

Abstract
Mechanisms of relaxation and contraction to protease-activated receptor- (PAR) tethered ligand peptides (SFLLRN/TFLLR, SLIGRL and GYPGKF (all C-terminally amidated) for PAR1, PAR2 and PAR4, respectively) and enzymes (thrombin and trypsin) were investigated in isolated segments of rat trachea, main and first order intrapulmonary bronchi.
In airway segments previously exposed to SLIGRL, SFLLRN caused contractions that were potentiated by indomethacin, but were independent of mast cell degranulation. Contractions to TFLLR in the intrapulmonary bronchi were similarly potentiated by indomethacin.
SLIGRL caused epithelium-dependent relaxations which were unaffected by NG-nitro-L-arginine, 1-H-oxodiazol-[1,2,4]-[4,3-a]quinoxaline-1-one or zinc-protoporphyrin-IX but were abolished by haemoglobin in all three regions of the airways. Relaxations to SLIGRL were markedly attenuated by indomethacin only in the main and intrapulmonary bronchi.
GYPGKF caused epithelium-dependent relaxations in all three regions of the airway which were only significantly inhibited by indomethacin in the intrapulmonary bronchi.
In general, thrombin and trypsin failed to cause any response in the airways tested.
Intense PAR2-immunoreactivity was observed on airway epithelium. PAR1-immunoreactivity was faint on airway epithelium and smooth muscle, but was prevalent in mast cells.
These findings indicate that PAR2 and possibly PAR4 present on rat airway epithelia mediate smooth muscle relaxation via cyclo-oxygenase-dependent and -independent mechanisms. PAR1-mediated contractions were most likely due to activation of smooth muscle receptors. The general failure of thrombin and trypsin to cause responses which may have been due to endogenous protease inhibitors, highlights the need for caution in assessing pathophysiological roles for PARs if only enzymes are used to activate PARs.
Keywords: Protease-activated receptors (PAR1, PAR2, PAR4); trypsin; thrombin; indomethacin; nitric oxide; prostanoid; rat airways; smooth muscle relaxation; haemoglobin
Introduction
Four members of the seven-transmembrane family of protease-activated receptors (PARs) are known (for review see Coughlin, 1994; 1999; Déry et al., 1998; Hollenberg, 1999). We (Cocks et al., 1999) and others (Lan et al., 2000) have shown that activation of epithelial PAR1, PAR2 and PAR4 in mouse airways with either the serine proteases thrombin and trypsin, respectively, or synthetic peptide mimetics of each receptor's tethered ligand sequence, cause powerful, cyclo-oxygenase-dependent relaxation of airway smooth muscle. Also, we demonstrated that the PAR2-selective synthetic peptide, SLIGRL caused similar epithelium-dependent relaxation in rat isolated bronchi as well as inhibition of the increase in airway resistance to intravenous administration of serotonin in anaesthetized rats (Cocks et al., 1999). These results together with our additional finding that PAR2 and its proteolytic activator, trypsin, are co-localized in human airways epithelium (Cocks et al., 1999), have led us to hypothesize that epithelial PARs act as sensors of early inflammation (Cocks & Moffatt, 2000). Given the importance of our in vivo rat studies in formulating this hypothesis, the aim of our present study was to examine the mechanisms underlying changes in reactivity to PAR activators in different regions of rat isolated airways.
Methods
Preparation of rat airways ring segments
Sprague-Dawley rats (200–300 g) of either sex were killed with a lethal dose of sodium pentobarbitone (120 mg kg−1, i.p.) and immediately exsanguinated to avoid blood clot formation in the lungs. The thorax was then opened and the lung and trachea removed and immersed in Krebs solution (composition in mM: Na+ 144, Cl− 128.7, HCO3− 25, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, H2PO4− 1.2, SO42− 1.2 and glucose 11) continuously bubbled with 95% O2, 5% CO2 to maintain a high pO2 and pH of 7.4. The trachea, main bronchi and the first order (intrapulmonary) bronchi from the left lobe of the lung were then dissected free from surrounding parenchyma and blood vessels.
Tracheal and bronchial rings (3–4 cartilage rings in length) were mounted in 5 or 10 ml organ baths containing Krebs solution (37°C) on 150 μm diameter stainless steel hooks with one hook connected to a micrometer-adjustable support rod and the other to a force transducer (Grass Instruments, Model FT03C, MA, U.S.A.). Segments of intrapulmonary bronchi 2 mm in length were mounted on 40 μm stainless steel wires in dual chamber Mulvany-Halpern myographs (JP Trading, Aarhus, Denmark) containing Krebs solution (37°C). Changes in circumferential isometric force were amplified and recorded on flat bed chart recorders (W & W Scientific Instruments, Basel, Switzerland). In some segments of trachea and bronchi the lumen was gently rubbed with a moistened tapered wooden stick. To similarly remove the epithelium from the intrapulmonary bronchi, Krebs solution containing 0.1% Triton X-100 was gently flushed through the intact lung then washed with Krebs solution before dissection (Cocks et al., 1999). The successful removal of the epithelium was confirmed histologically in haemotoxylin and eosin-stained 4 μm paraffin sections.
Rings of airways mounted in organ baths and myographs were allowed to equilibrate for 30–60 min before being stretched to a tension of 2, 1, and 0.5 g for trachea, main and intrapulmonary bronchi, respectively. When passive tension had stabilized the maximum contraction (Fmax) was determined for each ring with 30 μM acetylcholine (ACh). The preparations were then washed three times with Krebs solution and treated with 0.3 μM nifedipine to inhibit the development of spontaneous activity. Thirty minutes later, tissues were contracted with titrated concentrations of the stable muscarinic agonist, carbachol (0.1–1 μM), to achieve a maintained contraction of 40–60% Fmax.
