

Abstract
The aim of this study was to determine whether nifedipine could suppress an atherogenic process such as balloon-injured intimal thickening in vivo and the proliferation of vascular smooth muscle cells (VSMC) in vitro.
First, we examined the in vivo effect of nifedipine to determine whether it could suppress intimal thickening induced by balloon catheterization. Sprague-Dawley (SD) rats were divided into three groups (L, nifedipine 0.3 mg kg−1 day−1; H, nifedipine 3 mg kg−1 day−1; C, no nifedipine), and Alzet® osmotic pumps were implanted in their backs for continuous administration. The neointimal layers were completely occupied by proliferated VSMC, and the area ratios of neointima/media treated with nifedipine significantly decreased dose-dependently compared to those of the control. Neither blood pressure nor lipid levels changed among the three groups.
We next evaluated the in vitro effect of nifedipine on the proliferation of cultured rat VSMC. Nifedipine decreased the values of [3H]-thymidine incorporation and total cellular protein content as well as the levels of phosphorylated extracellular signal-regulated protein kinase (ERK) 1/2, mitogen-activated protein kinase kinase (MEK) 1/2, and even the phosphorylation of Pyk2, in dose-dependent fashions. In addition, nifedipine suppressed the levels of proliferative cell nuclear antigen (PCNA) dose-dependently in both VSMC and balloon-injured thoracic aortae.
These results indicate that nifedipine has an inhibitory effect on intimal thickening by attenuating intimal VSMC proliferation, suggesting that nifedipine could be effective for preventing the progression of atherosclerotic plaque as in restenosis after angioplasty.
Keywords: Nifedipine, vascular smooth muscle cell, rat, balloon catheterization, intimal thickening, PTCA, ERK1/2, Pyk2, PCNA
Introduction
Elevated blood pressure is one of the most crucial factors for the onset of serious cardiovascular and cerebrovascular diseases, congestive heart failure, and subsequent sudden death (Sagie et al., 1993; National High Blood Pressure Education Program Working Group, 1994; Staessen et al., 1997; Sacco et al., 1999). Over the past several years, numerous studies have demonstrated that adequate antihypertensive treatment is required to reduce these mortality and morbidity rates (SHEP Cooperative Research Group, 1991; Medical Research Council Working Party, 1992; Siscovick et al., 1994; Pahor et al., 1995; Psaty et al., 1999).
Nifedipine, one of the most common L-type dihydropyridine calcium antagonists, has been widely administered to hypertensive patients to reduce the prevalence of strokes, congestive heart failure, myocardial infarction, and sudden death (Loaldi et al., 1989). However, recent studies have shown that short-acting nifedipine neither prevents the progression of threatened myocardial infarction (Muller et al., 1984) nor decreases the total mortality of elderly hypertensive patients because of the reflex increase in sympathetic activity, which has such effects as the elevation of heart rate and oxygen consumption (Pahor et al., 1995; Furberg et al., 1995). These studies suggest that nifedipine may not be appropriate for average hypertensive therapy. On the other hand, nifedipine has been shown not to affect the structure and function of resistant arteries of hypertensive patients (Schiffrin & Deng, 1995) but to stimulate endothelial-derived nitric oxide release in porcine coronary endothelium (Berkels et al., 1996). In human studies, Gong et al. (1997) and Alderman et al. (1997) have reported that long-acting nifedipine treatment for elderly hypertensive patients diminished severe clinical events, and Staessen et al. (1997) have demonstrated that intensive nifedipine treatment can reduce the rate of cardiovascular complications in elderly systolic hypertensive patients. Recently, Jukema et al. (1998) have shown that the addition of calcium antagonists including nifedipine to statin therapy, but not each alone, acts synergistically to retard the angiographic progression of established coronary atherosclerosis. Thus, such diverse results indicate that the efficacy of nifedipine therapy for the prevention of atheromatous formation remains not to be fully established, since these studies have not addressed the biochemical and molecular aspects of the function of lesional cells during nifedipine treatment from a pharmacological point of view, which raises the question of whether nifedipine itself indeed initiates or suppresses a process occurring early in atherogenesis.
We believe that a better understanding of the precise molecular and cellular mechanisms in vascular wall cells is called for to evaluate the effect of nifedipine. Therefore, we attempted to determine whether nifedipine might be efficient for suppressing intimal thickening in vivo and inhibiting the proliferation of rat vascular smooth muscle cells (VSMC) in vitro.
Methods
Materials
Nifedipine was supplied by Bayer Yakuhin, Ltd. (Osaka, Japan). An ALZET® osmotic pump was purchased from ALZA Corp. (Mountain View, CA, U.S.A.), and a 2F Forgaty balloon catheter was from Baxter Healthcare (Santa Ana, CA, U.S.A.). The following items were obtained: anti-mouse α-smooth muscle actin antibody (SMA) from Sigma Chemical (St. Louis, MO, U.S.A.); an avidin-biotin-peroxidase complex (ABC) kit from Vectastains (Vector Laboratories, CA, U.S.A.); Dulbecco's modified Eagle medium (DMEM) and phosphate buffered saline (PBS) from Nissui Pharmaceutical Co. (Tokyo, Japan); foetal bovine serum (FBS) from Gibco BRL (Grand Island, NY, U.S.A.); antibodies specific or phosphospecific for extracellular-regulated protein kinase (ERK) 1/2 (p44/p42) and MAP kinase kinase 1/2 (MEK 1/2) from New England Biolab. (Beverly, MA, U.S.A.); antibodies specific for proliferative cell nuclear antigen (PCNA), Pyk2, and phosphotyrosine (PY20) from Santa Cruz Biotech. (Santa Cruz, CA, U.S.A.); [3H]-thymidine, PVDF membrane, peroxidase-conjugated immunoglobulins, and an ECL kit from Amersham (Arlington Heights, IL, U.S.A.); protein A-Sepharose 6MB from Pharmacia Biotech AB (Uppsala, Sweden); a cytotoxicity detection kit (LDH) from Roche (Mannheim, Germany). All other materials were from Wako Pure Chemical (Osaka, Japan), Nacalai Tesque (Kyoto, Japan), Bio-Rad Laboratories (Richmond, CA, U.S.A.), and Sigma Chemical.
Balloon catheterization into the thoracic aorta and the left common carotid artery of rat
Male Sprague-Dawley (SD) rats weighing approximately 400 g were used for this experiment. These rats were divided into three groups (L, nifedipine 0.3 mg kg−1 day−1 (n=9); H, nifedipine 3 mg kg−1 day−1 (n=9); C, no nifedipine (n=9)), and an Alzet® osmotic pump (model 2002) was implanted in the back of each rat under sodium pentobarbitone anaesthesia (30 mg kg−1 i.p.) for 17-day continuous administration (Wang et al., 1997). These pumps were filled with either a mixture of ethanol : polyethylene-glycol 400 (3 : 17) or with the same mixture containing a low or high concentration of nifedipine released at the rate of 12 μl day−1. On the 3rd day, balloon catheterization was performed as described previously (Igarashi et al., 1997). Briefly, under sodium pentobarbitone anaesthesia (30 mg kg−1 i.p.), the left femoral artery and the left common carotid artery were exposed through respective incisions in the left thigh and the left side of the neck. After a minor incision in the left femoral artery, a 2F Forgaty balloon catheter was reversely inserted into the thoracic aorta or the left common carotid artery. The inside space of the balloon was filled with water, and the intraluminal passages were filled with additive 0.02 or 0.06 ml of water three times to denude the endothelium of the thoracic aorta or the left common carotid artery, respectively.
