrations of tRA on the morphology and viability of CHMC. The left panel shows light microphotographs (upper panels) where apoptotic mesangial cells exhibits nuclear fragmentation into spherical structures and condensed cytoplasm, but they are still surrounded by a cell membrane, and fluorescent microphotographs (lower panels) where several nuclei with condensed chromatin are observed. The right panel shows the decrease in viable cells (as assessed by direct count under light microscopy of cells excluding trypan blue dye) after 24 h incubation with 25 μM tRA (Student's t-test ***P<0.001 vs control). (B) Effect of 9-cRA on DNA fragmentation in cultured human mesangial cells. Cells were incubated for 48 h in control conditions or with 50 μM 9-cRA. Then TUNEL staining was performed using terminal deoxynucleotidyl transferase, an enzyme which catalyzes incorporation of fluorescein-dUTP at sites of DNA breaks. The reaction was amplified by alkaline phosphatase enzyme. Apoptotic cells appear as red nuclei whereas non-apoptotic cells were non-stained. Micrographs correspond to control cells (left) or to 9-cRA incubated cells (right). (C) Effect 25 μM tRA on the catalase activity and reduced glutathione (GSH) content in CHMC. Cells were incubated for 48 h in control conditions or with 25 μM tRA. Then catalase activity and GSH content were measured as described in Methods. (Student's t-test ***P<0.001 vs control).
Time course profile of catalase activity and reduced glutathione content during tRA-induced apoptosis
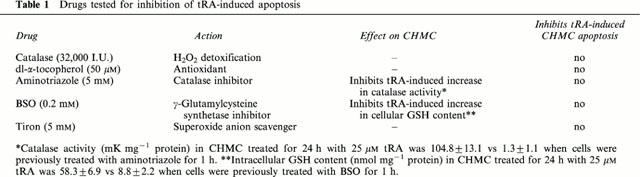
We have previously found (Moreno et al., 1999b) that non-apoptotic concentrations of tRA stimulate catalase activity and increase reduced glutathione content in a concentration-dependent, time-dependent manner. Here we found (Figure 2C) that when tRA is added at doses high enough to induce apoptosis, there is also an increase in the antioxidant defences coinciding with the development of the apoptotic cell death. We then studied whether there was a cause-effect relationship by exploring two opposite possibilities: first, that the increase in the antioxidant defences is a defensive cellular response to oxidative stress, which is relevant since oxygen free radicals are involved in several models of apoptosis (Buttke & Sandstrom, 1994). However, preincubation with the antioxidant dl-α-tocopherol, or with catalase, which specifically detoxifies H2O2, or with with the cell-permeable O2− scavenger tiron (Hein, 1998), did not inhibit tRA-induced CHMC apoptotic death. Secondly we studied the possibility that catalase and/or GSH were directly involved in the apoptotic signalling. Again, tRA-triggered apoptosis was not inhibited (Table 1) by aminotriazol, an irreversible inhibitor of catalase activity (Aebi, 1983), or by L-buthionine-(S-R)-sulphoximine (BSO), an irreversible inhibitor of γ-glutamylcysteine synthetase, the rate-limiting step of glutathione synthesis (Griffith, 1982).
Table 1.
Drugs tested for inhibition of tRA-induced apoptosis
Prevention of H2O2-induced cell death
H2O2 induces concentration-dependent damage in mesangial cells, characterized by apoptosis followed by secondary necrosis with plasma membrane damage resulting in loss of cytosolic LDH (Moreno et al., 1999b). H2O2-induced LDH release is clearly abolished by tRA, within a narrow concentration range: 10 μM tRA has the maximal protective action against H2O2 and 1 μM tRA has no effect at all (Moreno et al., 1999b). Figure 3A shows the results obtained when using retinoids at concentrations high enough to reach maximal prevention against H2O2. It is clear that maximal prevention with 9-cRA requires a 1000 fold lower concentration than that required with tRA. Since H2O2-induced cell death is apoptotic (Moreno et al., 1999a), we also confirmed by morphological examination with acridine orange-ethidium bromide staining that H2O2-induced apoptosis was suppressed by pretreatment with retinoids (results are not shown).
Figure 3.
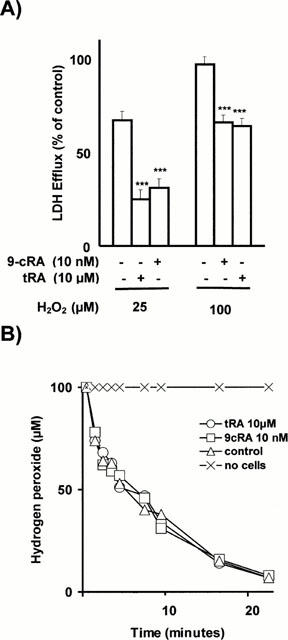
(A) Prevention by retinoids of H2O2-induced cytotoxicity. Confluent, quiescent CHMC cultured in 96-well microtiter plates, were incubated for 24 h with either 10 nM 9-cRA or 10 μM tRA. Cells were then washed and incubated for 24 h with either 25 or 100 μM H2O2. Control cells were incubated in parallel but without adding retinoids and H2O2. Cytotoxicity was quantified by measuring LDH efflux from the cytosol of the damaged cells to the supernatant. Results were expressed as the per cent of the control value (LDH released by control cells incubated with 2% Triton-X-100 was considered as 100% LDH efflux) and are mean±s.d. of three separate experiments in quadruplicate. As shown, 10 nM 9-cRA and 10 μM tRA prevented in a similar extension the H2O2-induced loss of cell viability (***P<0.001). No differences in LDH release were found beween control cells and cells incubated with 9-cRA or tRA (results are not shown in the figure). (B) Consumption of H2O2 by cells incubated with retinoids. Confluent, quiescent CHMC cultured in 35 mm Petri dishes, were incubated for 48 h with either 10 nM 9-cRA or 10 μM tRA in order to stimulate maximally antioxidant defences. Cells were then washed and incubated with 100 μM H2O2 from 0 to 22 min. Control cells were incubated in parallel but without adding retinoids. At the times shown in the figure, H2O2 was measured in an aliquot of medium by the horseradish peroxidase-mediated oxidation of phenol red method. Results are the mean±s.d. of three separate experiments in triplicate. No statistically significant differences were found in the consumption of H2O2 between the three experimental groups (ANOVA P>0.05).