Effect of PAR agonists
Sequential, cumulative concentration responses to the synthetic PAR-tethered ligand peptide sequences, SLIGRL (PAR2) and SFLLRN (PAR1) were obtained in carbachol-contracted tissues without washing between each curve, since the tissues spontaneously recovered to their initial levels of carbachol-induced force following the maximum response to SLIGRL. Similarly, sequential cumulative concentration responses were obtained to thrombin followed by trypsin. This protocol of sequential additions of agonists without washout resulted in complete desensitization of PAR2 with SLIGRL or PAR1 with thrombin (data not shown). Therefore, specific effects of the peptides and enzymes at their designated receptors could be evaluated without any potential receptor cross-over since in some tissues SFLLRN has been shown to activate PAR2 (Blackhart et al., 1996; Hollenberg et al., 1997) and trypsin can activate PAR1 (Vu et al., 1991; Molino et al., 1997). In other tissues, however, SFLLRN appears to selectively activate PAR1 (Hamilton & Cocks, 2000). There is no evidence that SLIGRL activates PAR1 or thrombin activates PAR2. Cumulative concentration responses were obtained to the PAR4 peptide, GYPGKF or the selective PAR1 peptide, TFLLR (Hollenberg et al., 1997) in separate tissues contracted with carbachol. In all cases, after responses to maximum concentrations of the PAR agonists were obtained, isoprenaline (3 μM) and 3-isobutyl-1-methylxanthine (IBMX, 100 μM) were added to obtain each tissue's maximum relaxation.
Mechanisms of responses to PAR-activating peptides
Prior to contracting with carbachol, some tissues were treated with various inhibitors for 30 min and run in parallel with untreated (control) tissues from the same animal. Inhibitors tested were indomethacin (3 μM), NG-nitro-L-arginine (L-NOARG, 100 μM), oxyhaemoglobin (oxyHb, 20 μM), metHb (20 μM), 1-H-oxodiazol-[1,2,4]-[4,3-a]quinoxaline-1-one (ODQ, 10 μM) and zinc-protoporphyrin-IX (ZnPP-IX, 10 μM). Also, the effect of mast cell degranulation on SFLLRN-induced contractions in rat bronchi was examined. These preparations were treated with indomethacin (3 μM) and compound 48/80 (3 μM) for 30 min prior to contraction with carbachol to 40–60% Fmax. When a stable level of contraction was achieved, SLIGRL (10 μM) was added before cumulative additions of SFLLRN, again to ensure that SFLLRN activated only PAR1.
Immunohistochemistry
Similar Sprague-Dawley rats as used in the functional studies were killed with an overdose of sodium pentobarbitone (120 mg kg−1, i.p.) and the lung perfused with phosphate-buffered 4% (w v−1) paraformaldehyde (pH 7.3). The lungs were removed and immersed overnight in the same fixative before being thoroughly washed in phosphate-buffered saline (PBS). Segments of trachea, main bronchi and intrapulmonary bronchi were then cryoprotected in a 15% (w v−1) sucrose solution in PBS for up to a week before being snap frozen in Tissue-Tek® OCT compound (Sakura Finetek, U.S.A.) and stored at −70°C until sectioned. Frozen sections (14 μm) were collected on 1% gelatin-coated slides.
PAR2 was localized using two different antibodies. Tissue sections were first exposed to a polyclonal rabbit antiserum raised against the C-terminus of the mouse PAR2 (CSVKTSY, PAR2-C, 1 : 500 dilution; gift from N. Bunnett, San Francisco, U.S.A.) for 48 h. Use of the PAR2-C antiserum was compared to controls where the antiserum was incubated (4°C, 24 h) with the immunizing peptide (10 μM). The PAR2 antiserum labelled tissues were then sequentially labelled with a biotinylated donkey anti-rabbit antiserum (1 : 100 dilution, 1 h; Amersham, U.K.) and fluorescein isothiocynate (FITC)-conjugated streptavidin (1 : 20 dilution, 1 h; Amersham, U.K.). The same sections were then exposed to a monoclonal mouse antibody raised against the N-terminus of human PAR2 (SLIGKVDGTSHVTG amino acid sequence, PAR2-N, 1 : 20 dilution; gift from L. Brass, Philadelphia, U.S.A.) for 24 h and then labelled with horse anti-mouse texas red (1 : 200 dilution, 1 h; Vector Lab, CA, U.S.A.). PAR1 was localized with a monoclonal mouse antiserum raised against the N-terminus (SFLLRNPNDKYEPF, 1 : 100 dilution, 48 h; gift from L. Brass) and sequentially labelled with biotinylated sheep anti-mouse antiserum (1 : 100 dilution, 1 h; Amersham, U.K.) and FITC-conjugated streptavidin. In between exposure to different reagents, the sections were washed repeatedly in PBS for 45 min. Sections were then mounted in filtered buffered glycerol and fluorescence observed under a confocal microscope (Biorad MRC 1000). Each image represents 3–5 z-sections of 0.5 μm thickness.
Analyses and statistics
All responses were measured as percentages of the carbachol-induced contractions. Where complete concentration response curves could be obtained, the sensitivity (pEC50) was determined by regression analysis (Kemp & Cocks, 1997). Unpaired t-tests were used to compare responses between two groups and a one-way analysis of variance (ANOVA) with a Tukey-Kramer post hoc test was used to compare maximum responses (Rmax) between multiple treatment groups within each airway branch. All results were expressed as mean±the standard error of the mean (s.e.mean). Responses were considered significantly different if P<0.05.