Histological and immunoblot analyses of balloon-injured common carotid artery
Fourteen days after balloon catheterization, the left common carotid arteries were rapidly removed and divided into four cross-sectional pieces at 3-mm intervals. These pieces were fixed with 4% paraformaldehyde for 6 h, and tissues were subsequently processed for paraffin embedding. They were stained with Elastica-Masson for the measurement of the area ratio of neointima/media (I/M ratio) and immunohistochemically stained with anti-SMA for VSMC detection by the ABC method (Hsu et al., 1981). The areas of the neointima, measured from the internal elastic lamina to the luminal surface, and the medial layer in each section of the left common carotid artery were calculated using a microscopical image-analysing system, XL-10 (Olympus, Tokyo, Japan). Concerning the ABC method, the sections were deparaffinized, and endogenous peroxidase was quenched by a 15-min incubation with 0.3% H2O2. The sections were then incubated with a 1 : 2000 dilution of anti-SMA at room temperature overnight. Bound antibody was visualized by the ABC method and 3,3′-diaminobenzidine tetrahydrochloride (DAB) as chromogen. In addition, the thoracic aortae were also rapidly removed and frozen with liquid nitrogen for the detection of PCNA.
Measurement of blood pressure and heart rate
The hypotensive effect of nifedipine was examined in all balloon-injured rats. Systolic and diastolic blood pressures and heart rate were measured by the tail-cuff method with an ultrasound device (BP98A, Softron, Tokyo, Japan) before the implantation of an Alzet® osmotic pump, 3, 7, and 14 days after balloon catheterization.
Cell culture
VSMC were harvested from the aortae of male SD rats (150–200 g) (Funabashi Farm, Chiba, Japan) by the medial explant technique and cultured in DMEM containing 10% FBS as described previously (Igarashi et al., 1997). Cells within 10 passages were used for the following studies.
Measurement of DNA content in VSMC
The assay was performed as described previously (Kanzaki et al., 1994) with minor modifications. VSMC were plated at a density of 5×104 cells per well on a 12-well culture plate and grown in DMEM containing 10% FBS for 48 h. Then, the medium was changed to DMEM containing 0.2% bovine serum albumin (BSA) for starvation. After 24 h of incubation, the media were aspirated, and the cells were cultured in DMEM containing 2% FBS either with dimethylsulphoxide (DMSO) only or DMSO containing various concentrations (final concentrations: 10−7, 10−6; 10−5, and 10−4 M) of nifedipine for 16 h. Then, 1 μCi of [3H]-thymidine was added to each well and incubated for another 4 h. At the end of incubation, the cells were washed, and a high-molecular mass of [3H]-thymidine was precipitated by 5% trichloroacetic acid at 4°C for 20 min. The precipitate was washed and solubilized in a mixture of 0.5 N NaOH and 0.1% sodium dodecyl sulphate (SDS), and the radioactivity was determined in a liquid scintillation counter. Protein concentrations were measured by the method of Lowry et al. (1950).
Growth assay in VSMC
The cells were prepared as mentioned above and starved with DMEM containing 0.2% BSA for 24 h. Then, the media were aspirated and cultured in DMEM containing 5% FBS with DMSO only or various concentrations (final concentrations: 10−7, 10−6, 10−5, and 10−4 M) of nifedipine solubilized in DMSO. After incubation for the indicated number of days (0, 1, 3 or 5 days), the media were aspirated, and the cells were solubilized in 1 ml of a mixture of 0.5% SDS and 0.5 N NaOH. After the solution had been neutralized with 500 μl of 1 N HCl, the measurement of total cellular protein content was performed by the method of Lowry et al. (1950). Those media were replaced every 24 h during this experiment.
Immunoblot analysis of ERK1/2, MEK 1/2, and Pyk2 in VSMC
After being starved with DMEM containing 0.2% BSA for 24 h at 37°C, the cells were cultured in DMEM containing 2% FBS for another 21 h. Then, either DMSO only or various concentrations (final concentrations: 10−7, 10−6, 10−5, and 10−4 M) of nifedipine solubilized in DMSO were added to each well and incubated for another 3 h. The following experimental procedure was carried out as described previously (Igarashi et al., 1999). The cells were washed and solubilized with 0.5 ml of a lysis buffer consisting of (mM): HEPES 20, MgCl2 5, KCl 25, Na3SO4 1, sodium molybdate 1, β-glycerophosphate 10, tetrasodium pyrophosphate 5, sucrose 250, adenosine 5′-triphosphate (ATP) 1, phenylmethylsulphonyl fluoride (PMSF) 2, 0.1 mg ml−1 aprotinin, and 1% Triton-X. The total cellular lysate was centrifuged at 14,000×g to remove insoluble materials, and then protein concentrations were determined by the method of Bradford (1976). After being solubilized in a Laemmli buffer, the samples were separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes. Alternatively, the above lysates were incubated with 10 μl of anti-Pyk2 antibody at 4°C overnight. Then, protein A-Sepharose 6MB was added and further incubated at 4°C for 2 h. The bound proteins were solubilized in a Laemmli buffer, and the samples were separated by 7.5% SDS–PAGE and transferred to PVDF membranes.
After blocking overnight, the membranes were incubated with the primary anti-phosphospecific antibodies at 4°C overnight or with the antibodies at room temperature for 1 h. The blots were washed and then incubated with peroxidase-conjugated immunoglobulins at room temperature. After washing, sites of antibody binding were visualized using the ECL Western blotting detection system and quantified with a densitometer.
Immunoblot analysis of PCNA in VSMC and balloon-injured thoracic aortae of rat
In the in vitro sample, after being starved with DMEM containing 0.2% BSA, the cells were cultured in DMEM containing 2% FBS with DMSO only or various concentrations (final concentrations: 10−7, 10−6, 10−5 and 10−4 M) of nifedipine solubilized in DMSO at 37°C for 24 h. The procedure for the extraction of nuclear protein was performed as described previously (Andrews et al., 1991). After being scraped with ice-cold PBS, the cell suspension was transferred to a microfuge and centrifuged at 14,000×g for 10 s. Then, the pellets were suspended in 400 μl of ice-cold buffer A (in mM: HEPES-KOH 10 (pH 7.9, 4°C), MgCl2 1.5, KCl 10, dithiothreitol (DTT) 0.5, PMSF 0.2), put on ice for 10 min, and centrifuged at 14,000×g for 10 s. The pellets were resuspended in 50 μl of ice-cold buffer C (mM): HEPES-KOH 20 (pH 7.9, 4°C), 25% glycerol, NaCl 420, MgCl2 1.5, ethylenediaminetetraacetic acid (EDTA) 0.2, DTT 0.5, PMSF 0.2, incubated on ice for 20 min, and centrifuged at 14,000×g for 2 min. In the in vivo sample, the balloon-injured thoracic aortae were pulverized and then homogenized with 1 ml of a lysis buffer consisting of (mM): HEPES 20, MgCl2 5, KCl 25, Na3SO4 1, sodium molybdate 1, β-glycerophosphate 10, tetrasodium pyrophosphate 5, sucrose 250, ATP 1, PMSF 2, 0.1 mg ml−1 aprotinin, and 1% Triton-X in a Dounce homogenizer for 30 strokes and centrifuged. The protein contents of these supernatants were determined by the method of Bradford (1976).
The samples were solubilized in a Laemmli buffer, separated by 12% SDS–PAGE, and transferred to PVDF membranes. After blocking overnight, the membranes were incubated with anti-PCNA antibody at room temperature for 1 h. The blots were treated to visualize the sites of antibody binding by the same method and quantified as described in the previous section.