Additional experiments were performed to assess if this protective effect was due to retinoid-induced stimulation of antioxidant defences. This possibility was raised from previous results (Moreno et al., 1999b) showing that catalase activity and GSH content are increased by 2.5 fold in tRA-incubated CHMC. We studied first the relative potency of tRA and 9-cRA in terms of maximal stimulation of the GSH system. Time-course experiments have previously shown (Moreno et al., 1999b) that a 48 h incubation with 10 μM tRA is needed to reach a maximal increase in the intracellular GSH content from CHMC. We have studied here the relative potency of the two retinoids and found in four separate experiments in triplicate that, after a 48 h incubation with 10 μM of either 9-cRA or tRA, the respective intracellular GSH contents (nmol mg−1 protein) were 68+5 and 53+4 (results are mean±s.d.; Student's t-test: P<0.001) whereas the control values were 23+3: that is to say, 9-cRA was a stronger inducer of the increase in GSH than tRA. However, stimulation of antioxidant defences by retinoids appears not to play a role in the preventive effect of retinoids against H2O2 since preincubation with aminotriazole (an irreversible inhibitor of catalase) or with BSO (an inhibitor of the synthesis of glutathione) did not abolish it (results are not shown) and, in fact, CHMC incubated with these retinoids did not exhibit a higher rate of catabolism of H2O2 exogenously added (Figure 3B).
Inhibition of monocyte adhesion to CHMC
The co-culture of monocytes with the FCS-stimulated CHMC resulted in time-dependent adhesion of monocytes to the underlying mesangial monolayer. Adhesion was substantially inhibited when CHMC were pretreated with 9-cRA (Figure 4) but within a narrow concentration range: maximal inhibition was found at 1 μM and the threshold inhibition appeared at 10 nM (results are not shown). Although we do not show the results, we found that tRA inhibited adhesion to a similar extension.
Figure 4.
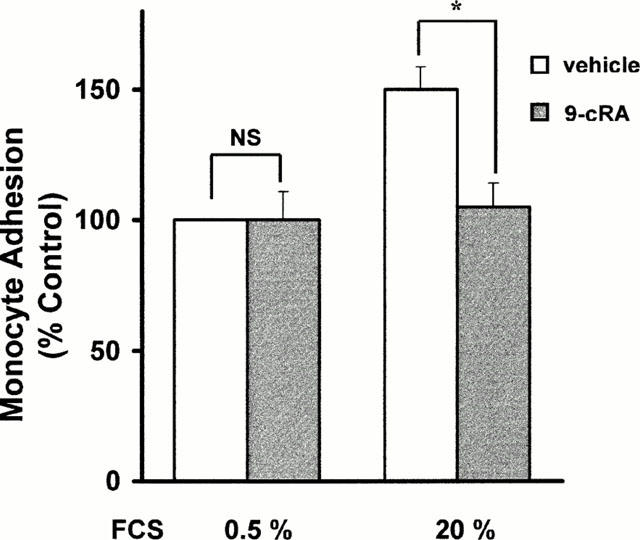
Inhibition by retinoids of the adhesion of monocytes to cultured human mesangial cells. Confluent CHMC were incubated for 24 h in 20% FCS containing 1 μM 9-cRA or vehicle. Cells were then washed and human monocytes, isolated from blood from healthy donors as described in Methods, were added to CHMC. After coincubation for 2 h, the adhesion of monocytes to CHMC was quantified as described in Methods. Data are mean±s.d. of three different experiments in triplicate. The actual value for 100% adhesion was 71±8 monocytes per well *P<0.05 vs vehicle.
Changes in the CHCM expression of FN, OP, ICAM-1 and VCAM-1 mRNA expression
We also studied the effect of 9-cRA on the mRNA and protein expression of four molecules which are involved in the adhesion of monocytes to mesangial cells: FN, OP, ICAM-1 and VCAM-1. These experiments were performed using identical experimental conditions to those used in the study of monocyte adhesion to CHMC, excepting that cells were stimulated with 10% FCS instead of 20% FCS. Results obtained may be classified into two categories: those related to molecules secreted by CHMC (i.e., FN and OP) and those involving cell adhesion molecules present in the cell surface of mesangial cells (i.e., ICAM-1 and VCAM-1). 9-cRA inhibited OP and FN mRNA and protein expression (Figures 5 and 6) in FCS-stimulated mesangial cells. On the contrary, 9-cRA had no effect on the ICAM-1 mRNA expression (data not shown) nor on the ICAM-1 protein expression (Figure 6). Finally, 9-cRA stimulated VCAM-1 mRNA expression (data not shown) but this effect did not result in increased VCAM-1 protein expression (Figure 6), this being probably due to the fact that post-transcriptional mechanisms may be relevant determinants of VCAM-1 protein expression (Lademarco et al., 1995; Pietersma et al., 1997). Although we do not show the results, tRA had similar effects on the expression of the four adhesion molecules.
Figure 5.