Drugs
Stock solutions of the PAR-activating peptides (10 mM), SFLLRN, TFLLR, SLIGRL and GYPGKF (single letter coded amino-acid sequence; Auspep, Melbourne, Australia; all C-terminally amidated) and the serine proteases, α-thrombin (1000 u ml−1, bovine serum; Sigma, U.S.A.), trypsin (1000 u ml−1, bovine pancreas; Worthington Biochem, U.S.A.), were dissolved in distilled water, aliquoted and stored at −20°C. Acetylcholine chloride, carbachol, (−)-isoprenaline (Sigma, U.S.A.), IBMX (Aldrich-Chemie, West Germany) and compound 48/80 (Sapphire Bioscience, Sydney, Australia) were dissolved in distilled water as 10 mM stock solutions. Stock solutions of L-NOARG (100 mM) and indomethacin (10 mM; both Sigma, U.S.A.) were prepared in 1 M NaHCO3 and 1 M Na2CO3, respectively whilst nifedipine (Biomol, U.S.A.) was prepared as a 1 mM stock solution in absolute ethanol. MetHb (bovine serum; Sigma, U.S.A.) was dissolved in 0.9% (w v−1) saline as a 1 mM solution and reduced with sodium dithionite (BDH Chemicals Ltd., U.K.). The reduced Hb (oxyHb) was then separated from excess dithionite and oxidized Hb (metHb) by gel filtration through a Sephadex PD-10 column (Pharmacia Biotech, Sweden). ODQ (Biomol, U.S.A.) and ZnPP-IX (Biomol, U.S.A.) were prepared as 10 mM stock solutions in dimethyl sulphoxide. All further dilutions of drugs were in distilled water.
Results
Responses to PAR-activating peptides
The PAR2-activating peptide, SLIGRL (0.1–100 μM), caused concentration-dependent relaxations with pEC50 and maxima of 5.1±0.1, 60.3±2.8%; 5.5±0.1, 47.8±5.6% and 5.1±0.2, 66.8±5.2% in the rat trachea, main and intrapulmonary bronchi, respectively (Figure 1), which in each region were abolished by the removal of the epithelium (data not shown). Similar concentration- and epithelium-dependent relaxations were observed in response to the PAR4-activating peptide, GYPGKF (1–300 μM) in the trachea, main and intrapulmonary bronchi with responses at 300 μM of 41.2±5.9, 32.5±1.6 and 36.4±3.0%, respectively (Figure 1). By contrast, the PAR1-activating peptide, SFLLRN (0.1–30 μM) caused only concentration-dependent contractions in the trachea, main and intrapulmonary bronchi with responses at 30 μM of 19.2±5.6, 59.5±14.1 and 49.3±12.9%, respectively (Figure 1). The more selective PAR1-activating peptide, TFLLR (30 μM) produced no responses in the intrapulmonary bronchi (n=5) but caused 19.5±5.5% relaxation (n=8) in the trachea (data not shown).
Figure 1.
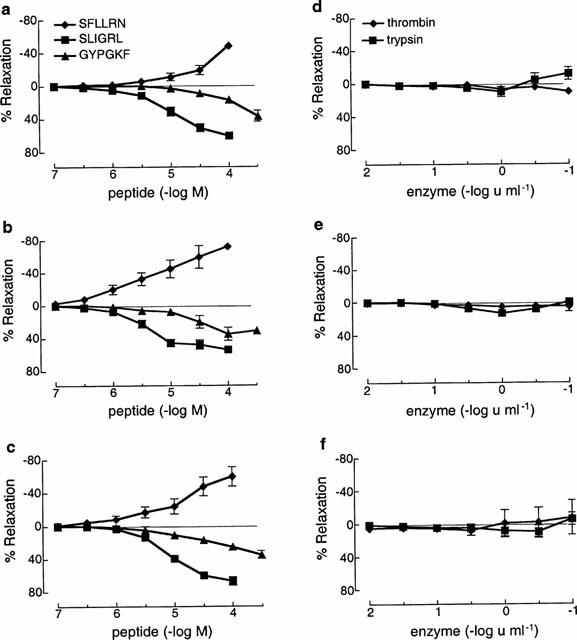
Cumulative concentration-response curves to the PAR1, PAR2 and PAR4-activating peptides, SFLLRN, SLIGRL and GYPGKF, respectively and the PAR-activating enzymes, thrombin and trypsin in isolated rat trachea (a,d), main bronchi (b,e) and intrapulmonary bronchi (c,f) contracted to ≈50% Fmax with carbachol. Values are expressed as percentage (mean±s.e.mean) relaxations or contractions of the carbachol-induced contraction (n=6–12).
Responses to enzymes
The PAR-activating serine proteases, trypsin and thrombin (0.01–10 u ml−1) failed to produce any response in all three regions of the airways examined from five animals (Figures 1 and 2). In preparations from one rat, however, concentration-dependent relaxations to both thrombin and trypsin were observed but only in the intrapulmonary bronchi (Figure 2). In two further animals the effects of higher concentrations of trypsin and thrombin were examined in the trachea and main bronchi. In both cases, concentrations of trypsin ⩾30 u ml−1 caused slowly developing relaxations that were at least partly the result of tissue damage, since active tension fell below the level of passive tension in the preparations (data not shown). Similar high concentrations of thrombin had either no effect or caused weak contractions (data not shown).
Figure 2.