Evaluation of cytotoxicity
The cytotoxic effect of nifedipine was assessed by measuring the release of lactate dehydrogenase (LDH) in VSMC as described previously (Ellouk-Achard et al., 1995). The cells were plated at a density of 5×104 cells per well on a 24-well culture plate and cultured in DMEM containing 10% FBS for 48 h. After being starved with DMEM containing 0.2% BSA for 24 h, the cells were cultured in DMEM containing 2 or 5% FBS with DMSO only or various concentrations (final concentrations: 10−7, 10−6, 10−5, and 10−4 M) of nifedipine solubilized in DMSO. After being incubated for 24 h, 100 μl of the supernatants was transferred into the wells of a 96-well microtiter plate. To determine the LDH activity in the supernatants, the reaction mixture was added to each well and incubated for 30 min at room temperature. Then, absorbance of the samples to evaluate the percentage of cytotoxicity by nifedipine was measured at 490 nm by using an ELISA reader. The average values were calculated as follows:
where AV treated: the absorbance value in the supernatant with addition of either DMSO or various concentrations of nifedipine solubilized with DMSO; AV untreated: the absorbance value in the supernatant without addition of DMSO or nifedipine; AV cell: the absorbance value in the cell lysate without addition of DMSO or nifedipine, solubilized with 100 μl of 1% Triton-X. In addition, the cell viability was determined by the exclusion of 0.2% Trypan blue.
Other measurements
Blood samples were taken from the inferior vena cava after overnight starvation, and sera were obtained by centrifugation for the measurement of total cholesterol (T-Cho), triglyceride (TG), and high-density lipoprotein-cholesterol (HDL-C). T-Cho and TG levels were determined by the enzymatic method, and HDL-C was measured by the precipitation method modified by the phosphotungstate-MgCl2 method.
Statistical analysis
Results are expressed as mean±s.e.mean. Statistical significance was estimated by one-way analysis of variance (ANOVA) for the comparison of several groups, and the differences were considered to be significant at P<0.05.
Results
Body weight and levels of serum lipid, blood pressure, and heart rate
The treatment with nifedipine modified neither rat body weight (C, 409.3±2.9 g; L, 421.0±5.7 g; H, 418.3±5.6 g) nor the levels of serum T-Cho (C, 66.9±4.9 mg dl−1; L, 68.3±4.5 mg dl−1; H, 57.1±2.4 mg dl−1), TG (C, 89.4±11.1 mg dl−1; L, 103.4±14.7 mg dl−1; H, 112.9±9.6 mg dl−1), or HDL-C (C, 39.0±2.7 mg dl−1; L, 40.2±3.1 mg dl−1; H, 34.3±1.7 mg dl−1) in the three groups. In addition, the treatment with nifedipine slightly lowered the level of systolic (C, 133.8±1.9 mmHg; L, 131.7±1.0 mmHg, H, 127.0±3.8 mmHg) and/or diastolic blood pressure (C, 106.3±3.0 mmHg; L, 102.7±2.5 mmHg; H, 103.3±2.4 mmHg) or increased the heart rate (C, 364.5±5.9 min−1; L, 378.8±9.7 min−1; H, 377.5±5.5 min−1) during the procedures, but no significant differences were observed among the three groups.
I/M ratio on the left carotid artery after balloon catheterization
During the procedures, no rat died in any of the three groups. Figure 1 shows the typical Elastica Masson stain (C, (A); L, (B); H, (C)). The regression of intimal thickening in the nifedipine groups could be confirmed by the position of the internal elastic lamina. Differences in I/M ratio are shown in Figure 2. Treatment with nifedipine resulted in a distinct change in the size of intimal thickening in a clear dose-dependent fashion compared to that of the control. The I/M ratio and intimal area in the C group were significantly lower than those in the L group (C, 7.88±0.72 mm2 vs L, 6.41±0.37 mm2, respectively, P<0.05). Furthermore, these values in the H group (5.22±0.24 mm2) were considerably lower than those of the C (P<0.01) and the L (P<0.05) groups. The area of the medial layer was not significantly different among the three groups (C, 7.27±0.30 mm2; L, 7.22±0.28 mm2; H, 7.25±0.24 mm2). These findings indicate that nifedipine appears to have an inhibitory effect on the intimal thickening induced by balloon catheterization.
Figure 1.
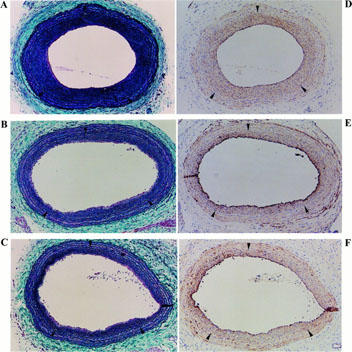
Cross-sections of rat left common carotid artery 14 days after balloon catheterization. EM stains (A–C) and immunohistological stainings with anti-SMA antibody and developed by the ABC method (D–F). 0.4 mg ml−1 of DAB was used as the chromogen of the ABC method, and the antibody-positive staining is shown as brown. (A,D) C: mixture of ethanol and polyethylen-glycol 400 only; (B,E) L: treatment with 0.3 mg kg−1 day−1 of nifedipine; (C,F) H: treatment with 3 mg kg−1 day−1 of nifedipine. Arrowheads indicate the position of the internal elastic lamina. The preparations were examined under ×20 magnification.
Figure 2.
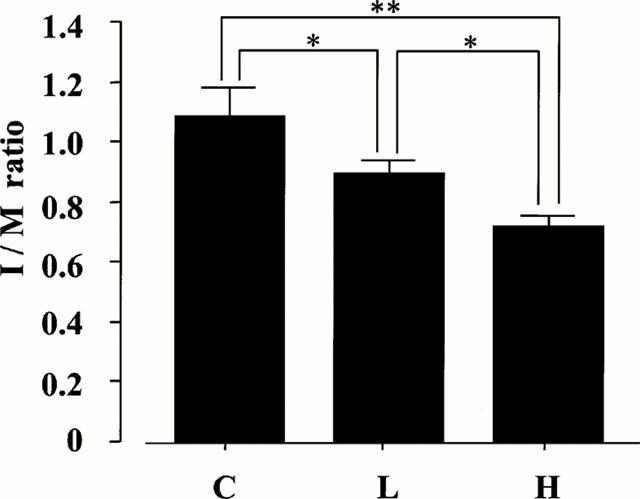
Effect of nifedipine on the value of I/M ratio from rat left common carotid artery 14 days after balloon catheterization. C: mixtur of ethanol and polyethylen-glycol 400 only (n=9); L: treatment with 0.3 mg kg−1 day−1 of nifedipine (n=9); H: treatment with 3 mg kg−1 day−1 of nifedipine (n=9). Values are the mean±s.e.mean. **P<0.01; *P<0.05 vs C.
Immunohistochemical findings on balloon catheterization
Immunohistological stainings with anti-SMA antibody revealed diffuse SMA-positive immuno-reactivities throughout the intimal thickening layers of all preparations (Figure 1D–F). These areas were equivalent to the values of the I/M ratios because of their wide distribution in the neointima. This finding demonstrated that the intimal thickening layers in this experiment were composed of proliferated and migrated VSMC and that the amount of VSMC in those layers was clearly attenuated by the treatment with nifedipine in a distinct dose-dependent manner.