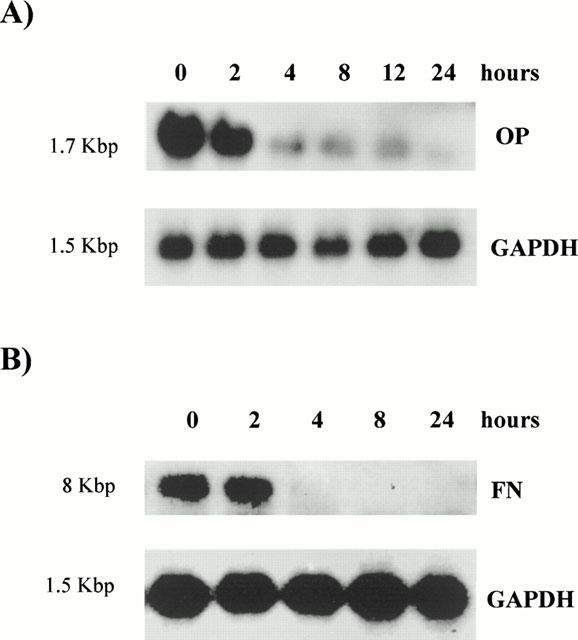
Effect of 9-cRA on osteopontin and fibronectin mRNA expression in human mesangial cells. Confluent human mesangial cells were incubated up to 24 h in 10% FCS with 1 μM 9-cRA and total RNA was extracted. Northern blotting for OP (A) and FN (B) was carried out as described in Methods. The blots were stripped and rehybridized with a cDNA probe for the housekeeping gene GAPDH to adjust for small variations in RNA loading and transfer to the filter. The autoradiograms shown are representative of three separate experiments.
Figure 6.
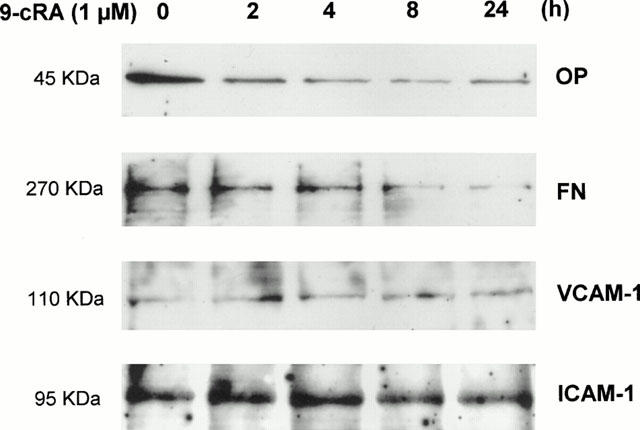
Effect of 9-cRA on the protein expression of osteopontin, fibronectin, VCAM-1 and ICAM-1 in human mesangial cells. Confluent human mesangial cells were incubated up to 24 h in 10% FCS with 1 μM 9-cRA and total protein was extracted. Twenty μg of every protein extract was electrophoresed and transferred onto nitrocellulose. Membranes were probed with primary antibodies against OP, FN, VCAM and ICAM-1 and blots were developed using secondary antibodies conjugated with horseradish peroxidase for viewing enhanced chemiluminescence as described in Methods.
Expression of mRNA for RARs and RXRs in mesangial cells and its modulation by 9-cRA
We characterized the steady state mRNA levels of all six retinoid receptors as well as their regulation by 9-cRA. As shown in Figure 7 all major RARs isotypes were expressed in CHMC regardless of the presence or absence of 9-cRA. Transcripts to RAR-α have been previously found both in normal human renal tissue (De Thé et al., 1989) and in 12 human renal cell carcinoma lines (Hoffman et al., 1996). In these lines 13-cis retinoic acid failed to increase RAR-α expression (Hoffman et al., 1996) this result being in contrast with the 9-cRA-triggered induction of this receptor in CHMC (Figure 7). RAR-γ mRNA expression was also present in CHMC and it was not modified by 9-cRA. Although previous studies also showed that the renal expression of this receptor may be modulated by nuclear hormones such as androgens (Huang et al., 1997), the retinoid 13-cis retinoic acid did not modify the RAR-γ expression in renal cell carcinoma lines (Hoffman et al., 1996). Low but reproducibly detectable transcripts to RAR-β were also present in CHMC and the addition of 9-cRA increased their levels. Increased expression of RAR-β mRNA has also been documented in several cell lines after 9-cRA treatment (Lovat et al., 1997; Levin & Davis, 1997) and it has been related to the antiproliferative effects of 13-cis-retinoic acid in human renal cell carcinomas (Hoffman et al., 1996) and to the response of renal cancer patients to 13-cis retinoic acid (Berg et al., 1999). The identification of a retinoic acid response element located on the promoter of the RAR-β gene suggests that the RAR-β gene might amplify the response to retinoids in target tissues by raising its own receptor expression level (Orfanos et al., 1997).
Figure 7.