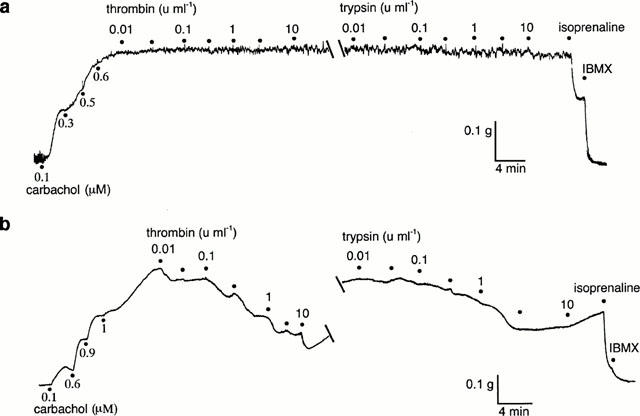
Digitized traces of original chart recordings showing responses to cumulative additions of thrombin and trypsin in two rings of rat intrapulmonary bronchi from different rats contracted to ≈50% Fmax with carbachol. Maximum relaxations were obtained with isoprenaline (3 μM) and IBMX (100 μM). (a) A typical preparation that failed to respond to either trypsin or thrombin. (b) One of six preparations in which concentration-dependent relaxations to both enzymes could be obtained. The breaks in the traces represent 20 min without washout of thrombin.
Effect of inhibitors of cyclo-oxygenase, nitric oxide (NO) and haem oxygenase on responses to PAR-activating peptides
Indomethacin (3 μM) significantly inhibited the relaxations to SLIGRL in the main (Rmax: 24.9±5.7%) and intrapulmonary bronchi (Rmax: 32.8±6.8%) but had no effect on relaxations to SLIGRL in the trachea (Rmax: 47.1±6.0%; Figure 3). Relaxations to SLIGRL in all segments of the airways were abolished by either oxyHb (Figure 3) or metHb (data not shown) yet were unaffected by either the NO synthase inhibitor, L-NOARG (Figure 3), the guanylyl cyclase inhibitor, ODQ (n=4, data not shown) or the haem oxygenase inhibitor, ZnPP-IX (10 μM; n=4, data not shown). Like SLIGRL, epithelium-dependent relaxations to GYPGKF were unaffected by indomethacin in the trachea, but were inhibited in the intrapulmonary bronchi (Figure 4). Also, there was a trend for indomethacin to inhibit relaxations to GYPGKF in the main bronchi (Figure 4). By contrast, contractions to SFLLRN were significantly potentiated by indomethacin in each region of the airways (Table 1), but were unaffected following treatment of bronchi with 3 μM compound 48/80 (Figure 5). Indomethacin unmasked a 32.8±8.9% (n=6) contraction to TFLLR in the intrapulmonary bronchi and abolished the relaxation to TFLLR in the trachea (7.2±5.6%, n=8).
Figure 3.
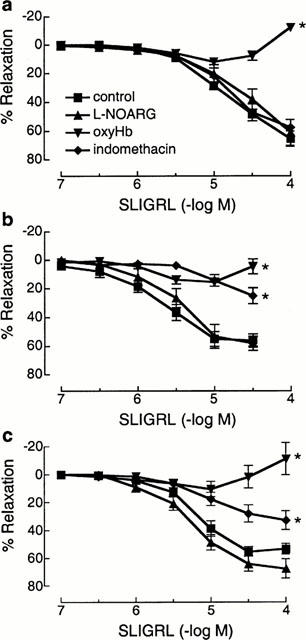
Cumulative concentration-relaxation curves to the PAR2-activating peptide, SLIGRL in the presence of either NG-nitro-L-arginine (L-NOARG, 100 μM), oxyhaemoglobin (oxyHb, 20 μM) or indomethacin (3 μM) in rat isolated trachea (a), main bronchi (b) and intrapulmonary bronchi (c) contracted to ≈50% Fmax with carbachol. Values are expressed as percentage (mean±s.e.mean) relaxations of the carbachol-induced contraction (n=6–12). *Denotes maximum values significantly different from controls (P<0.05).
Figure 4.
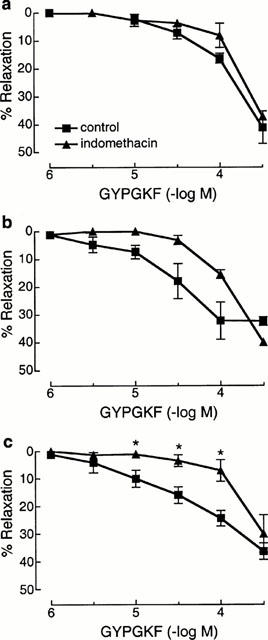
Cumulative concentration relaxation curves to the PAR4-activating peptide, GYPGKF in the rat trachea (a), main bronchi (b) and intrapulmonary bronchi (c) in the absence and presence of indomethacin (3 μM). Values are expressed as percentage (mean±s.e.mean) relaxations of the carbachol-induced contraction (n=3–6). Note that the control curves are reproduced from Figure 1. *Denotes significant differences (P<0.05) from control responses.
Table 1.
Effect of indomethacin (3 μM) on contractile responses (mean±s.e.mean % of the carbachol-induced contraction; n=5–8) to SFLLRN (30 μM) in rat airway preparations
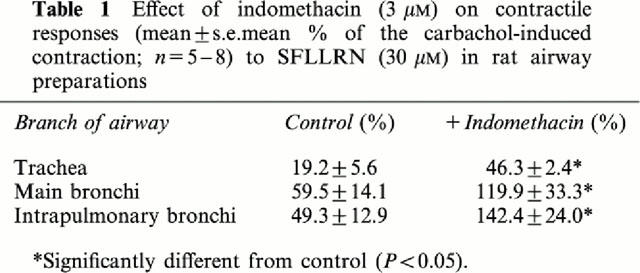
Figure 5.