Effect of nifedipine on VSMC growth
To characterize the effect of nifedipine on rat VSMC growth in vitro, we examined the mitogenic response by a [3H]-thymidine incorporation assay. As shown in Figure 3, the values of [3H]-thymidine incorporation were inhibited by the treatment with nifedipine dose-dependently. At 10−6 M, 10−5 M, and 10−4 M of nifedipine, the values were significantly decreased to 63.82±4.31%, 22.68±1.22%, and 5.45±3.26% of those of the control, respectively (P<0.01).
Figure 3.
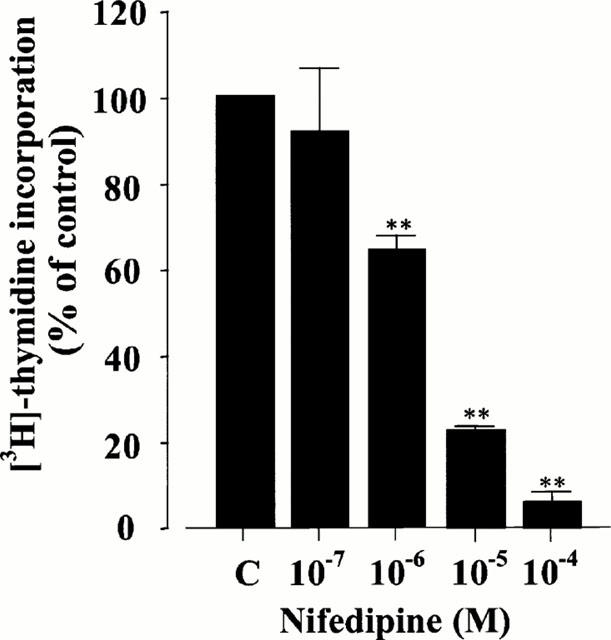
Effect of nifedipine on DNA synthesis in rat VSMC. The results were derived from five separate experiments, and each experiment was performed in triplicate. Each bar represents the mean±s.e.mean. **P<0.01 vs control.
To support the inhibitory effect of nifedipine on rat VSMC growth, we further examined the changes of total cellular protein concentration (Figure 4). The values in the control group were gradually increased to 58.33±5.69 μg ml−1 on day 3 and 64.53±5.94 μg ml−1 on day 5 by the stimulation of 5% FBS. Compared with those of the control, the values of the groups treated with nifedipine decreased time- and dose-dependently. The values at 10−5 and 10−4 M of nifedipine were significantly decreased even on day 1 and at 10−6 M on day 5 (P<0.05), indicating that nifedipine has an inhibitory effect on rat VSMC growth.
Figure 4.
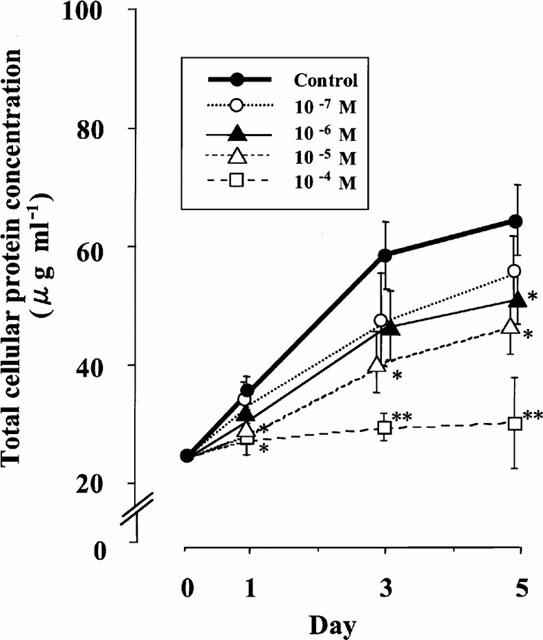
Effect of nifedipine on the changes of total cellular protein concentrations in rat VSMC. The results were derived from five separate experiments, and each experiment was performed in triplicate. Each bar represents the mean±s.e.mean. *P<0.05, **P<0.01 vs control.
Effect of nifedipine on the phosphorylation of ERK1/2 and its upstream, MEK 1/2, in VSMC
We examined whether nifedipine could affect the activation of ERK 1/2 (p44/42) in rat VSMC by immunoblot analysis. The upper panel of Figure 5A shows the effect of nifedipine on the phosphorylation of ERK1/2. Compared with the control basal band, the levels of phosphorylated ERK1/2 were dose-dependently decreased by the treatment with nifedipine and significantly suppressed to 69.6±14.6% at 10−6 M, 48.8±3.3% at 10−5 M, and 42.8±5.9% at 10−4 M (Figure 5B). In contrast, the protein levels of ERK 1/2 were not changed by the treatment with nifedipine (the lower panel of Figure 5A).
Figure 5.
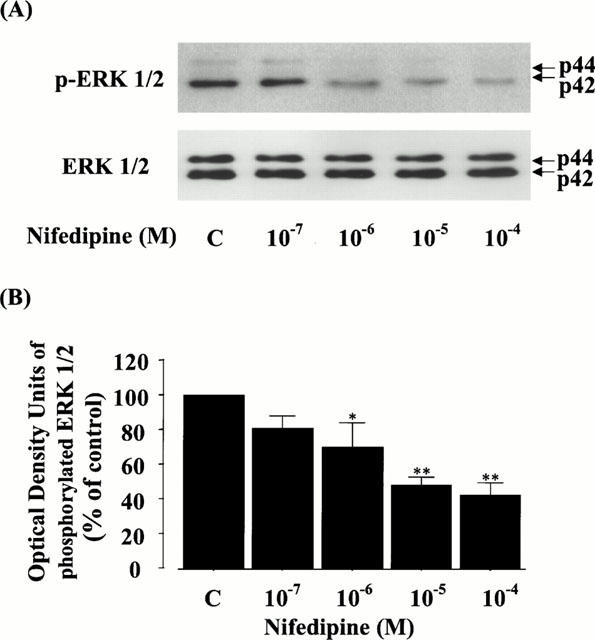
Effect of nifedipine on the phosphorylation and protein level of ERK 1/2 (p44/42) in rat VSMC. (A) The phosphorylation or protein level of ERK 1/2 was detected by immunoblot analysis using antibody phosphospecific or specific for ERK 1/2, as described in Methods. p-ERK 1/2 or ERK 1/2 shows the phosphorylation or the protein level of ERK 1/2, respectively. (B) The quantitation of the phosphorylated ERK 1/2 from three separate experiments is shown, and each bar represents the mean±s.e.mean. *P<0.05, **P<0.01 vs control.
To confirm the identity of the effect of nifedipine on the signal transduction pathway of ERK, we further examined the phosphorylation of MEK 1/2, which is an upstream kinase of ERK 1/2 (Seger & Krebs, 1995), in rat VSMC. As in Figure 6, the levels of phosphorylated MEK 1/2 were also decreased by the treatment with nifedipine dose-dependently (the upper panel of Figure 6A) and significantly suppressed to 81.0±1.6% at 10−6 M, 59.2±2.6% at 10−5 M, and 54.6±3.3% at 10−4 M (Figure 6B), but the protein levels of MEK 1/2 were not changed in any of the conditions (the lower panel of Figure 6A). These data clearly demonstrate that nifedipine can prevent cell growth via a MEK-ERK cascade in rat VSMC.
Figure 6.