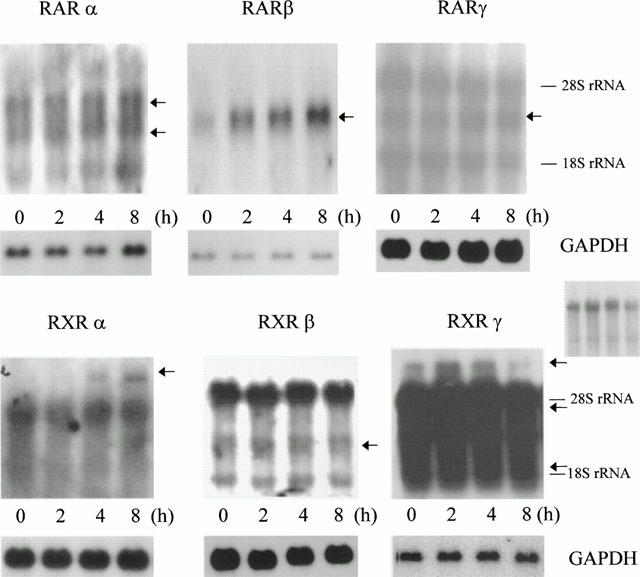
Expression of mRNA for RARs and RXRs and its modulation by 9-cRA. Quiescent, confluent human mesangial cells were incubated up to 8 h with 1 μM 9-cRA and total RNA was extracted. Northern blot for the subtypes α, β and γ of either RAR or RXR was carried out as described in Methods. The blots were stripped and rehybridized with a cDNA probe for the housekeeping gene GAPDH to adjust for small variations in RNA loading and transfer to the filter. The autoradiograms shown are representative of three separate experiments. A second RXRγ autoradiogram with lower exposition is also shown (centre right) in order to improve the observation of the two bands between 28S and 18S rRNA.
On the other hand, all major RXRs isotypes were also expressed in CHMC regardless of the presence or absence of 9-cRA. Regarding RXR-β and RXR-γ, these findings are in good agreement with previous reports showing that rat glomeruli express RXR-β transcripts (Yang et al., 1999) and RXR-γ protein (Sugawara et al., 1997). However, these studies failed to find transcripts to RXR-α and RXR-α protein in glomeruli and this contrasts with our results since we have found mRNA for RXR-α in glomerular CHMC. Interestingly, RXR-α transcripts have also been found in cultured rat mesangial cells (Q. Xu & M. Kitamura, unpublished observation). 9-cRA increased the expression of RXR-α and inhibited the expression of RXR-γ (Figure 7). Regulation of RXRs may be particularly relevant since they have been shown to function as coregulators that enhance the binding of other nuclear hormone receptors (including RARs, thyroid hormone receptor, vitamin D receptor and peroxisome proliferator-activated receptors) to their cognate response elements (reviewed by Giguere, 1999).
Discussion
Glomerular cells and especially mesangial cells play an active part in the inflammatory response to glomerular injury (Pfeilschifter, 1994). This work is mainly a prospective study on the possibility that 9-cRA may act as an anti-inflammatory agent at the mesangial cell level. The approach involved the study of three relevant effects: inhibition of CHMC proliferation, prevention of H2O2-induced mesangial cell death, and inhibition by retinoids of monocyte adhesion to CHMC.
Glomerular mesangial cell proliferation plays an important role in the pathogenesis of many types of progressive glomerular disease (Pfeilschifter, 1994) and its inhibition has, therefore, a potential therapeutic interest. 9-cRA had antiproliferative action on CHMC (Figure 1), a well known effect of retinoids on many normal and transformed cells (Giguere, 1994). tRA also inhibited CHMC proliferation–although the concentration required was 500 fold higher–and this effect has been attributted to the inhibition of activating protein-1 activity in CHMC (Simonson, 1994). Ligand-activated RARs, which are able to inhibit activating-protein-1 activity (Giguere, 1994), have been traditionally linked to the antiproliferative effect of retinoids. Therefore this could be one of the mechanisms through which 9-cRA, an RAR ligand (Allenby et al., 1994), exerts its antiproliferative effect on CHMC. However, a contribution of RXRs can not be ruled out: recent work suggest that activation of RXRs by selective retinoids can also mediate growth inhibitory effects (Wan et al., 1998) and 9-cRA activates both RARs and RXRs (Heyman et al., 1992; Levin et al., 1992).
The potent growth-inhibitory properties of 9-cRA on serum-stimulated CHMC are not associated with cytotoxicity. However, we asked if higher concentrations are toxic for CHMC and we found that concentrations of 9-cRA higher than 50 μM induced apoptosis (Figure 2). This concentration is much greater than the concentrations required for the effects with a potential anti-inflammatory value (see Results). In contrast, apoptosis in CHMC was found with 25 μM tRA (that is to say, a concentration slightly higher than that needed to obtain anti-inflammatory effects), although the onset of tRA-induced apoptosis was associated with a strong increase in both catalase activity and GSH. Further studies using several modulators (see the Results section) did not confirm our effect suggesting that the oxidation/antioxidation balance might be involved in the mechanism of retinoid-induced apoptosis. RARs are, on the contrary, likely to play a role in the mechanism of retinoid-induced CHMC apoptosis since TTNPB, another RAR pan-agonist like tRA (Orfanos et al., 1997), induced apoptosis at concentrations as low as 10 μM (F.J. Lucio, unpublished data).
The results obtained for the prevention by retinoids of H2O2-induced mesangial cell death are exactly equivalent to those found in the experiments of inhibition of CHMC proliferation: maximal prevention with 9-cRA requires a 1000 fold lower concentration than that required with tRA (Figure 3A). Recently we have found that the apoptotic signal of H2O2 on CHMC (Ishikawa et al., 1997) is blocked by tRA inhibiting both c-Jun NH2-terminal kinase and activating protein-1 expression (Moreno et al., 1999a). This is the most likely mechanism involved in the preventive effect of retinoids against H2O2-induced cell death since our results (see Figure 3B and Results section) exclude any role of antioxidant defences.