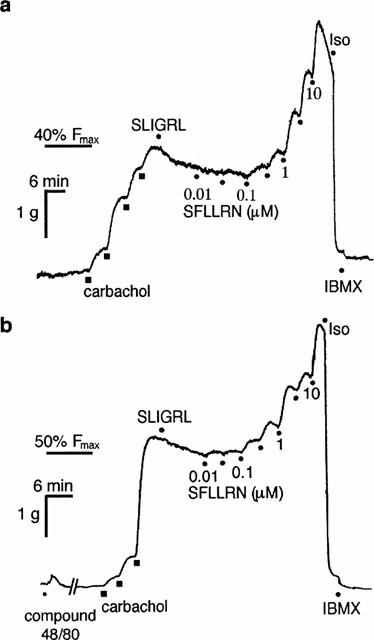
Digitized traces of original chart recordings showing concentration-dependent contractions to SFLLRN in the absence (a) and presence (b) of compound 48/80 (3 μM). Both tissues were treated with indomethacin (3 μM) and nifedipine (0.3 μM) prior to contracting to 40–50% Fmax with carbachol and subsequently treated with SLIGRL (10 μM) before cumulative additions of SFLLRN. Maximum relaxations were obtained with isoprenaline (Iso, 3 μM) and IBMX (100 μM). The break in (b) represents 30 min. Representative of three similar experiments.
Immunohistochemical localization of PAR2 and PAR1
Immunoreactivities to PAR2-C and PAR2-N antibodies co-localized and were most commonly observed on the apical surface of ciliated columnar epithelial cells in the rat trachea, bronchi and its first branch (Figure 6). In some preparations PAR2 immunofluorescence was observed on the plasma membrane and cytoplasm of non-ciliated epithelial cells and on airways smooth muscle. Immunofluorescence in sections labelled with PAR2-C-antiserum preabsorbed with the immunizing peptide was abolished (data not shown). Very weak PAR1 immunoreactivity was observed on epithelial and smooth muscle cells of the trachea, main bronchi and intrapulmonary bronchi, but the cytoplasm of mast cells was intensely immunofluorescent (Figure 7).
Figure 6.
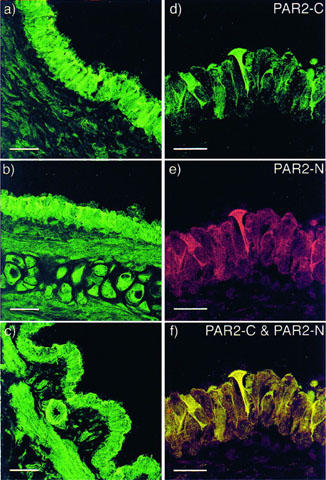
Confocal microscopic images showing immunohistochemical localization of PAR2 in rat trachea (a), main bronchi (b) and intrapulmonary bronchi (c) with the PAR2-C antibody (green). Higher magnification images (d,e,f) of a single section of the main bronchus to show co-localization of PAR2-like immunofluorescence with two separate PAR2 antibodies; (d) PAR2-C antibody (green), (e) PAR2-N antibody (red) and (f) superimposed images of (d) and (e) showing co-localization as yellow. Scale bars for left panel (a–c) represent 30 μm; whilst the right panel (d–f) scale bars represent 15 μm.
Figure 7.
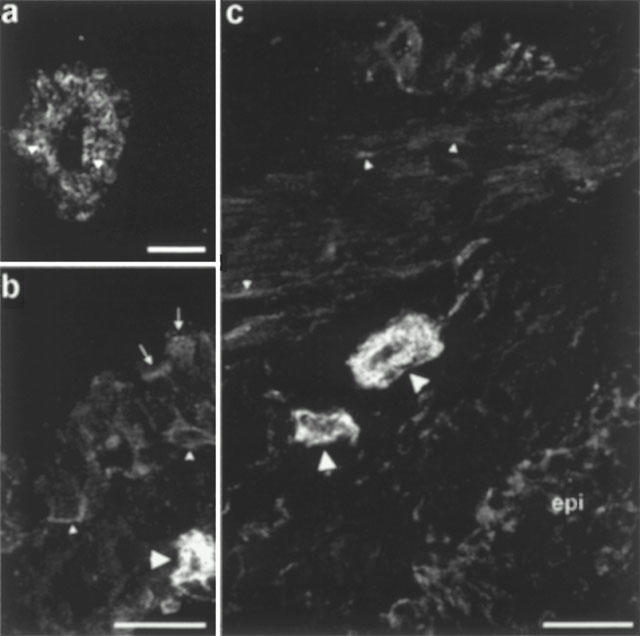
Confocal microscopic images showing PAR1 immunofluorescence in rat bronchi, which is representative of the other two airway regions used in the functional studies. (a) A mast cell showing intense cytoplasmic immunofluorescence with distinct granules (arrowheads) showing no staining. (b) Faint staining of ciliated (arrows) and basal (small arrowheads) epithelial cells. A mast cell (large arrowhead) is shown to illustrate the low level of epithelial fluorescence. (c) Comparison of PAR-1 immunofluorescence on smooth muscle (small arrowheads), mast cells (large arrowheads) and the epithelium (epi). Only a few smooth muscle cells show patchy surface staining while most cells appear to show very weak cytoplasmic staining. The scale bar in (a) represents 10 μm, whilst those in (b) and (c) represent 25 μm.
Discussion
The main finding of this study was that although the PAR1-, PAR2- and PAR4-activating peptides, SFLLRN, SLIGRL and GYPGKF, respectively caused contraction (PAR1) and epithelium-dependent relaxation (PAR2 and PAR4) in three discrete regions of the rat isolated airways, the enzyme activators of these receptors, thrombin and trypsin (Nystedt et al., 1994; Saifeddine et al., 1996; Molino et al., 1997; Xu et al., 1998) were mostly ineffective. Also, another novel and puzzling result was that SLIGRL-induced relaxations were inhibited by haemoglobin regardless of the redox state of its haem prosthetic group (i.e. Fe2+/ Fe3+) but not by other conventional NO inhibitors. Thus, the inhibition by Hb most likely occurred via a mechanism independent of its NO- or CO-scavenging properties (Bowman et al., 1982; Martin et al., 1985).