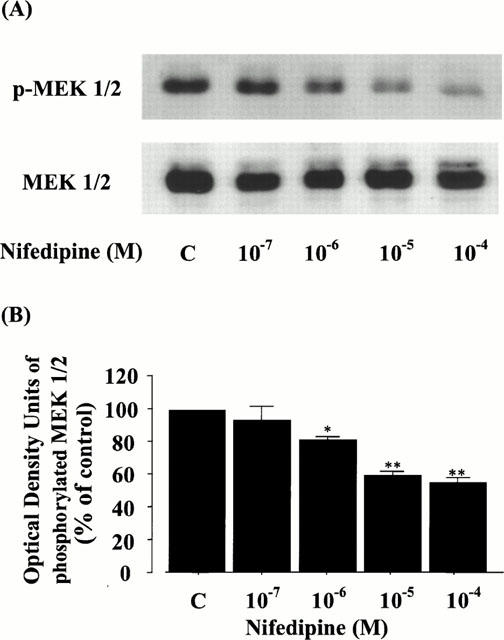
Effect of nifedipine on the phosphorylation and protein level of MEK 1/2 in rat VSMC. (A) The phosphorylation or protein level of MEK 1/2 was detected by immunoblot analysis using antibody phosphospecific or specific for MEK 1/2, as described in Methods. p-MEK 1/2 or MEK 1/2 shows the phosphorylation or the protein level of MEK 1/2, respectively. (B) The quantitation of the phosphorylated MEK 1/2 from three separate experiments is shown, and each bar represents the mean±s.e.mean. *P<0.05, **P<0.01 vs control.
Effect of nifedipine on the phosphorylation of Pyk2 in VSMC
To evaluate the effect of nifedipine on the upstream level of a MEK-ERK cascade in rat VSMC, we further characterized the role of Pyk2, which activates a MEK-ERK signal transduction pathway in response to the elevation of intracellular calcium concentrations (Tokiwa et al., 1996; Ganju et al., 1998). Interestingly, compared with the control basal band, the levels of phosphorylated Pyk2 were also decreased by the treatment with nifedipine dose-dependently and significantly suppressed to 70.9±2.9% at 10−6 M, 61.1±11.9% at 10−5 M, and 28.9±15.8% at 10−4 M without any changes in the protein levels (Figure 7A,B).
Figure 7.
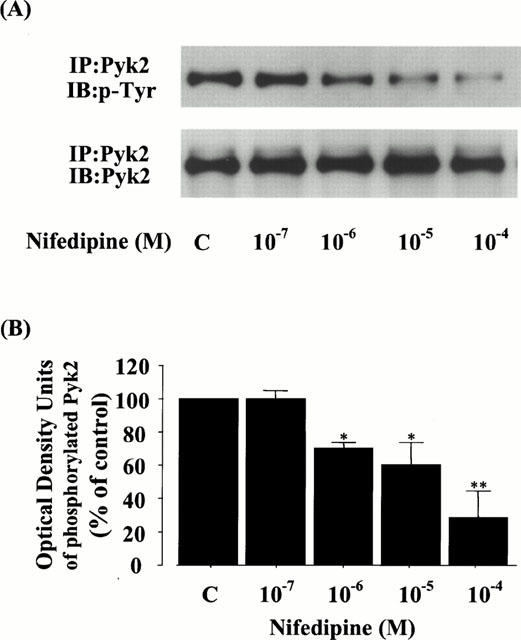
Effect of nifedipine on the phosphorylation and protein level of Pyk2 in rat VSMC. (A) The phosphorylation or protein level of Pyk2 was detected by immunoblot analysis using antibody specific for phosphotyrosine (PY20) or for Pyk2, as described in Methods. p-Pyk2 or Pyk2 shows the phosphorylation or protein level of Pyk2, respectively. (B) The quantitation of the phosphorylated Pyk2 from three separate experiments is shown, and each bar represents the mean±s.e.mean. *P<0.05, **P<0.01 vs control.
Effect of nifedipine on the expression of PCNA in VSMC and balloon-injured thoracic aortae
To estimate the inhibitory effect of nifedipine on VSMC growth in the nuclear signalling level, we further investigated the expression of PCNA in vitro and in vivo. Interestingly, the protein level of PCNA was dose-dependently attenuated by the treatment with nifedipine in balloon-injured rat thoracic aortae (Figure 8A) and VSMC (Figure 8B). In rat aortae, compared to the control, the PCNA levels in the L and H groups were significantly decreased to 73.9±7.8% and 48.8±8.8%, respectively (the lower panel of Figure 8A). In VSMC, the levels were also significantly decreased to 68.9±16.4% at 10−6 M, 58.8±6.0% at 10−5 M, and 25.8±9.0% at 10−4 M (the lower panel of Figure 8B).
Figure 8.
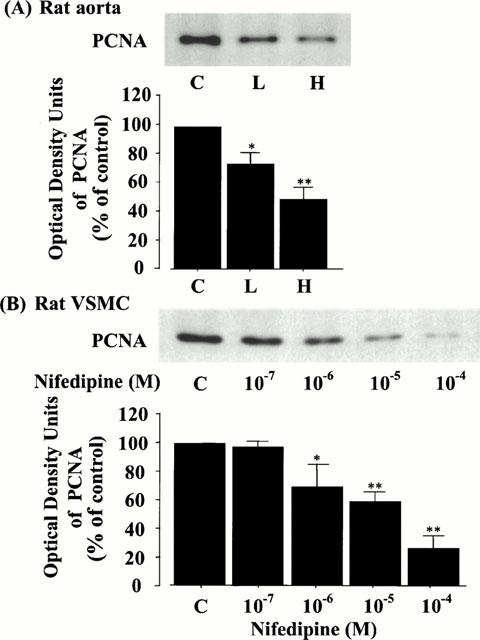
Effect of nifedipine on the levels of PCNA in rat thoracic aorta (A) and VSMC (B) by immunoblot analysis. The quantitation of PCNA from three separate experiments is shown in each lower panel, and each bar represents the mean±s.e.mean. *P<0.05, **P<0.01 vs control.
Cytotoxic effect of nifedipine in VSMC
The release of LDH in the supernatant was not significantly changed by the treatment with various concentrations of nifedipine for 24 h in rat VSMC (Table 1). In addition, the cell viability with or without addition of nifedipine was more than 98% as determined by the exclusion of 0.2% Trypan blue.
Table 1.
Cytotoxic effect of nifedipine by measuring the release of LDH in rat VSMC

Discussion
Considerable evidence supports the view that hypertension is a major risk factor for the progression of atherosclerosis (SHEP Cooperative Research Group, 1991; Medical Research Council Working Party, 1992; Sagie et al., 1993; National High Blood Pressure Education Program Working Group, 1994; Siscovick et al., 1994; Pahor et al., 1995; Staessen et al., 1997; Psaty et al., 1999; Sacco et al., 1999). The major consequence of atherosclerotic changes is the obstruction and/or occlusion of one or more arteries, leading to strokes (Sacco et al., 1999) or acute coronary syndromes such as unstable angina and acute myocardial infarction (Libby, 1995). The mechanism(s) of arterial obstruction remains unclear, but it is generally considered that (1) increased vasoconstriction responses, (2) decreased antithrombic properties, and (3) abnormal proliferation and migration of VSMC are implicated in the development of atherosclerotic plaque (Ross, 1986).
In this study, we have clearly demonstrated that nifedipine has an inhibitory effect even on the early events of atheromatous formation by direct suppression of VSMC growth via a MEK-ERK pathway coupled with Pyk2 in a strict dose-dependent manner and that the effect is independent of the level of blood pressure or serum cholesterol. These favourable results suggest that nifedipine could be one of the most promising agents for preventing the progression of vascular complications such as restenosis after percutaneous transluminal coronary angioplasty (PTCA).