9-cRA also inhibited monocyte adhesion to FCS-stimulated CHMC (Figure 4). Western blot analysis of adhesion molecules revealed that 9-cRA inhibited the expression of FN and OP (Figure 6) but not VCAM-1 and ICAM-1 expression. The inhibitory effect of the retinoid on the mRNA expression of FN and OP (Figure 5) is an effect likely responsible for the inhibition of the expression of the respective proteins. Although VCAM-1, ICAM-1, FN and OP are involved in monocyte adhesion to mesangial cells (Chana & Wheeler, 1999; Nagasaki et al., 1997; Denton et al., 1991; Khachigian et al., 1997) it is worthy of remark that increased monocyte adhesion does not necessarily correlate to increased VCAM-1 or ICAM-1 expression. For instance, in the model of low-density lipoprotein-stimulated monocyte adhesion to cultured human mesangial cells–characterized by increased expression of VCAM-1, ICAM-1 and FN–monocyte adhesion is inhibited by treatment of CHMC with the antibody anti-fibronectin but not with antibodies anti-VCAM-1 or anti-ICAM-1 (Chana & Wheeler, 1999). In the same way, monocyte entry in human atherosclerotic lesions does not correlate with the endothelial areas of increased VCAM-1 expression but with increased expression of connecting segment-1 of FN (Shih et al., 1999). Therefore, it may be speculated that the inhibition of the expression of FN and OP might be sufficient to explain the retinoid-induced inhibition of monocyte adhesion to CHMC. However, specific studies must be undertaken to confirm this point.
Retinoids are known to exert their biological effects by binding to specific nuclear receptors. Each subtype of nuclear retinoid receptors is thought to regulate the expression of distinct genes because the subtypes exhibit specific patterns of expression during embryonal development and different distribution in adult tissues (Giguere, 1994). Therefore we characterized the RAR/RXR mRNA expression pattern in CHMC (Figure 7). Several interesting observations evolved from this characterization. First, the presence of all three RARs and all three RXRs, implies that mesangial cells can respond to exogenously administered retinoids in a receptor-mediated manner. Taking into account that endogenous ligand activity of 9-cRA has been detected in kidney (Heyman et al., 1992), the presence of RARs and RXRs in mesangial cells suggests that these receptors are likely to play a significant role in controlling mesangial-specific gene expression and mesangial cell phenotype in vivo. This notion is supported by our data showing that CHMC are highly responsive to 9-cRA in terms of modulation of RARs and RXRs: 9-cRA treatment stimulated expression of RAR-α, RAR-β, and RXR-α genes, and it inhibited the expression of RXR-γ. These changes may be critical for the effects of 9-cRA. Upon binding to RARs and RXRs, retinoids regulate cellular processes by directly modulating the expression of responsive genes. Therefore the changes induced by 9-cRA on mRNA expression of the RARs and RXRs may be critical for the effects of this retinoid on CHMC. On the other hand, the changes in RXRs could trigger a broad spectrum of phenotypic modifications given that the regulation of the expression of target genes by nuclear hormone receptors, including RARs, VDR (vitamin D receptors), TR (thyroid receptors) and PPARs (peroxisome proliferator activated receptors), usually requires prior ligand-activated nuclear hormone receptor heterodimerization with RXRs (Giguere, 1994). It is important to emphasize, however, that our studies have not assigned a retinoid receptor-mediated process to any action of 9-cRA in CHMC. Such studies will require dominant negative retinoid receptors, synthetic retinoids that specifically activate one given receptor or that bind specific receptors without activating them, or the cultivation of glomerular mesangial cells from knock-out mice deficient in specific retinoid receptors.
The glomerular capillary network is unique in that circulating phagocytes may potentially adhere to both endothelial and mesangial cells, as the latter may be found interposed between endothelial cells (Schena et al., 1993). The fact that 9-cRA inhibited monocyte adhesion to FCS-stimulated CHMC is particularly important (FCS resembles an inflammatory situation, given that it contains a high number of active factors present during inflammation) taking into account that monocytes can be considered as the real effectors of the immune system in the pathogenesis of glomerular diseases (Schena et al., 1997). The same applies to the prevention of mesangial cell death by H2O2 – that may be released from activated monocytes/macrophages (Schena et al., 1997)–and to the inhibition of FCS-induced mesangial cell proliferation. The potential of 9-cRA for the treatment of inflammatory glomerular diseases is even more evident if we take into account the fact that the concentration of 9-cRA used in our studies is comparable to the blood levels found in cancer patients receiving this retinoid (Rizvi et al., 1998). However, the relevance of the findings presented here needs to be confirmed by in vivo studies in animal models of glomerulonephritis.
Acknowledgments
This work was supported by grants SAF99-0085 from the Spanish Comisión Interministerial de Ciencia y Tecnología and 08.06/0014/99 from the Comunidad de Madrid. V. Moreno Manzano is supported by a grant from the Comunidad de Madrid. Juan Carlos Sepúlveda is supported by a grant of the Spanish Ministerio de Educacion, Cultura y Deportes. We gratefully acknowledge Ana Morales Entrena for expert technical assistance. We are also grateful to Productos Roche S.A. for providing the retinoids and to Dr P. Chambon for providing the RARs and RXRs probes, to Dr Mushtaq Ahmad for providing the VCAM-1 cDNA probe, to Dr C.M. Giachelli for providing the OP cDNA probe and to Dr Jennifer Laird for careful reading of the manuscript.