One explanation for the failure of trypsin and thrombin to mimic the responses observed to the PAR-activating peptides could be that the ligands were acting independent of the cloned PARs. Activation of PAR1 and PAR2 by the activating peptides is supported by immunohistochemical localization of these receptors on rat airway epithelia (and smooth muscle). Moreover, precise co-localization of immunofluorescence with two distinct PAR2 antibodies validates labelling of the cloned PAR2. Although similar immunohistochemical techniques were not used in this study to localize PAR4, evidence currently available suggests that GYPGKF selectively activates PAR4 (Hollenberg et al., 1999; Lan et al., 2000). Therefore, responses observed to this peptide in the rat airways were most likely mediated by PAR4. One of the PAR2 antibodies used was raised against part of the N-terminal of the human PAR2 which includes ser37 of the arg36-ser37 cleavage site (D'Andrea et al., 1998) and the other raised against the C-terminal of the mouse PAR2 (Cocks et al., 1999; Corvera et al., 1999). Thus, unless the rat airway epithelial PAR2 contains a mutation at the specific cleavage site for trypsin, it is difficult to explain why trypsin failed to cleave the receptor and cause relaxation. The inability of enzymes to activate both epithelial and smooth muscle PARs in rat airways may be related to the presence of endogenous inhibitors of serine proteases (e.g. serpins: Potempa et al., 1994; Wright, 1996; Whisstock et al., 1998). For example, the main regulator of extracellular trypsin-like activity in both the airways (Cichy et al., 1997) and gut (Potempa et al., 1994) is α1-antitrypsin (α1-AT) which is also able to inhibit thrombin (Bonniec et al., 1995). Relative high concentrations of trypsin were required to stimulate ion transport across rat intestinal epithelium compared to a cloned PAR2 assay (Vergnolle et al., 1998). These authors, however, argued that both the high concentrations of trypsin and the reduced potency of peptide PAR2 activators required to stimulate ion transport in the rat intestinal assay indicated that a separate PAR2-like, but relatively trypsin-insensitive receptor, was present in the rat jejunum (Vergnolle et al., 1998). However, in the present study SLIGRL and SFLLRN had similar potencies as reported previously for mouse airways (Cocks et al., 1999; Lan et al., 2000). Therefore, we suggest that the PARs present in rat airways are similar to the cloned PARs and that the inability of both trypsin and thrombin to activate PAR1, PAR2 and PAR4 is most likely due to the presence of broad-spectrum anti-proteases, like α1-AT, in levels sufficient to block the activity of exogenously applied enzymes.
Although PAR1 immunoreactivity was weak in the epithelium and smooth muscle of rat airways, mast cells were strongly PAR1 immunoreactive. It is unlikely, however, that mast cell degranulation products such as histamine and serotonin accounted for the contractions to SFLLRN observed since degranulation of mast cells with compound 48/80 (Ennis et al., 1984; Levi-Schaffer & Riesel-Yaron, 1990) failed to affect the contractions to SFLLRN. Furthermore, compound 48/80 caused much weaker contractions than SFLLRN (see Figure 5). The finding that the contractions to SFLLRN were potentiated by indomethacin in all sections of the rat airway at first suggested the involvement of a basally-released inhibitory prostanoid. However, this is unlikely since contractions to serotonin were not similarly potentiated by indomethacin (Chow & Cocks, unpublished data). Also, the enhanced contractile responses to SFLLRN after treatment with indomethacin was not due to the inhibition of prostanoid-mediated relaxations to this ligand acting non-specifically on PAR2 (Blackhart et al., 1996; Hollenberg et al., 1997; Moffatt & Cocks, 1998; Al-Ani et al., 1999; Kawabata et al., 1999) since PAR2 were desensitized with SLIGRL prior to the addition of SFLLRN. The contractions unmasked by indomethacin in the intrapulmonary bronchi by the more selective but less potent PAR1-activating peptide, TFLLR (Hollenberg et al., 1997) support the conclusion here that similar contractions to SFLLRN in the rat airways were PAR1-mediated. Any role for PAR2-induced release of inhibitory prostanoids in the response to SFLLRN is further unlikely as indomethacin failed to affect the relaxations to SLIGRL in the trachea, yet the contractions to SFLLRN in this preparation were potentiated as in the other regions of the airways. Therefore, although PAR1 immunofluorescence was only weakly detected on epithelial and smooth muscle cells, we suggest that epithelial PAR1 were activated by SFLLRN to release inhibitory prostanoids in sufficient amounts to blunt the contractile response to simultaneous activation of smooth muscle PAR1. This is further supported by concentration-dependent relaxations observed to TFLLR in the trachea which were inhibited by indomethacin. The failure of the cyclo-oxygenase inhibitor to have any effect on the epithelium-dependent relaxations to SLIGRL and GYPGKF in the trachea suggests that unlike epithelial PAR1, epithelial PAR2 and PAR4 are not coupled to cyclo-oxygenase in this region of the rat airways.