First, we examined the in vivo effect of nifedipine to determine whether it could suppress rat intimal thickening induced by balloon catheterization. The I/M ratio in the L and H groups treated with nifedipine was significantly decreased by 17 and 34%, respectively, compared to the control. In addition, the ratio in the H group was significantly lower than that in the L group. Furthermore, the lesions were composed mainly of SMA-positive cells, which suggested that nifedipine has a significant inhibitory effect on the content of VSMC in the neointima and the subsequent intimal thickening. In agreement with our results, Jackson et al. (1989) have shown that nifedipine can prevent the proliferation of VSMC occurring in response to balloon catheter injury, suggesting that nifedipine might inhibit the phenotypic modulation of VSMC and the subsequent uptake of lipids. In addition, Kerry et al. (1999) have recently reported that another L-type dihydropyridine calcium antagonist, nicardipine, also suppresses the collar-induced intimal thickening in rabbit carotid artery without affecting the changes in vascular reactivity. In contrast, Üstünes et al. (1996) have shown that even a near lethal dose of verapamil, another subclass of calcium antagonists, cannot prevent the collar-induced intimal thickening in rabbits. Although the precise mechanism(s) is not clear, it is likely that the discrepancy may result from the different types of agents, i.e., the L-type dihydropyridine calcium antagonists may have a rather sensitive property to suppress the intimal thickening by inhibiting the proliferation of VSMC or the synthesis of the extracellular matrix.
Neither systolic nor diastolic blood pressure was affected by the treatment with nifedipine in this study. Recently, Bult et al. (1999) have suggested that calcium antagonists can modulate nitric oxide bioactivity. The precise mechanism is obscure, but several investigators have previously reported that nifedipine treatment did not necessarily result in a significant reduction of blood pressure in normal rats compared to spontaneous hypertensive rats (Rakusan et al., 1994). In addition, it might be considered that continuous subcutaneous administration of nifedipine by means of an osmotic pump affects its performance as a long-acting agent. It is more likely that reduction of contractile VSMC in the medial layer by balloon injury might result in the decrease of hypotensive effect by the agent. Furthermore, neither lipid levels nor the heart rate was changed in any of the three groups, indicating that blood pressure can be considered insufficient to influence the inhibition of intimal thickening.
Based on the above in vivo findings, we next examined the in vitro effect of nifedipine on the proliferation of rat VSMC. Similarly to the in vivo findings, the values of both [3H]-thymidine incorporation and total protein content were reduced by the addition of nifedipine in dose-dependent fashions. In addition, nifedipine affected neither the cell viability nor the value of LDH in the supernatant in VSMC for a 24-h culture (Table 1), indicating that nifedipine itself can inhibit VSMC proliferation but not by cytotoxicity. In agreement with our findings, Rabkin & Kong (2000) have reported that nifedipine does not induce cytotoxicity at 10−4 M in chick embryonic cardiomyocytes.
Recently, it was revealed that the MAP kinase superfamily plays a crucial role in cell growth, differentiation, or even programmed cell death in response to diverse extracellular stimuli in eukaryotic cells (Seger & Krebs, 1995). Extensive studies have clarified that a variety of growth factors and hormones can activate ERK1/2 (p44/42) signal transduction pathways through the GTPase-activating protein of Ras (Ras-GTP), leading to cellular proliferation and differentiation by stimulating transcription factors that induce the expression of c-fos and other growth-responsive genes (Daub et al., 1996; Lopez-Ilasaca, 1998). As shown in Figure 5, nifedipine inhibited the levels of phosphorylated ERK 1/2 (p44/42) in dose-dependent fashions and significantly reduced the levels at more than 10−6 M. In addition, the levels of phosphorylated MEK 1/2, the upstream of ERK 1/2, were also inhibited by the agent in the same fashion as those of ERK 1/2, suggesting that nifedipine may have an effect on the change(s) of signal transduction through Ras-GTP to a MEK-ERK pathway. In agreement with our findings, Benes et al. (1998) have shown that nifedipine can inhibit the activation of ERK 1/2 in the MIN6 pancreatic β cell line.
Pyk2 (also known as RAFTK) has recently been characterized as one of the focal adhesion kinases (Lev et al., 1995; Tokiwa et al., 1996; Ganju et al., 1998). Pyk2 is activated by various extracellular signals that increase intracellular calcium concentrations and subsequently activate the signalling pathway from Ras-GTP to MAP kinase (Lev et al., 1995). Interestingly, our results clearly demonstrated that the levels of phosphorylated Pyk2 were decreased dose-dependently by the treatment with nifedipine and also significantly suppressed at more than 10−6 M (Figure 7), suggesting that nifedipine itself can suppress the proliferation of VSMC via a MEK-ERK pathway coupled with Pyk2 by reducing the intracellular calcium ion concentration. In contrast, Soma et al. (1998) have shown that the (R)-enantiomer of lercanidipine, which has a very low affinity for calcium channels, can inhibit collar-induced intimal VSMC proliferation and cholesterol-induced fatty streak formation in rabbits, suggesting that these anti-atherosclerotic effects are independent of the inhibition of calcium channels. It is plausible to assume that the discrepancy is based on the difference of the experimental condition or the essential properties of the agents. Soma et al. (1998) adapted the cholesterol-fed rabbit model and administered the lercanidipine and its (R)-enantiomer for 10 weeks, which is quite different from our study, since we used SD rats whose lipid levels were remarkably lower than theirs. In addition, they reported that lercanidipine (R)-enantiomer itself shares the antioxidant properties.
Although we did not evaluate an inhibitory effect of nifedipine on calcium influx in VSMC, Bialecki et al. (1991) have shown that 10% serum-stimulated calcium influx is potentiated 3.7 fold in cholesterol-enriched VSMC and that these increases are significantly attenuated by even 1 μM of nifedipine. Lucchesi et al. (1996) have recently reported that angiotensin II stimulates ERK 1/2 phosphorylation in a calcium-dependent manner in rat VSMC, and that the phosphorylation is stronger in spontaneously hypertensive rats than in Wistar-Kyoto rats. These findings may support the possibility that elevated intracellular calcium concentration in VSMC causes the intimal thickening and nifedipine directly prevents the progression of atheromatous formation by inhibiting calcium accumulation in VSMC.
PCNA is known to be synthesized in the early G1 and S phases of the cell cycle and to behave as a marker for proliferating cells (Tsuboi et al., 1996; Capron et al., 1997). In this study, we could show that nifedipine can suppress the level of PCNA in both VSMC and balloon-injured thoracic aorta in dose-dependent fashions (Figure 8), suggesting that nifedipine can directly inhibit VSMC growth even at the nuclear level both in vitro and in vivo by the same mechanisms.
In conclusion, in this study, we have established that nifedipine has an inhibitory effect on intimal thickening by attenuating intimal VSMC proliferation via a MEK-ERK pathway coupling with Pyk2, suggesting that intracellular calcium kinetics play an important role in the pathogenesis of atherosclerosis and that nifedipine could be effective for preventing the progression of atherosclerotic plaque as in restenosis after PTCA. These results provide a new insight into the potential cellular and molecular mechanisms whereby nifedipine treatment may have clinical benefits and contribute to the prevention of atherosclerosis.
Acknowledgments
We are grateful to Prof Masao Endoh, from the Department of Pharmacology, Yamagata University School of Medicine, for his helpful suggestions during the preparation of this manuscript.