Abbreviations
- BSO
L-buthionine-(S-R)-sulphoximine
- CHMC
cultured human mesangial cells
- FCS
foetal calf serum
- FN
fibronectin
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSH
reduced glutathione
- ICAM-1
intercellular molecule adhesion-1
- LDH
lactate dehydrogenase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OP
osteopontin
- RARs
retinoic acid receptors
- RXRs
retinoid X receptors
- tRA
all-trans-retinoic acid
- VCAM-1
vascular cell adhesion molecule-1
- 9-cRA
9-cis retinoic acid
References
- AEBI H.E.Catalase Methods of Enzymatic Analysis vol III. Enzymes I: Oxidoreductases, transferases 1983Basel: Verlag-Chemie; 273–285.ed. Bergmeyer H.U. pp [Google Scholar]
- ALLENBY G., JANOCHA R., KAZMER S., SPECK J., GRIPPO J.F., LEVIN A.A. Binding of 9-cis-retinoic acid and all-trans-retinoic acid to retinoic acid receptors α, β and γ. J. Biol. Chem. 1994;269:16689–16695. [PubMed] [Google Scholar]
- BERG W.J., NANUS D.M., LEUNG A., BROWN K.T., HUTCHINSON B., MAZUMDAR M., XU X., LOTAN R., REUTER V.E., MOTZER R.J. Up-regulation of retinoic acid receptor β expression in renal cancers in vivo correlates with reponse to 13-cis-retinoic acid and interferon-α-2a. Clin. Cancer Res. 1999;5:1671–1675. [PubMed] [Google Scholar]
- BRAND N., PETKOVICH M., KRUST A., CHAMBON P., DE THE H., MARCHIO A., TIOLLAIS P., DEJEAN A. Identification of a second human retinoic acid receptor. Nature. 1988;332:850–853. doi: 10.1038/332850a0. [DOI] [PubMed] [Google Scholar]
- BUTTKE T.M, SANDSTROM P.A. Oxidative stress as a mediator of apoptosis. Immunol. Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- CHANA R.S., WHEELER D.C. Fibronectin augments monocyte adhesion to low-density lipoprotein-stimulated mesangial cells. Kidney Int. 1999;55:179–188. doi: 10.1046/j.1523-1755.1999.00250.x. [DOI] [PubMed] [Google Scholar]
- CHOMCZYNSKI P., SACCHI N. Single-step method of RNA isolation by guanidinium thiocyanate-pheno-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- DE THE H., MARCHIO A., TIOLLAIS P., DEJEAN A. Differential expression and ligand regulation of retinoic acid receptor α and β genes. EMBO J. 1989;8:429–433. doi: 10.1002/j.1460-2075.1989.tb03394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENTON M.D., MARSDEN P.A., LUSCINSKAS F.W., BRENNER B.M., BRADY H.R. Cytokine-induced phagocyte adhesion to human mesangial cells: role of CD11/CD18 integrins and ICAM-1. Am. J. Physiol. 1991;261:F1071–F1079. doi: 10.1152/ajprenal.1991.261.6.F1071. [DOI] [PubMed] [Google Scholar]
- DÍEZ M.L., GARCÍA-ESCRIBANO C., MEDINA J., BOYANO M.C., ARILLA E., TORRECILLA G., RODRÍGUEZ D., RODRÍGUEZ M. Effects of somatostatin on cultured human mesangial cell. Endocrinology. 1995;136:3444–3451. doi: 10.1210/endo.136.8.7628380. [DOI] [PubMed] [Google Scholar]
- GIACHELLI C.M., BAE N., LOMBARDI D., MAJESKY M., SCHWARTZ S.M. Molecular cloning and characterization of 2B7, a rat mRNA which distinguishes smooth muscle phenotypes in vitro and is identical to osteopontin (secreted phosphoprotein I, 2aR) Biochem. Biophys. Res. Commun. 1991;177:867–873. doi: 10.1016/0006-291x(91)91870-i. [DOI] [PubMed] [Google Scholar]
- GIACHELLI C.M., SCHWARTZ S.M., LIAW L. Molecular and cellular biology of osteopontin: Potential role in cardiovascular disease. Trends Cardiovasc. Med. 1995;5:88–95. doi: 10.1016/1050-1738(95)00005-T. [DOI] [PubMed] [Google Scholar]
- GIGUÈRE V. Retinoic acid receptors and cellular retinoid binding proteins: Complex interplay in retinoid signaling. Endocrine Rev. 1994;15:61–79. doi: 10.1210/edrv-15-1-61. [DOI] [PubMed] [Google Scholar]
- GIGUERE V. Orphan nuclear receptors: from gene to function. Endocrine Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- GRIFFITH U.W. Mechanism of action, metabolism and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J. Biol. Chem. 1982;257:13704–13712. [PubMed] [Google Scholar]
- HEIN T.W., KUO L. LDL impair vasomotor function of the coronary microcirculation: role of superoxide anions. Circ. Res. 1998;83:404–414. doi: 10.1161/01.res.83.4.404. [DOI] [PubMed] [Google Scholar]
- HEINDENREICH S., SCHMIDT M., BACHMANN J., HARRACH B. Apoptosis of monocytes cultured from long-term hemodialysis patients. Kidney Int. 1996;49:792–799. doi: 10.1038/ki.1996.110. [DOI] [PubMed] [Google Scholar]
- HEYMAN R.A., MANGELSDORF D.J., DYCK J.A., STEIN R.B., EICHELE G., EVANS R.M., THALLER C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- HISSIN P.J., HILF R. A fluorimetric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- HOFFMAN A.D., ENGELSTEIN D., BOGENRIEDER T, PAPANDREOU C.N., STECKELMAN E., DAVE A., MOTZER R.J., DMITROVSKY E., ALBINO A.P., NANUS D.M. Expression of retinoic acid receptor ß in human renal cell carcinomas correlates with sensitivity to the antiproliferative effects of 13-cis-retinoic acid. Clin. Cancer Res. 1996;2:1077–1082. [PubMed] [Google Scholar]