The dependence of the relaxant responses to SLIGRL and GYPGKF on cyclo-oxygenase products appeared to increase with decreasing airway calibre, since indomethacin caused greater inhibition of responses to each peptide in the bronchi but had no effect in the trachea. We considered it likely that the non-prostanoid components of these responses were mediated by NO, since similar epithelium-dependent relaxations to substance P in rat airways have been shown to be mediated by both prostaglandin E2 (PGE2) and NO (Szarek et al., 1998; Mhanna et al., 1999). Whilst relaxations to SLIGRL were abolished by either oxyHb or metHb, this was unlikely due to removal of NO or CO since inhibitors of NO synthase (L-NOARG: Moore et al., 1990; Rees et al., 1990), guanylyl cyclase (ODQ: Garthwaite et al., 1995) and haem oxygenase (ZnPP-IX: Ny et al., 1996) had no effect on the responses to SLIGRL in any region of the airways studied. In addition, any direct interaction between Hb and SLIGRL or PGE2 seems unlikely for two reasons. First, as we have previously reported, oxyHb had no effect on similar epithelium- and PGE2-dependent relaxations to SLIGRL in the mouse isolated bronchi (Cocks et al., 1999) and second, oxyHb did not affect the relaxations to exogenously applied PGE2 in both the mouse and rat bronchi (Chow & Cocks, unpublished observations). Therefore, although we have not determined the nature and mechanism of the mediator inducing indomethacin-insensitive relaxations to the PAR-activating peptides, SLIGRL (and GYPGKF), the most likely site of action for Hb is proximal to the production of mediator(s) from the epithelium, since cyclo-oxygenase-derived products contributed differentially to relaxations in each region of the airway despite universal inhibition of responses with Hb.
In conclusion, this study indicates that rat airway epithelial PAR2 and PAR4 as well as smooth muscle PAR1 appear to be activated by their respective synthetic tethered ligands but not enzymatically by either trypsin or thrombin. If this inactivity of trypsin and thrombin is due to endogenous anti-proteases then our study highlights the need to consider species differences in the environments in which PARs are expressed when assessing physiological roles for these receptors.
Acknowledgments
The authors would like to thank Dr Nigel Bunnett and Dr Lawrence Brass for providing the PAR antibodies. The experiments were funded by the National Health and Medical Research Council of Australia and the Western Healthcare Network.
Abbreviations
- Hb
haemoglobin
- IBMX
iso-butyl-methylxanthine
- L-NOARG
NG-nitro-L-arginine
- ODQ
1-H-oxodiazol-[1,2,4]-[4,3-a]quinoxaline-1-one
- PAR
protease-activated receptors
- ZnPP-IX
zinc-protoporphyrin-IX
References
- AL-ANI B., SAIFEDDINE M., KAWABATA A., RENAUX B., MOKASHI S., HOLLENBERG M.D. Proteinase-activated receptor 2 (PAR2). Development of a ligand-binding assay correlating with activation of PAR2 by PAR1- and PAR2- derived peptide ligands. J. Pharm. Exp. Ther. 1999;290:753–760. [PubMed] [Google Scholar]
- BLACKHART B.D., EMILSSON K., NGUYEN D., TENG W., MARTELLI A.J., NYSTEDT S., SUNDELIN J., SCARBOROUGH R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- BONNIEC B.F.L., GUINTO E.R., STONE S.R. Identification of thrombin residues that modulate its interactions with antithrombin III and α1-antitrypsin. Biochemistry. 1995;34:12241–12248. doi: 10.1021/bi00038a019. [DOI] [PubMed] [Google Scholar]
- BOWMAN A., GILLESPIE J.S., POLLACK D. Oxyhaemoglobin blocks nonadrenergic inhibition in the bovine retractor penis muscle. Eur. J. Pharmacol. 1982;85:221–224. doi: 10.1016/0014-2999(82)90470-8. [DOI] [PubMed] [Google Scholar]
- CICHY J., POTEMPA J., TRAVIS J. Biosynthesis of α1-proteinase inhibitor by human lung-derived epithelial cells. J. Biol. Chem. 1997;272:8250–8255. doi: 10.1074/jbc.272.13.8250. [DOI] [PubMed] [Google Scholar]
- COCKS T.M., FONG B., CHOW J.M., ANDERSON G.P., FRAUMAN A.G., GOLDIE R.G., HENRY P.J., CARR M.J., HAMILTON J.R., MOFFATT J.D. A protective role for protease-activated receptors in the airways. Nature. 1999;398:156–160. doi: 10.1038/18223. [DOI] [PubMed] [Google Scholar]
- COCKS T.M., MOFFATT J.D. Protease-activated receptors: sentries for inflammation. Trends Pharmacol. Sci. 2000;21:103–108. doi: 10.1016/s0165-6147(99)01440-6. [DOI] [PubMed] [Google Scholar]
- CORVERA C.U., DERY O., MCCONALOGUE K., GAMP P., THOMA M., AL-ANI B., CAUGHEY G.H., HOLLENBERG M.D., BUNNETT N.W. Thrombin and mast cell tryptase regulate guinea-pig myenteric neurons through proteinase-activated receptors-1 and -2. J. Physiol. 1999;517:741–756. doi: 10.1111/j.1469-7793.1999.0741s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUGHLIN S.R. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9200–9202. doi: 10.1073/pnas.91.20.9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUGHLIN S.R. How the protease thrombin talks to cells. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11023–11027. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'ANDREA M.R., DERIAN C.K., LETURCQ D., BAKER S.M., BRUNMARK A., LING P., DARROW A.L., SANTULLI R.J., BRASS L.F., ANDRADE-GORDON P. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J. Histochem. Cytochem. 1998;46:157–164. doi: 10.1177/002215549804600204. [DOI] [PubMed] [Google Scholar]
- DÉRY O., CORVERA C.U., STEINHOFF M., BUNNETT N.W. Proteinase-activated receptors: novel mechanism of signalling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- ENNIS M., BARROW S.E., BLAIR I.A. Prostaglandin and histamine release from stimulated rat peritoneal mast cells. Agents Actions. 1984;14:397–400. doi: 10.1007/BF01973836. [DOI] [PubMed] [Google Scholar]