Abbreviations
- ABC
avidin-biotin-peroxidase complex
- ATP
adenosine 5′ - triphosphate
- BSA
bovine serum albumin
- DAB
3,3′-diaminobenzidine tetrahydrochloride
- DMEM
Dulbecco's modified Eagle medium
- DMSO
dimethylsulphoxide
- DTT
dithiothreitol
- EDTA
ethylenediamine-tetraacetic acid
- ERK
extracellular signal-regulated protein kinase
- FBS
foetal bovine serum
- HDL-C
high-density lipoprotein-cholesterol
- LDH
lactate dehydrogenase
- MAP
mitogen-activated protein
- MEK
MAP kinase kinase
- PBS
phosphate buffered saline
- PCNA
proliferative cell nuclear antigen
- PMSF
phenylmethylsulphonyl fluoride
- PTCA
percutaneous transluminal coronary angioplasty
- SD
Sprague-Dawley
- SDS–PAGE
sodium dodecylsulphate–polyacrylamide gel electrophoresis
- SMA
α-smooth muscle actin
- T-Cho
total cholesterol
- TG
triglyceride
- VSMC
vascular smooth muscle cells
References
- ALDERMAN M.H., COHEN H., ROQUE R., MADHAVAN S. Effect of long-acting and short-acting calcium antagonists on cardiovascular outcomes in hypertensive patients. Lancet. 1997;349:594–598. doi: 10.1016/S0140-6736(96)08359-6. [DOI] [PubMed] [Google Scholar]
- ANDREWS N.C., FALLER D.V. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENES C., ROISIN M.P., TAN H.V., CREUZET C., MIYAZAKI J., FAGARD R. Rapid activation and nuclear translocation of mitogen-activated protein kinases in response to physiological concentration of glucose in the MIN6 pancreatic β cell line. J. Biol. Chem. 1998;273:15507–15513. doi: 10.1074/jbc.273.25.15507. [DOI] [PubMed] [Google Scholar]
- BERKELS R., BERTSCH A., BREITENBACH T., KLAUS W., ROSEN R. The calcium antagonist nifedipine stimulates endothelial NO release in therapeutical concentrations. Pharm. Pharmacol. Lett. 1996;6:75–78. [Google Scholar]
- BIALECKI R.A., TULENKO T.N., COLUCCI W.S. Cholesterol enrichment increases basal and agonist-stimulated calcium influx in rat vascular smooth muscle cells. J. Clin. Invest. 1991;88:1894–1900. doi: 10.1172/JCI115512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BULT H., HERMAN A.G., MATTHYS K.E. Antiatherosclerotic activity of drugs in relation to nitric oxide function. Eur. J. Pharmacol. 1999;375:157–176. doi: 10.1016/s0014-2999(99)00328-3. [DOI] [PubMed] [Google Scholar]
- CAPRON L., JARNET J., JOSEPH-MONROSE D., BRUNEVAL P. Repeated balloon injury of rat aorta. A model of neointima with attenuated inhibition by heparin. Arterioscler. Thromb. Vasc. Biol. 1997;17:1649–1656. doi: 10.1161/01.atv.17.9.1649. [DOI] [PubMed] [Google Scholar]
- DAUB H., WEISS F.U., WALLASCH C., ULLICH A. Role of transactivation of EGF receptor in signaling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- ELLOUK-ARCHARD S., MAWET E., THIBAULT N., DETERTRE-CATELLA H., THEVENIN M., CLAUDE J.R. Protective effect of nifedipine against cytotoxicity and intracellular calcium alterations induced by acetaminophen in rat hepatocyte cultures. Drug Chem. Toxicol. 1995;18:105–117. doi: 10.3109/01480549509014315. [DOI] [PubMed] [Google Scholar]
- FURBERG C.D., PSATY B.M., MEYER M.S. Nifedepine: Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326–1331. doi: 10.1161/01.cir.92.5.1326. [DOI] [PubMed] [Google Scholar]
- GANJU R.K., DUTT P., WU L., NEWMAN W., AVRAHAM H., AVRAHAM S., GROOPMAN J.E. β-chemokine receptor CCR5 signals via the novel tyrosine kinase RAFTK. Blood. 1998;91:791–797. [PubMed] [Google Scholar]
- GONG L., ZHANG W., ZHU Y., ZHU J., 11 COLLABORATING CENTRES IN THE SHANGHAI AREA. KONG D., PAGE V., GHADIRIAN P., LELORIER J., HAMET P. Shanghai trial of nifedipine in the elderly (STONE) J. Hypertens. 1997;14:1237–1245. doi: 10.1097/00004872-199610000-00013. [DOI] [PubMed] [Google Scholar]
- HSU S.M., RAINE L., FANGER H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase technique: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- IGARASHI M., TAKEDA Y., ISHIBASHI N., TAKAHASHI K., MORI S., TOMINAGA M., SAITO Y. Pioglitazone reduces smooth muscle cell density of rat carotid arterial intima induced by balloon catheterization. Horm. Metab. Res. 1997;29:444–449. doi: 10.1055/s-2007-979074. [DOI] [PubMed] [Google Scholar]
- IGARASHI M., WAKASAKI H., TAKAHARA N., ISHII H., JIANG Z.Y., YAMAUCHI T., KUBOKI K., MEIER M., RHODES C.J., KING G.L. Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J. Clin. Invest. 1999;103:185–195. doi: 10.1172/JCI3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JACKSON C.L., BUSH R.C., BOWYER D.E. Mechanism of antiatherogenic action of calcium antagonists. Atherosclerosis. 1989;80:17–26. doi: 10.1016/0021-9150(89)90063-4. [DOI] [PubMed] [Google Scholar]
- JUKEMA J.W., VAN BOVEN A.J., ZWINDERMAN A.H., VAN DER LAARSE A., BRUSCHKE A.V.G. Proposed synergistic effect of calcium channel blockers with lipid-lowering therapy in retarding progression of coronary atherosclerosis. Cardiovasc. Drugs Ther. 1998;12:111–118. doi: 10.1023/a:1007733311487. [DOI] [PubMed] [Google Scholar]
- KANZAKI T., SHINOMIYA M., UEDA S., MORISAKI N., SAITO Y., YOSHIDA S. Enhanced arterial intimal thickening after balloon catheter injury in diabetic animals accompanied by PDGF β-receptor overexpression of aortic media. Eur. J. Clin. Invest. 1994;24:377–381. doi: 10.1111/j.1365-2362.1994.tb02179.x. [DOI] [PubMed] [Google Scholar]
- KERRY Z., YASA M., AKPINAR R., SEVIN G., YETIK G., TOSUN M., ÖZDEMIR N., ERHAN Y., ÜSTÜNES L., ÖZER A. Effects of nicardipine on collar-induced intimal thickening and vascular reactivity in the rabbit. J. Pharm. Pharmacol. 1999;51:441–447. doi: 10.1211/0022357991772493. [DOI] [PubMed] [Google Scholar]
- LEV S., MORENO H., MARTINEZ R., CANOLL P., PELES E., MUSACCHINO J.M., PLOWMAN G.D., RUDY B., SCHLESSINGER J. Protein tyrosine kinase PYK2 involved in Ca++-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- LIBBY P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- LOALDI A., POLESE A., MONTORSI P., DE CESARE N., FABBIOCCHI F., RAVAGNANI P., GUAZZI M.D. Comparison of nifedipine, propranolol, and isosorbide dinitrate on angiographic progression and regression of coronary arterial narrowings in angina pectoris. Am. J. Cardiol. 1989;64:433–439. doi: 10.1016/0002-9149(89)90417-7. [DOI] [PubMed] [Google Scholar]