- HUANG H.F.S., LI M.T., VON HAGEN S., ZHANG Y.F., IRWIN R.J. Androgen modulation of the messenger ribonucleic acid of retinoic acid receptors in the prostate, seminal vesicles and kidney in the rat. Endocrinology. 1997;138:553–559. doi: 10.1210/endo.138.2.4945. [DOI] [PubMed] [Google Scholar]
- ISHIKAWA Y., YOKOO T., KITAMURA M. c-jun/AP-1, but not NF-κB, is a mediator for oxidant initiated apoptosis in glomerular mesangial cells. Biochem. Biophys. Res. Commun. 1997;240:496–501. doi: 10.1006/bbrc.1997.7665. [DOI] [PubMed] [Google Scholar]
- KASHGARIAN M., STERZEL R.B. The pathobiology of the mesangium. Kidney Int. 1992;41:524–529. doi: 10.1038/ki.1992.74. [DOI] [PubMed] [Google Scholar]
- KHACHIGIAN L.M., COLLINS T., FRIES J.W.U. N-Acetylcysteine blocks mesangial VCAM-1 and NF-κB expression in vivo. Am. J. Pathol. 1997;151:1225–1229. [PMC free article] [PubMed] [Google Scholar]
- KOSMEHL H., BERNDT A., KATENKAMP D. Molecular variants of fibronectin and laminin: structure, physiological occurrence and histopathological aspects. Virchows Arch. A Pathol. Anat. Histopathol. 1996;429:311–322. doi: 10.1007/BF00198435. [DOI] [PubMed] [Google Scholar]
- KRUST A., KASTNER P., PETKOVICH M., ZELENT A., CHAMBON P. A third human retinoic acid receptor, hRAR-γ. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5310–5314. doi: 10.1073/pnas.86.14.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LADEMARCO M.F., BARKS J.L., DEAN D.C. Regulation of vascular cell adhesion molecule-1 expression by IL-4 and TNF-α in cultured endothelial cells. J. Clin. Invest. 1995;95:264–271. doi: 10.1172/JCI117650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAN H.Y., YU X.Q., YANG N., NIKOLIC-PATERSON D.J., MU W., PICHLER R., JOHNSON R.J., ATKINS R.C. De novo glomerular osteopontin expression in rat crescentic glomerulonephritis. Kidney Int. 1998;53:136–145. doi: 10.1046/j.1523-1755.1998.00748.x. [DOI] [PubMed] [Google Scholar]
- LEID M., KASTNER P., LYONS R., NAKSHARI H., SAUNDERS M., ZACHAREWSKI T., CHEN J., STAUB A., GARNIER J.M., MADER S., CHAMBON P. Purification, cloning and RXR identity of the HeLa cell factor with which RAR or TR heterodimerizes to bind target sequences efficiently. Cell. 1992;68:377–395. doi: 10.1016/0092-8674(92)90478-u. [DOI] [PubMed] [Google Scholar]
- LEVIN A.A., STURZENBECKER L.J., KAZMER S., BASOKOWSKI T., HUSELTON C., ALLENBY G., SPECK J., KRATZEISEN C., ROSENBERG M., LOVEY A., GRIPPO J.F. 9-cis retinoic acid stereoisomer binds and activates the nuclear receptor RXRα. Nature. 1992;355:359–361. doi: 10.1038/355359a0. [DOI] [PubMed] [Google Scholar]
- LEVIN M.S., DAVIS A.E. Retinoic acid increases cellular retinol binding protein II mRNA and retinol uptake in the human intestinal Caco-2 cell line. J. Nutr. 1997;127:13–17. doi: 10.1093/jn/127.1.13. [DOI] [PubMed] [Google Scholar]
- LOVAT P.E., IRVING H., ANNICCHIARICO-PETRUZZELLI M., BERNASSOLA F., MALCOM A.J., PEARSON A.D., MELINO G., REDFERN C.P. Retinoids in neuroblastoma therapy: distinct biological properties of 9-cis- and all-trans-retinoic acid. Eur. J. Cancer. 1997;33:2075–2080. doi: 10.1016/s0959-8049(97)00242-6. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH K.F., FARR A.G., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;19:265–275. [PubMed] [Google Scholar]
- MCGAHON A.J., MARTIN S.J., BISSONNETTE R.P., MAHBOUBI A., SHI Y., MOGIL R.J., NISHIOKA W.K., GREEN D.R.The end of the (cell) line: Methods for the study of apoptosis in vitro Methods in Cell Biology: Vol 46. Cell Death 1995New York: Academic Press; 153–187.ed. Schwartz, L.M. and Osborne, B.A. pp [DOI] [PubMed] [Google Scholar]
- MEYERS J.C. Differential expression of type I collagen and cellular fibronectin. Kidney Int. 1993;43:45–52. doi: 10.1038/ki.1993.9. [DOI] [PubMed] [Google Scholar]
- MORENO V, ISHIKAWA Y., LUCIO F.J., KITAMURA M. Suppression of apoptosis by all-trans-retinoic acid: dual intervention via inhibition of c-Jun NH2-terminal kinase and AP-1 expression. J. Biol. Chem. 1999a;274:20251–20258. doi: 10.1074/jbc.274.29.20251. [DOI] [PubMed] [Google Scholar]
- MORENO V., RODRIGUEZ M., RODRIGUEZ D., LUCIO F.J. Tretinoin prevents age-related renal changes and stimulates antioxidant defenses in cultured renal mesangial cells. J. Pharmacol. Exp. Ther. 1999b;289:123–132. [PubMed] [Google Scholar]