- GARTHWAITE J., SOUTHAM E., BOULTON C.L., NIELSON E.B., SCHMIDT K., MAYER B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- HAMILTON J.R., COCKS T.M. Heterogenous mechanisms of endothelium-dependent relaxation to thrombin and peptide activators of protease-activated receptor-1 in porcine isolated coronary artery. Br. J. Pharmacol. 2000;130:181–188. doi: 10.1038/sj.bjp.0703146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLLENBERG M.D. Protease-activated receptors: PAR4 and counting: how long is the count. Trends Pharmacol. Sci. 1999;20:271–273. doi: 10.1016/s0165-6147(99)01333-4. [DOI] [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., AL-ANI B., GUI Y. Proteinase-activated receptor 4 (PAR4): action of PAR4-activating peptides in vascular and gastric tissue and lack of cross-reactivity with PAR1 and PAR2. Can. J. Physiol. Pharmacol. 1999;77:458–464. [PubMed] [Google Scholar]
- HOLLENBERG M.D., SAIFEDDINE M., AL-ANI B., KAWABATA A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can. J. Physiol. Pharmacol. 1997;75:832–841. [PubMed] [Google Scholar]
- KAWABATA A., SAIFEDDINE M., AL-ANI M., LEBLOID L., HOLLENBERG M.D. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: Activation of PAR2 by PAR1-targeted ligands. J. Pharm. Exp. Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- KEMP B.K., COCKS T.M. Evidence that mechanisms dependent and independent of nitric oxide mediate endothelium-dependent relaxation to bradykinin in human small resistance-like coronary-arteries. Br. J. Pharmacol. 1997;120:757–762. doi: 10.1038/sj.bjp.0700928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAN R.S., STEWART G.A., HENRY P.J. Modulation of airway smooth muscle tone by protease activator receptor-1, -2, -3 and -4 in trachea isolated from influenza A virus-infected mice. Br. J. Pharmacol. 2000;129:63–70. doi: 10.1038/sj.bjp.0703007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVI-SCHAFFER F., RIESEL-YARON N. Effects of prolonged incubation of rat peritoneal mast cells with compound 48/80. Eur. J. Immunol. 1990;20:2609–2613. doi: 10.1002/eji.1830201213. [DOI] [PubMed] [Google Scholar]
- MARTIN W., VILLANI G.M., JOTHIANANDAN D., FURCHGOTT R.F. Selective blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation by hemoglobin and methylene blue in rabbit aorta. J. Pharm. Exp. Ther. 1985;232:708–716. [PubMed] [Google Scholar]
- MHANNA M.J., DRESHAJ I.A., HAXHIU M.A., MARTIN R.J. Mechanism for substance P-induced relaxation of precontracted airway smooth muscle during development. Am. J. Physiol. 1999;276:L51–L56. doi: 10.1152/ajplung.1999.276.1.L51. [DOI] [PubMed] [Google Scholar]
- MOFFATT J.D., COCKS T.M. Endothelium-dependent and -independent responses to protease-activated receptor-2 (PAR-2) activation in mouse isolated renal arteries. Br. J. Pharmacol. 1998;125:591–594. doi: 10.1038/sj.bjp.0702157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLINO M., WOOLKALIA M.J., REAVEY-CANTWELL J., PRATICO D., ANDRADE-GORDON P., BARNATHAN E.S., BRASS L.F. Endothelial cell thrombin receptors and PAR-2. J. Biol. Chem. 1997;272:11133–11141. doi: 10.1074/jbc.272.17.11133. [DOI] [PubMed] [Google Scholar]
- MOORE P.K., AL-SWAYEH O.A., CHONG N.W.S., EVANS R.A., GIBSON A. L-NG-nitro arginine (L-NOARG), a novel, L-arginine-reversible inhibitor of endothelium-dependent vasodilatation in vitro. Br. J. Pharmacol. 1990;99:408–412. doi: 10.1111/j.1476-5381.1990.tb14717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NY L., EKSTRÖM P., LARSSON B., GRUNDEMAR L., ANDERSSON K.E. Localization and activity of haem oxygenase and functional effects of carbon monoxide in the feline lower oesophageal sphincter. Br. J. Pharmacol. 1996;118:392–399. doi: 10.1111/j.1476-5381.1996.tb15415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYSTEDT S., EMILSSON K., WAHLESTEDT C., SUNDELIN J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POTEMPA J., KORZUS E., TRAVIS J. The serpin superfamily of proteinase inhibitors: Structure, function, and regulation. J. Biol. Chem. 1994;269:15957–15960. [PubMed] [Google Scholar]
- REES D.D., PALMER R.M.J., SCHULTZ R., HODSON H.F., MONCADA S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAIFEDDINE M., AL-ANI B., CHENG C.-H., WANG L., HOLLENBERG M.D. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br. J. Pharmacol. 1996;118:521–530. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZAREK J.L., SPURLOCK B., GRUETTER C.A., LEMKE S. Substance P and capsaicin release prostaglandin E2 from rat intrapulmonary bronchi. Am. J. Physiol. 1998;275:L1006–L1012. doi: 10.1152/ajplung.1998.275.5.L1006. [DOI] [PubMed] [Google Scholar]
- VERGNOLLE N., MACNAUGHTON W.K., AL-ANI B., SAIFEDDINE M., WALLACE J.L., HOLLENBERG M.D. Proteinase-activated receptor 2 (PAR2)-activating peptides: Identification of a receptor distinct from PAR2 that regulates intestinal transport. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7766–7771. doi: 10.1073/pnas.95.13.7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VU T.-K.H., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- WHISSTOCK J., SKINNER R., LESK A.M. An atlas of serpin conformations. Trends Biochem. Sci. 1998;23:63–67. doi: 10.1016/s0968-0004(97)01172-9. [DOI] [PubMed] [Google Scholar]
- WRIGHT H.T. The stuctural puzzle of how serpin serine proteinase inhibitors work. BioEssays. 1996;18:453–464. doi: 10.1002/bies.950180607. [DOI] [PubMed] [Google Scholar]
- XU W-F., ANDERSEN H., WHITMORE T.E., PRESNELL S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., FOSTER D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]