- LOPEZ-ILASACA M. Signaling from G-protein-coupled receptors to mitogen-activated protein (MAP) kinase cascades. Biochem. Pharmacol. 1998;56:269–277. doi: 10.1016/s0006-2952(98)00059-8. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1950;193:265–275. [PubMed] [Google Scholar]
- LUCCHESI P.A., BELL J.M., WILLIS L.S., BYRON K.L., CORSON M.A., BERK B.C. Ca2+-dependent mitogen-activated protein kinase activation in spontaneously hypertensive rat vascular smooth muscle defines a hypertensive signal transduction phenotype. Circ. Res. 1996;78:962–970. doi: 10.1161/01.res.78.6.962. [DOI] [PubMed] [Google Scholar]
- MEDICAL RESEARCH COUNCIL WORKING PARTY Medical Research Council trial of treatment of hypertension in older adults: Principal results. BMJ. 1992;304:405–412. doi: 10.1136/bmj.304.6824.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLER J.E., MORRISON J., STONE P.H., RUDE R.E., ROSNER B., ROBERTS R., PEARLE D.L., TURI Z.G., SCHNEIDER J.F., SERFAS D.H., TATE C., SCHEINER E., SOBEL B.E., HENNEKENS C.H., BRAUNWALD E. Nifedipine therapy for patients with threatened and acute myocardial infarction: A randomized, double-blind, placebo-controlled comparison. Circulation. 1984;69:740–747. doi: 10.1161/01.cir.69.4.740. [DOI] [PubMed] [Google Scholar]
- NATIONAL HIGH BLOOD PRESSURE EDUCATION PROGRAM WORKING GROUP National High Blood Pressure Education Working Group Report on hypertension in the elderly. Hypertension. 1994;23:275–285. [PubMed] [Google Scholar]
- PAHOR N., GURALNIK J.M., CORTI M.-C., FOLEY D.J., CARBONIN P., HAVLIK R.J. Long-term survival and use of antihypertensive medications in older persons. J. Am. Geriatr. Soc. 1995;43:1191–1197. doi: 10.1111/j.1532-5415.1995.tb07393.x. [DOI] [PubMed] [Google Scholar]
- PSATY B.M., WEISS N.S., FURBERG C.D., KOEPSELL T.D., SISCOVICK D.S., ROSENDAAL F.R., SMITH N.L., HECKBERT S.R., KAPLAN R.C., LIN D., FLEMING T.R., WAGNER E.H. Surrogate end points, health outcomes, and the drug-approval process for the treatment of risk factors for cardiovascular disease. JAMA. 1999;282:786–790. doi: 10.1001/jama.282.8.786. [DOI] [PubMed] [Google Scholar]
- RABKIN S.W., KONG J.Y. Nifedipine does not induce but rather prevents apoptosis in cardiomyocytes. Eur. J. Pharmacol. 2000;388:209–217. doi: 10.1016/s0014-2999(99)00880-8. [DOI] [PubMed] [Google Scholar]
- RAKUSAN K., CICUTTI N., KAZDA S., TUREK Z. Effect of nifedipine on coronary capillary geometry in normotensive and hypertensive rats. Hypertension. 1994;24:205–211. doi: 10.1161/01.hyp.24.2.205. [DOI] [PubMed] [Google Scholar]
- ROSS R. The pathogenesis of atherosclerosis – An update. N. Eng. J. Med. 1986;314:488–500. doi: 10.1056/NEJM198602203140806. [DOI] [PubMed] [Google Scholar]
- SACCO R.L., WOLF P.A., GORELICK P.B. Risk factors and their management for stroke prevention: Outlook for 1999 and beyond. Neurology. 1999;53:S15–S24. [PubMed] [Google Scholar]
- SAGIE A., LARSON M.G., LEVY D. The natural history of borderline isolated systolic hypertension. N. Eng. J. Med. 1993;329:1912–1917. doi: 10.1056/NEJM199312233292602. [DOI] [PubMed] [Google Scholar]
- SCHIFFRIN E.L., DENG L.Y. Structure and function of resistance arteries of hypertensive patients treated with a β-blocker or a calcium-channel antagonist. J. Hypertens. 1995;14:1247–1255. doi: 10.1097/00004872-199610000-00014. [DOI] [PubMed] [Google Scholar]
- SEGER R., KREBS E.G. The MAPK signaling cascade. FASEB J. 1995;9:726–798. [PubMed] [Google Scholar]
- SHEP COOPERATIVE RESEARCH GROUP Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: Final results of Systolic Hypertension in the Elderly Program (SHEP) JAMA. 1991;265:3255–3264. [PubMed] [Google Scholar]
- SISCOVICK D.R., RAGHUNATHAN T.E., PSATY B.M., KOEPSELL T.D., WICKLUND K.G., LIN X., COBB L., RAUTAHARJU P.M., COPASS M.K., WAGNER E.H. Diuretic therapy for hypertension and the risk of primary cardiac arrest. N. Eng. J. Med. 1994;330:1852–1857. doi: 10.1056/NEJM199406303302603. [DOI] [PubMed] [Google Scholar]
- SOMA M.R., NATALI M., DONETTI E., BAETTA R., FARINA P., LEONARDI A., COMPARATO C., BARBERI L., CATAPANO A.L. Effect of lercanidipine and its (R)-enantiomer on atherosclerotic lesions induced in hypercholesterolemic rabbits. Br. J. Pharmacol. 1998;125:1471–1476. doi: 10.1038/sj.bjp.0702221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAESSEN J.A., FAGARD R., THIJS L., CELIS H., ARABIDZE G.G., BIRKENHAGER W.H., BULPITT C.J., DELEEUW P.W., DOLLEY C.T., FLETCHER A.E., FORETTE A.E., LEONETTI G., NACHEV C., O'BRIEN E.T., ROSENFELD J., RODICIO J.L., TOUMILEHTO J., ZANCHETTI A. Randomized double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension: The Systolic Hypertension in Europe (Sys-Eur) Trial Investigators. Lancet. 1997;350:757–764. doi: 10.1016/s0140-6736(97)05381-6. [DOI] [PubMed] [Google Scholar]
- TOKIWA G., DIKIC I., LEV S., SCHLESSINGER J. Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science. 1996;273:792–794. doi: 10.1126/science.273.5276.792. [DOI] [PubMed] [Google Scholar]
- TSUBOI Y., SHANKLAND S.J., GRANDE J.P., WALKER H.J., JOHNSON R.J., DOUSA T.P. Suppression of mesangial proliferative glomerulonephritis development in rats by inhibitors of cAMP phosphodiesterase isozymes types III and IV. J. Clin. Invest. 1996;98:262–270. doi: 10.1172/JCI118788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ÜSTÜNES L., YASA M., KERRY Z., ÖZDEMIR N., BERKAN T., ERHAN Y., ÖZER A. Effect of verapamil on intimal thickening and vascular reactivity in the collared carotid artery of the rabbit. Br. J. Pharmacol. 1996;118:1681–1688. doi: 10.1111/j.1476-5381.1996.tb15592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Y., PERFETTI R., GREIG N.H., HOLLOWAY H.W., DEORE K.A., MONTROSE-RAFIZADEH C., ELAHI D., EGAN J.M. Glucagon-like peptide-1 can reverse the age-related decline in glucose tolerance in rats. J. Clin. Invest. 1997;99:2883–2889. doi: 10.1172/JCI119482. [DOI] [PMC free article] [PubMed] [Google Scholar]