- NAGASAKI T., ISHIMURA E., SHIOI A., JONO S., INABA M., NISHIZAWA Y., MORII H., OTANI S. Osteopontin gene expression and protein synthesis in cultured rat mesangial cells. Biochem. Biophys. Res. Commun. 1997;233:81–85. doi: 10.1006/bbrc.1997.6399. [DOI] [PubMed] [Google Scholar]
- NATH K.A., FISCHEREDER M., HOSTETTER T.H. The role of oxidants in progressive renal injury. Kidney Int. 1994;45 Suppl. 45:S111–S115. [PubMed] [Google Scholar]
- ORFANOS C.E., ZOUBOULIS C.C., ALMOND-ROESLER B., GEILEN C.C. Current use and future potential role of retinoids in dermatology. Drugs. 1997;53:358–388. doi: 10.2165/00003495-199753030-00003. [DOI] [PubMed] [Google Scholar]
- OSBORN L., HESSION C., TIZARD R., VASSALLO C., LUHOWSKYJ S., CHI R.G., LOBB R. Direct expression cloning of vascular cell adhesion molecule-1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. 1989;52:1203–1211. doi: 10.1016/0092-8674(89)90775-7. [DOI] [PubMed] [Google Scholar]
- PASSLICK B., FLIEGER D., ZIEGLER-HEITBROCK H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–2534. [PubMed] [Google Scholar]
- PETKOVICH N., BRAND N., KRUST A., CHAMBON P. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature. 1987;330:444–450. doi: 10.1038/330444a0. [DOI] [PubMed] [Google Scholar]
- PFEILSCHIFTER J. Mesangial cells orchestrate inflammation in the renal glomerulus. News Physiol. Sci. 1994;9:271–276. [Google Scholar]
- PICK E., KEISARI Y. A simple colorimetric method for the measurement of hydrogen peroxide produced by cells in culture. J. Immunol. Meth. 1980;38:161–170. doi: 10.1016/0022-1759(80)90340-3. [DOI] [PubMed] [Google Scholar]
- PIETERSMA A., TILLY B.C., GAESTEL M., JONG N., LEE J.C., KORSTER J.F., SLUITER W. P38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem. Biophys. Res. Commun. 1997;230:44–48. doi: 10.1006/bbrc.1996.5886. [DOI] [PubMed] [Google Scholar]
- RICARDO S.D., BERTRAM J.F., RYAN G.B. Reactive oxygen species in puromycin aminonucleoside nephrosis: In vitro studies. Kidney Int. 1994;45:1057–1069. doi: 10.1038/ki.1994.142. [DOI] [PubMed] [Google Scholar]
- RIZVI N.A., MARSHALL J.L., NESS E., YOE J., GILL G.M., TRUGLIA J.A., LOEWEN G.R., JAUNAKIS D., ULM E.H., HAWKINS M.J. Phase I study of 9-cis retinoic acid (ALRT1057 capsules) in adults with advanced cancer. Clin. Cancer. Res. 1998;4:1437–1442. [PubMed] [Google Scholar]
- SCHENA F.P., GESUALDO L., GRANDALIANO G., MONTINARO V. Progression of renal damage in human glomerulonephritides: is there sleight of hand in winning the game. Kidney Int. 1997;52:1439–1457. doi: 10.1038/ki.1997.475. [DOI] [PubMed] [Google Scholar]
- SCHENA F.P., MONTINARO V., GESUALDO L.Glomerular mesangial and epithelial cells Immunopharmacology of the Renal System 1993London: Academic Press; 49–86.ed. Tetta, C. pp [Google Scholar]
- SHIH P.T., ELICES M.J., FANG Z.T., UGAROVA T.P., STRAHL D., TERRITO M.C., FRANK J.S., KOVACH N.L., CABANAS C., BERLINER J.A., VORA D.K. Minimally modified low-density lipoproteins induces monocyte adhesion to endothelial connecting segment-1 by activating β1-integrin. J. Clin. Invest. 1999;103:613–625. doi: 10.1172/JCI5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMIZU A., KITAMURA H., MASUDA Y., ISHIZAKI M., SUGISAKI Y., YAMANAKA K. Apoptosis in the repair process of experimental proliferative glomerulonephritis. Kidney Int. 1995;47:114–121. doi: 10.1038/ki.1995.13. [DOI] [PubMed] [Google Scholar]
- SIMONSON M.S. Anti-AP-1 activity of all-trans retinoic acid in glomerular mesangial cells. Am. J. Physiol. 1994;267:F805–F815. doi: 10.1152/ajprenal.1994.267.5.F805. [DOI] [PubMed] [Google Scholar]
- SPRINGER T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- STAUTON D.E., DUSTIN M.L., SPRINGER T.A. Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature. 1989;339:61–64. doi: 10.1038/339061a0. [DOI] [PubMed] [Google Scholar]
- SUGAWARA A., SANNO N., TAKAHASHI N., OSAMURA R.Y., ABE K. Retinoid X receptors in the kidney: their protein expression and functional significance. Endocrinology. 1997;138:3175–3180. doi: 10.1210/endo.138.8.5351. [DOI] [PubMed] [Google Scholar]
- WAN H., DAWSON M.I., HONG W.K., LOTAN R. Overexpressed activated retinoid X receptors can mediate inhibitory effects of retinoids in human carcinoma cells. J. Biol. Chem. 1998;273:26915–26922. doi: 10.1074/jbc.273.41.26915. [DOI] [PubMed] [Google Scholar]
- YANG T., MICHELE D.E., PARK J., SMART A.M., LIN Z., BROSIUS F.C., III, SCHNERMANN J.B., BRIGGS J.P. Expression of peroxisomal proliferator-activated receptors and retinoid X receptors in the kidney. Am. J. Physiol. 1999;277:F966–F973. doi: 10.1152/ajprenal.1999.277.6.F966. [DOI] [PubMed] [Google Scholar]